

Abstract
Varicella zoster virus (VZV) is a highly neurotropic human herpesvirus. Primary infection usually causes varicella (chicken pox), after which virus becomes latent in ganglionic neurons along the entire neuraxis. VZV reactivation results in zoster (shingles) which is frequently complicated by chronic pain (postherpetic neuralgia). VZV reactivation also causes meningoencephalitis, myelitis, ocular disorders, and vasculopathy, all of which can occur in the absence of rash. This review focuses on the association of VZV and stroke, and on the widening spectrum of disorders produced by VZV vasculopathy in immunocompetent and immunocompromised individuals, including recipients of varicella vaccine. Aside from ischemic stroke, VZV infection of cerebral arteries may lead to development of intracerebral aneurysms, with or without hemorrhage. Moreover, recent clinical-virological case reports and retrospective pathological-virological analyses of temporal arteries positive or negative for giant cell arteritis (GCA) indicate that extracranial VZV vasculopathy triggers the immunopathology of GCA. While many patients with GCA improve after corticosteroid treatment, prolonged corticosteroid use may potentiate VZV infection, leading to fatal vasculopathy in the brain and other organs.
Keywords: Zoster, Stroke, Varicella zoster virus vasculopathy, Aneurysm, Giant cell arteritis
Introduction
Stroke associated with varicella zoster virus (VZV) was first described in 1896 [1•]. It is now known that VZV vasculopathy is caused by productive VZV infection of cerebral arteries, most often leading to ischemic as well as hemorrhagic stroke, aneurysm with and without subarachnoid and intracerebral hemorrhage, dolichoectasia, dissection, and venous sinus thrombosis. The clinical diagnosis of VZV vasculopathy is verified by the detection of VZV DNA in CSF, the presence of anti-VZV IgG or anti-VZV IgM antibody in CSF, or the presence of anti-VZV IgM antibody in serum. In recent years, the spectrum of intracerebral VZV vasculopathy has expanded to include extracranial VZV vasculopathy as a cause of giant cell arteritis (GCA), and multiple epidemiological analyses have revealed an increased risk of stroke after zoster.
VZV Vasculopathy
VZV, a highly neurotropic member of the herpesvirus family, is the only human virus that has been shown to replicate in arteries and produce disease. VZV vasculopathy is characterized by clinical features of transient ischemic attacks or stroke. Magnetic resonance imaging (MRI) of VZV vasculopathy typically shows ischemic lesions, some of which enhance, primarily at gray-white matter junctions. Angiography reveals large vessel stenosis and post-stenotic dilatation. Recently, high-resolution MRI (HRMR) analysis of six patients with virologically verified VZV vasculopathy revealed various patterns of stenosis, vessel wall thickening, and enhancement, predominantly in terminal internal carotid artery segments and the M1 segment of the middle cerebral arteries; importantly, follow-up HRMR at multiple times after treatment showed improvement of stenosis, with reduced enhancement and reduced vessel wall thickening, indicating the potential value of HRMR in the management of VZV vasculopathy [2•].
A study of 30 subjects with virologically confirmed VZV vasculopathy [3] revealed rash in 63 % and CSF pleocytosis in 67 %, brain MRI or CTabnormalities in 97 %, and four-vessel angiography or MRA abnormalities in 70 %; large and small arteries were involved in 50 %, small arteries in 37 %, and large arteries in only 13 % of subjects The CSF of only 30 % of subjects contained VZV DNA, whereas 93 % had anti-VZV IgG antibody in CSF, with a reduced serum/CSF ratio of anti-VZV IgG that confirmed intrathecal synthesis of anti-VZV IgG. While both PCR and detection of antibody to VZV in CSF are highly specific, detection of anti-VZV IgG antibody in CSF is the most reliable test to diagnose VZV vasculopathy [4]. Importantly, diagnosis of this cause of stroke, which is treatable with intravenous acyclovir, is often missed due to no history of zoster rash in 1/3 of subjects, normal CSF in 1/3 of subjects, an average 4.2-month delay from zoster to neurological symptoms and signs, and the frequent absence of VZV DNA in CSF [5].
Association of VZV and Stroke
While the exact incidence of VZV vasculopathy is unknown, recent epidemiological studies from Taiwan, Denmark, and the UK have all revealed an increased risk of stroke after zoster [6]. Multiple studies from the UK, Europe, and Asia have shown that stroke incidence after zoster is greater than in age-matched control subjects. Analysis of the Taiwan National Health Research Institute records revealed a 30 % increased risk of stroke within 1 year after zoster and a fourfold increased risk after ophthalmic-division zoster [7]. Equivalent studies of records from the Danish national registry indicated a 126 % increased risk of stroke within 2 weeks after zoster, a 17 % increased risk from 2 weeks to 1 year after zoster, and a 5 % increased risk of stroke after the first year [8]. Studies from the UK Health Improvement Network General Practice Database showed that transient ischemic attacks and myocardial infarctions were greater in patients with zoster; unexpectedly, the increased risk was greatest in zoster patients under age 40. The mechanism(s) of VZV-associated transient ischemic attacks and myocardial infarctions are unknown but most likely involves virus effect, as well as immune-mediated pathological vascular remodeling. The most recent analysis of the UK General Practice Research Database [9] showed a decreasing risk over time after zoster in all dermatomes, with statistically significant age-adjusted incidences of stroke found at 1–4 weeks after zoster (1.63), 5–12 weeks after zoster (1.42), and 13–26 weeks after zoster (1.23), but no increase at later times. In patients with ophthalmic-distribution zoster, the risk of stroke was increased threefold at 5–12 weeks after zoster. Finally, among 55 % of zoster patients who received oral antiviral therapy, the stroke risk was reduced compared to that in untreated zoster patients [10], indicating the value of antiviral treatment in reducing stroke incidence after zoster.
Pathogenesis
After reactivation from cranial nerve ganglia, VZV likely spreads transaxonally to the outermost adventitial layer of the artery wall; infected cerebral arteries contain a thickened intima composed of myofibroblasts, a disrupted internal elastic lamina, and a paucity of smooth muscle cells [11]. Inflammatory cells (primarily CD4 and CD8 T cells and CD68 macrophages) are present predominantly in the adventitia and, to a lesser degree, in the luminal surface of the thickened intima [12]. Early VZV vasculopathy reveals a striking number of neutrophils in the adventitia. A remarkable finding in a VZV-infected artery is the association of inflammation with a thickened intima, supporting findings in the cardio-and pulmonary-vascular fields that inflammation is heavily involved in vascular remodeling.
Cerebral Aneurysm Due to VZV Vasculopathy
Not surprisingly, VZV vasculopathy can lead to cerebral aneurysm, with or without hemorrhage. A CNS aneurysm produced by VZV infection was initially reported in a 24-year-old healthy woman who developed herpes zoster ophthalmicus (HZO) with ipsilateral peripheral facial palsy and contralateral hemiplegia [13]. An initial angiogram was normal, but 4 weeks after onset, she developed decreased hearing and left-sided Horner’s syndrome. Repeat angiography revealed an aneurysm in the intrapetrosal region of the left internal carotid artery. Two other described cases of aneurysms after HZO were in a 67-year-old man who developed a right anterior choroidal artery aneurysm 3 weeks after right HZO [14] and in a 33-year-old woman who developed a left anterior cerebral artery aneurysm 4 months after HZO [14]. The first case demonstrating VZV antigen and viral particles in the aneurysmal wall was in a 70-year-old man who developed painful skin lesions in the occipital region and died 2 months later ostensibly due to cardiac arrest and subarachnoid hemorrhage; postmortem examination of the single fusiform basilar artery aneurysm revealed granulomatous angiitis, with positive immunohistochemical staining for VZV antigen in the wall of the aneurysm and electron microscopic detection of herpesvirus particles [15]. A second study demonstrating VZV in aneurysm walls was of a 6-year-old HIV-positive girl who presented with subarachnoid hemorrhage with aneurysms in the internal carotid, middle cerebral, anterior cerebral, and basilar arteries; postmortem examination revealed VZV antigen in dilated vessels and smaller vessels of the brain parenchyma [16]. Five additional cases of aneurysm caused by VZV infection in patients with HIV were subsequently reported [17–19]. Another report described a 42-year-old renal transplant recipient who developed a vertebral artery aneurysm with dissection; VZV DNA was present in CSF, and the patient was treated successfully with embolization and acyclovir [20]. A more recent case was a 6-year-old girl who presented with varicella followed 1 month later by a left basal ganglia infarction; 3 years later, she developed a saccular anterior communicating artery aneurysm, with increased anti-VZV IgG in CSF [21]. Most remarkable is the recent report of a patient with zoster in multiple dermatomes who presented with a severe headache and a normal magnetic resonance angiogram but, 2 months later, displayed nine anterior circulation aneurysms upon four-vessel digital subtraction angiography; antiviral treatment resulted in clinical improvement, size reduction of most aneurysms, and complete resolution of the two largest aneurysms (Fig. 1) [22•].
Fig. 1.
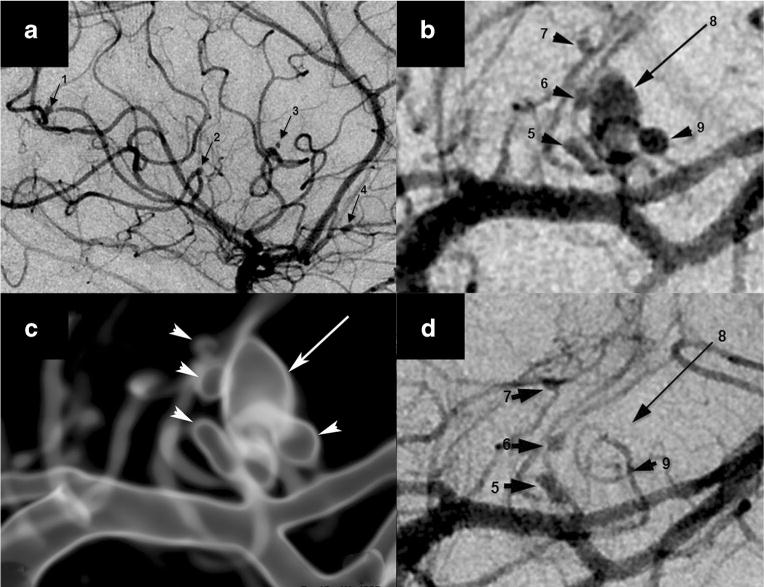
Nine aneurysms in varicella zoster virus vasculopathy. Digital subtraction angiography (DSA) demonstrated a total of nine mycotic aneurysms, four of which arose from the right middle cerebral artery (MCA) M3 segments (a, arrows 1–4) in the sylvian fissure and a cluster of left MCA lateral lenticulostriate perforator aneurysms, configured as a dominant oval aneurysm (b, long arrow 8), surrounded by four smaller lesions (b, arrowheads 5–7 and 9). A translucent rendered model from an angiographic 3D spin acquisition (c) further illustrates the complex arrangement of the left lateral lenticulostriate aneurysms. Repeat DSA after antiviral treatment and corona radiata perforator infarction showed interval thrombosis of the dominant aneurysm (d, long arrow 8) and one of the satellite lesions (d, arrowhead 9) that are no longer visible. Permission from Wolters Kluwer Health, Inc., Neurology 2014;82:2139–41
Vaccine-Associated VZV Vasculopathy
Although both the Varivax vaccine used to prevent varicella and the Zostavax vaccine to prevent zoster contain live attenuated virus, VZV vasculopathy does not occur after administration of Zostavax in adults. The likely reason is that vasculopathy develops after reactivation of virus latent in ganglia, followed by transaxonal spread to cerebral arteries; because Zostavax is administered to adults who have immunity to VZV, the attenuated VZV in the vaccine does not become latent. In contrast, Varivax VZV does become latent in children who are not immune to VZV, with the potential to reactivate and cause zoster and other neurological disorders, including stroke. Not surprisingly, multiple reports describe zoster and meningitis [23–26] as well as VZV vasculopathy in Varivax recipients, with two immunocompetent children developing ischemic stroke 5 days and 3 weeks, respectively, after varicella immunizations [27], and a third case in a 6-year-old boy immunocompromised by DOCK8 deficiency who developed a multifocal large artery vasculopathy involving both anterior cerebral arteries and the right posterior and left middle cerebral arteries; PCR amplified VZV DNA in his CSF [28].
Association of Giant Cell Arteritis with VZV Infection
The first report linking VZV with GCA was of a 77-year-old woman who developed right-sided temporal headache and ophthalmic-distribution zoster. Two months later, when temporal pain recurred along with a bilateral internuclear ophthalmoplegia and right middle cerebral artery stroke, a temporal artery (TA) biopsy was positive for GCA, and in situ hybridization detected VZV DNA in the GCA-positive TA [29]. The same group then described an 80-year-old man who developed right-sided ophthalmic-distribution zoster and granulomatous anterior uveitis, followed 4 months later by temporal headache and loss of vision; a right TA biopsy was GCA-positive, and immunohistochemistry detected VZV antigen in the GCA-positive TA [30]. A few years later, a third report described a 74-year-old woman who developed right temple pain and jaw claudication, followed 5 days later by blurred vision in the right eye; 3 days later, erythrocyte sedimentation rate (ESR) was elevated and she developed right-sided ophthalmic-distribution zoster 4 h after a TA biopsy. Although clinical and laboratory features were typical of GCA, the TA biopsy was GCA-negative but did reveal inflammation in the adventitia [31], as recently found in many patients with suspected GCA [32••].
The link between VZV and GCA was further established in three patients who presented with ischemic optic neuropathy (ION) that led to a TA biopsy. GCA pathology was not found in any of the TAs, but VZV antigen was present in the TAs of all three patients. The first patient was an 80-year-old man with left ophthalmic-distribution zoster, followed 1 month later by a left ION. Ipsilateral TA biopsy revealed extensive inflammation that was mistakenly identified as GCA. He was treated with steroids but did not improve. After the diagnosis of VZV vasculopathy was confirmed virologically, he was treated with intravenous acyclovir and his vision improved [33]. The second patient was a 75-year-old woman who presented with clinical symptoms and signs of a left ION and no past history of zoster [34]. ESR and C-reactive protein (CRP) were elevated. She was treated with intravenous methylprednisolone followed by oral prednisone without improvement. The left TA revealed no pathological abnormalities, and brain MRI with gadolinium was negative. On day 9, pain and vision worsened. On day 11, orbital CT and head CT angiography were negative. On day 15, visual acuity was 20/400 OS, with relative left afferent pupillary defect. On day 17, she was blind OS and the left pupil did not react to light; fundus examination was obscured by vitreous hemorrhage. CSF contained 8 WBCs/mm3, protein 72 mg%, and glucose 54 mg%. A diagnosis of VZV ION was made, and she was treated with intravenous acyclovir, 10 mg/kg q8h for 7 days. On day 31, the CSF contained anti-VZV IgG but not anti-HSV IgG antibody, and the serum-to-CSF ratio of anti-VZV IgG was reduced compared to ratios for total IgG and albumin. Immunohistochemistry and pathology revealed VZV antigen and neutrophils in the original left TA specimen, and she was treated with oral valacyclovir, 1 g TID for 6 weeks; prednisone was reduced to 20 mg daily and tapered 5 mg/week. Six weeks later, pain resolved, and visual acuity improved to finger-counting. The left optic nerve was pale, with clear margins and resolution of hemorrhage. The third patient was a 54-year-old diabetic woman who developed scalp pain, jaw claudication, and an ION followed by acute retinal necrosis and multiple areas of focal venous beading [35]. Vitreous fluid contained amplifiable VZV DNA but not herpes simplex virus-1 (HSV-1), cytomegalovirus, or toxoplasma DNA. TA biopsy was pathologically negative for GCA but contained VZV antigen. In all three patients, multifocal VZV vasculopathy manifested clinically as ION, and VZV was present in the ipsilateral TA. Only one of the three patients had a recent history of zoster. Importantly, the vision of two patients failed to improve with steroids but did improve after antiviral treatment.
To further address the incidence of VZV infection in GCA biopsy-negative patients, we examined archived biopsy-negative TAs from subjects with clinically suspected GCA for the presence of VZV antigen. Immunohistochemical analysis of formalin-fixed, paraffin-embedded (FFPE) TAs that were pathologically negative for GCA as well as normal TAs revealed VZV, but not HSV-1 antigen, in 5 (21 %) of 24 TAs from patients with clinically suspect but biopsy-negative GCA [36], while 13 normal TAs contained neither VZV nor HSV-1 antigen. Importantly, all five subjects whose TAs contained VZV antigen presented with clinical and laboratory features of GCA and early visual disturbances, reinforcing the notion that multifocal VZV vasculopathy can present with the full spectrum of features that characterize GCA.
Further direct evidence that VZV infection triggers the inflammatory cascade characteristic of GCA came from analysis of FFPE GCA-positive TA biopsies, including adjacent skeletal muscle, and normal TA biopsies from subjects >50 years of age for the presence and distribution of VZV antigen. VZV antigen was detected immunohistochemically in 61/82 (74 %) GCA-positive TAs (Fig. 2) compared with 1/13 (8 %) normal TAs, and most GCA-positive TAs contained viral antigen in skip areas [37••]. The most likely explanation for the 21 TAs with no VZV antigen is that VZV may have been missed because it is present in skip lesions. VZV antigen was present mostly in adventitia, followed by media and intima. Hematoxylin-eosin staining revealed VZV antigen in 12/32 (38 %) skeletal muscles adjacent to VZV antigen-positive TAs. While formalin fixation can interfere with PCR amplification of DNA extracted from fixed tissue, PCR was still able to detect VZV DNA in 18/45 (40 %) GCA-positive VZV antigen-positive TAs, in 6/10 (60 %) VZV antigen-positive skeletal muscles, and in one VZV antigen-positive normal TA. Ultrastuctural analysis revealed VZ virions in a GCA-positive TA (Fig. 2). GCA pathology in sections adjacent to those containing VZV was seen in 89 % of GCA-positive TAs, but in none of 18 adjacent sections from normal TA. Most GCA-positive TAs contained VZV in skip areas that correlated with adjacent GCA pathology, supporting the notion that VZV triggers GCA immunopathology.
Fig. 2.
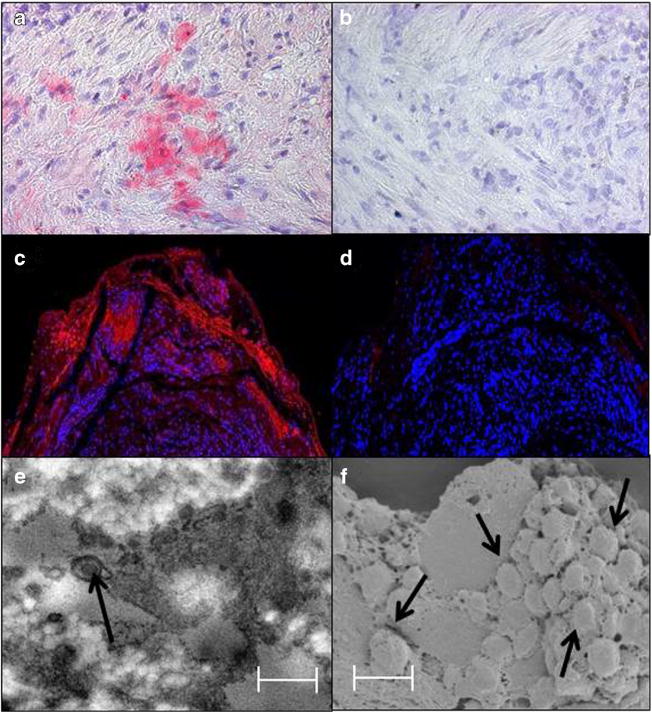
Immunofluorescence staining and ultrastructural imaging of a varicella zoster virus-infected temporal artery. Immunohistochemical staining with rabbit anti-VZV IE63 antibody revealed VZV antigen in the media (a, pink color), but not after staining with rabbit anti-HSV-1 antibody (b). Immunofluorescence staining with a different mouse anti-VZV IgG antibody than that used in panel a revealed VZV antigen in the adventitia (c, red color), but not when primary antibody was omitted (d). Transmission electron microscopy of sections adjacent to those containing VZV antigen revealed an enveloped virus particle (e, arrow), while both scanning and transmission electron microscopy of these sections showed a cluster of virus particles in the adventitia egressing through an outer cell wall (f, arrows). Virus particles appear slightly larger than 200 nm because they were sputter-coated with a gold alloy. In panels e and f, scale bars = 300 nm. Permission from Wolters Kluwer Health, Inc., Neurology 2015;84:1948–55
The association between GCA and TA infection by VZV was further analyzed in archival TAs of patients with clinically suspected GCA whose TAs were histopathologically negative and in normal TAs removed postmortem from age-matched subjects. Archival FFPE TA sections were examined immunohistochemically, while regions adjacent to those containing VZV were examined by hematoxylin-eosin staining. Immunohistochemistry identified inflammatory cells and cell types around nerve bundles containing VZV. VZV antigen was detected in 45/70 (64 %) GCA-negative TAs compared with 11/49 (22 %) normal TAs; since VZV is present in skip lesions, VZV antigen may have been missed in the other 36 % of TAs. Furthermore, extension of our earlier study revealed VZV antigen in 68/93 (73 %) of GCA-positive TAs compared with 11/49 (22 %) normal TAs [32••]. VZV antigen was more likely to be present in adventitia of both GCA-negative TAs and GCA-positive TAs than in normal TA adventitia. In GCA-negative subjects whose TAs contained VZV antigen, adventitial inflammation was seen in 36 % adjacent to viral antigen, but not in any normal TAs. Overall, the prevalence of VZV in TAs of subjects with clinically suspected GCA was independent of whether they were GCA-negative or GCA-positive pathologically.
Corticosteroids and VZV Vasculopathy
It is important for clinicians to recognize that prolonged steroid use is associated with increased VZV reactivation and vasculopathy. The risk of zoster was reportedly increased in 1465 systemic lupus erythematosus patients treated with prednisone [38] and in 60 patients with autoimmune diseases when corticosteroids were added to cyclophosphamide [39]; additional treatment with prednisone in 28,852 rheumatoid arthritis patients treated previously or currently with methotrexate increased the risk of zoster by 81 % [40]. Determining the role of corticosteroids on VZV antigen expression in GCA-positive and GCA-negative TAs in prior studies above was not possible since all TAs were de-identified.
Prolonged corticosteroid treatment has been shown to lead to serious complications of zoster. For example, a 50-year-old diabetic woman who had been treated for months with prednisolone developed zoster and, upon continued corticosteroid therapy, disseminated zoster followed by fatal VZV meningoencephalitis [41]. More recently, prolonged corticosteroid use in a 60-year-old man with thoracic-distribution zoster reportedly led to impaired wound healing, with VZV DNA found in the ulcerated skin 3 months after zoster and sudden death 2 months later; at autopsy, VZV antigen was widespread in organs and arteries, most abundant in the coronary arteries, aorta, and bundle of His, raising the possibility of VZV-induced fatal arrhythmia. Although no neurological disease developed, VZV antigen was also found in his posterior cerebral artery, revealing for the first time subclinical VZV vasculopathy [42].
While corticosteroids are the mainstay of treatment for GCA, a 75-year-old woman with pathologically verified GCA in both TAs continued to deteriorate and developed a fatal myelopathy after treatment with high-dose corticosteroids [43]. Postmortem examination revealed spinal cord infarction secondary to extensive necrotizing granulomatous arteritis of spinal arteries. Further studies based on the association between VZV infection and GCA revealed VZV antigen in the TA, aorta, left carotid artery, and an unidentified artery from the circle of Willis; the presence of VZV in the cerebral artery was further confirmed by the detection of VZV DNA in the same section containing VZV antigen [44]. Overall, prolonged corticosteroid use was shown to potentiate VZV infection.
Conclusion
In the past few years, the spectrum of VZV vasculopathy has expanded to include stroke in immunocompetent and immunocompromised children vaccinated with attenuated VZV, the development of multiple intracranial aneurysms, and extracranial arterial infection manifesting as GCA. High-resolution MRI has been shown to assist in diagnosis and treatment of VZV vasculopathy (Fig. 3), while prolonged corticosteroid treatment of GCA can potentiate VZV infection, leading to fatal VZV vasculopathy.
Fig. 3.
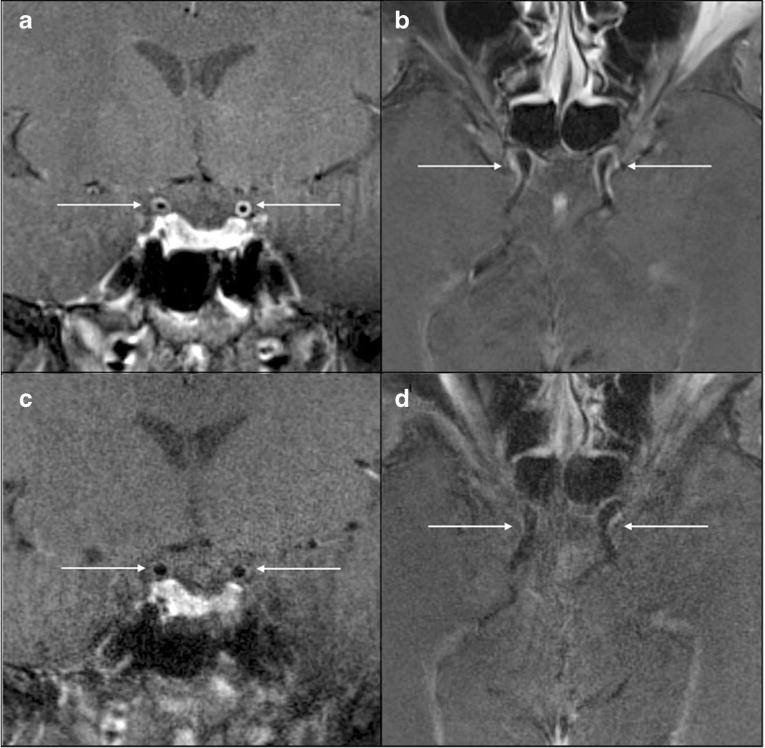
Contrast-enhanced high-resolution MRI (HRMR) over time in a patient with VZV vasculopathy. At presentation, coronal (a) and axial (b) HRMR revealed vessel wall thickening and enhancement of both terminal ICA segments; 3 months later, coronal (c) and axial (d) HRMR showed resolution of thickening and enhancement. Permission from Elsevier, J Neurol Sci 2015;351;168–73
Acknowledgments
The study was supported by grants from the National Institutes of Health (NS093716 to Dr. Gilden, AG032958 to Dr. Gilden and Dr. Nagel, and NS094758 to Dr. Nagel). We thank Marina Hoffman for the editorial review and Cathy Allen for the manuscript preparation.
Don Gilden has received the following grants: NIH/NIA PPG, Molecular Pathogenesis of Varicella Zoster Virus Infection, and NIH/NINDS R01, Neurobiology of Varicella Zoster Virus.
Footnotes
This article is part of the Topical Collection on Infection
Conflict of Interest Maria A. Nagel has received the following grant: NIH/NINDS R01, Purinergic Signaling in Varicella Zoster Virus Vasculopathy.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1•.Baudouin E, Lantuejoul P. Les troublecas moteurs dans le zona. Gazette des Hopitaux. 1919 The first description of stroke associated with VZV. [Google Scholar]
- 2•.Cheng-Ching E, Jones S, Hui FK, et al. High-resolution MRI vessel wall imaging in varicella zoster virus vasculopathy. J Neurol Sci. 2015;351:168–73. doi: 10.1016/j.jns.2015.02.017. A study demonstrating decreased vessel wall thickening and decreased enhancement after antiviral treatment of VZV vasculopathy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagel MA, Cohrs RJ, Mahalingam R, et al. The varicella zoster virus vasculopathies: clinical, CSF, imaging and virological features. Neurology. 2008;70:853–60. doi: 10.1212/01.wnl.0000304747.38502.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagel MA, Forghani B, Mahalingam R, et al. The value of detecting anti-VZV IgG antibody in CSF to diagnose VZV vasculopathy. Neurology. 2007;68:1069–73. doi: 10.1212/01.wnl.0000258549.13334.16. [DOI] [PubMed] [Google Scholar]
- 5.Gilden D, Nagel MA, Cohrs RJ, Mahalingam R. The variegate neurological manifestations of varicella zoster virus infection. Curr Neurol Neurosci Rep. 2013;13:374. doi: 10.1007/s11910-013-0374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin HC, Cw C, Ho JD. Herpes zoster ophthalmicus and the risk of stroke: a population-based follow-up study. Neurology. 2010;74:792–7. doi: 10.1212/WNL.0b013e3181d31e5c. [DOI] [PubMed] [Google Scholar]
- 7.Kang JH, Ho JD, Chen YH, et al. Increased risk of stroke after a herpes zoster attack: a population-based follow-up study. Stroke. 2009;40:3443–8. doi: 10.1161/STROKEAHA.109.562017. [DOI] [PubMed] [Google Scholar]
- 8.Sreenivasan N, Basit S, Wohlfahrt J, et al. The short- and long-term risk of stroke after herpes zoster—a nationwide population-based cohort study. PLoS One. 2013;8:e69156. doi: 10.1371/journal.pone.0069156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breuer J, Pacou M, Gauthier A, Brown MM. Herpes zoster as a risk factor for stroke and TIA: a retrospective cohort study in the UK. Neurology. 2014;82:206–12. doi: 10.1212/WNL.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langan SM, Minassian C, Smeeth L, Thomas SL. Risk of stroke following herpes zoster: a self-controlled case-series study. Clin Infect Dis. 2014;58:1497–503. doi: 10.1093/cid/ciu098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagel MA, Traktinskiy I, Azarkh Y, et al. Varicella zoster virus vasculopathy: analysis of virus-infected arteries. Neurology. 2011;77:364–70. doi: 10.1212/WNL.0b013e3182267bfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagel MA, Traktinskiy I, Stenmark KR, Frid MG, Choe A, Gilden D. Varicella zoster virus vasculopathy: immune characteristics of virus-infected arteries. Neurology. 2013;80:62–8. doi: 10.1212/WNL.0b013e31827b1ab9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gursoy G, Aktin E, Bahar S, et al. Post-herpetic aneurysm in the intrapetrosal portion of the internal carotid artery. Neuroradiology. 1980;9:279–82. doi: 10.1007/BF00347809. [DOI] [PubMed] [Google Scholar]
- 14.O’Donohue JM, Enzmann DR. Mycotic aneurysm in angiitis associated with herpes zoster ophthalmicus. AJNR Am J Neuroradiol. 1987;8:615–9. [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumoto S, Kinjo M, Hokamura K, et al. Subarachnoid hemorrhage and granulomatous angiitis of the basilar artery: demonstration of the varicella-zoster virus in the basilar artery lesions. Stroke. 1986;17:1024–8. doi: 10.1161/01.str.17.5.1024. [DOI] [PubMed] [Google Scholar]
- 16.Fulmer BB, Dillard SC, Musulman EM, et al. Two cases of cerebral aneurysms in HIV+ children. Pediatr Neurosurg. 1998;28:31–4. doi: 10.1159/000028615. [DOI] [PubMed] [Google Scholar]
- 17.Saraya T, Shimura C, Wada H, et al. Evidence for vascular spread of varicella zoster-associated vasculopathy. Ann Intern Med. 2006;144:535–7. doi: 10.7326/0003-4819-144-7-200604040-00022. [DOI] [PubMed] [Google Scholar]
- 18.de Broucker T, Verollet D, Schoindre Y, et al. Cerebral vasculitis with aneurysms caused by varicella-zoster virus infection during AIDS: a new clinicoangiographical syndrome. Rev Neurol (Paris) 2008;164:61–71. doi: 10.1016/j.neurol.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Yasuda C, Okada K, Ohnari N, et al. Cerebral infarction and intra-cranial aneurysm related to the reactivation of varicella zoster virus in a Japanese acquired immunodeficiency syndrome (AIDS) patient. Rinsho Shinkeigaku. 2013;53:701–5. doi: 10.5692/clinicalneurol.53.701. [DOI] [PubMed] [Google Scholar]
- 20.Bhayani N, Ranade P, Clark NM, et al. Varicella-zoster virus and cerebral aneurysm: case report and review of the literature. Clin Infect Dis. 2008;47:e1–3. doi: 10.1086/588842. [DOI] [PubMed] [Google Scholar]
- 21.Kawatani M, Nakai A, Okuno T, et al. A case of intracranial saccular aneurysm after primary varicella zoster virus infection. Brain Dev. 2012;34:80–2. doi: 10.1016/j.braindev.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 22•.Liberman AL, Nagel MA, Hurley MC, et al. Rapid development of nine cerebral aneurysms in varicella zoster virus vasculopathy. Neurology. 2014;82:2139–41. doi: 10.1212/WNL.0000000000000503. A study of aneurysms associated with VZV vasculopathy and improvement with antiviral therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wise RP, Salive ME, Braun MM, et al. Postlicensure safety surveillance for varicella vaccine. JAMA. 2000;284:1271–9. doi: 10.1001/jama.284.10.1271. [DOI] [PubMed] [Google Scholar]
- 24.Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella-zoster virus vaccine. J Infect Dis. 2008;198:1444–7. doi: 10.1086/592452. [DOI] [PubMed] [Google Scholar]
- 25.Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792–5. doi: 10.1016/j.annemergmed.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 26.Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266–8. doi: 10.1097/INF.0b013e3181f63cf9. [DOI] [PubMed] [Google Scholar]
- 27.Wirrell E, Hill MD, Jadavji T, et al. Stroke after varicella vaccination. J Pediatr. 2004;145:845–7. doi: 10.1016/j.jpeds.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Sabry A, Hauk PJ, Jing H, et al. Vaccine strain varicella-zoster virus-induced central nervous system vasculopathy as the presenting feature of DOCK8 deficiency. J Allergy Clin Immunol. 2014;133:1225–7. doi: 10.1016/j.jaci.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Abdulla NA, Rismondo V, Minkowski JS, et al. Herpes zoster vasculitis presenting as giant cell arteritis with bilateral internuclear ophthalmoplegia. Am J Ophthalmol. 2002;134:912–4. doi: 10.1016/s0002-9394(02)01811-1. [DOI] [PubMed] [Google Scholar]
- 30.Al-Abdulla NA, Kelley JS, Green WR, et al. Herpes zoster vasculitis presenting as giant cell arteritis with choroidal infarction. Retina. 2003;23:567–9. doi: 10.1097/00006982-200308000-00027. [DOI] [PubMed] [Google Scholar]
- 31.de Castro LE, Peterson AM, Givre SJ, et al. Herpes zoster ophthalmicus: presenting as giant-cell arteritis. Clin Experiment Ophthalmol. 2005;33:636–8. doi: 10.1111/j.1442-9071.2005.01111.x. [DOI] [PubMed] [Google Scholar]
- 32••.Nagel MA, White T, Khmeleva N, et al. Analysis of varicella-zoster virus in temporal arteries biopsy-positive and −negative for giant cell arteritis. JAMA Neurol. 2015;72:1281–7. doi: 10.1001/jamaneurol.2015.2101. An extensive, retrospective study of temporal artery biopsies showing that most GCA-negative and GCA-positive arteries contain VZV antigen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salazar R, Russman AN, Nagel MA, et al. Varicella zoster virus ischemic optic neuropathy and subclinical temporal artery involvement. Arch Neurol. 2011;68:517–20. doi: 10.1001/archneurol.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagel MA, Khmeleva N, Boyer PJ, et al. Varicella zoster virus in the temporal artery of a patient with GCA. J Neurol Sci. 2013;335:229–30. doi: 10.1016/j.jns.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathias M, Nagel MA, Khmeleva N, et al. VZV multifocal vasculopathy with ischemic optic neuropathy, acute retinal necrosis and temporal artery infection in the absence of zoster rash. J Neurol Sci. 2013;325:180–2. doi: 10.1016/j.jns.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagel MA, Bennett JL, Khmeleva N, et al. Multifocal VZV vasculopathy with temporal artery infection mimics giant cell arteritis. Neurology. 2013;80:2017–21. doi: 10.1212/WNL.0b013e318294b477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Gilden D, White T, Khmeleva N, et al. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology. 2015;84:1948–55. doi: 10.1212/WNL.0000000000001409. The first extensive, retrospective study of temporal artery biopsies showing a causal link between VZVand biopsy-positive giant cell arteritis, as evidenced by the presence of VZV antigen, VZV DNA and herpesvirus particles in the temporal artery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakravarty EF, Michaud K, Katz R, et al. Increased incidence of herpes zoster among patients with systemic lupus erythematosus. Lupus. 2013;22:238–44. doi: 10.1177/0961203312470186. [DOI] [PubMed] [Google Scholar]
- 39.Cavallasca JA, Costa CA, Mdel M, et al. Severe infections in patients with autoimmune diseases treated with cyclophosphamide. Reumatol Clin. 2015;11:221–3. doi: 10.1016/j.reuma.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Pappas DA, Hooper MM, Kremer JM, et al. Herpes zoster reactivation in patients with rheumatoid arthritis: analysis of disease characteristics and disease modifying anti-rheumatic drugs. Arthritis Care Res (Hoboken) 2015 doi: 10.1002/acr.22628. [DOI] [PubMed] [Google Scholar]
- 41.Tako J, Rado JP. Zoster meningoencephalitis in a steroid-treated patient. Arch Neurol. 1962;12:610–2. doi: 10.1001/archneur.1965.00460300058007. [DOI] [PubMed] [Google Scholar]
- 42.Nagel MA, Lenggenhager D, White T, et al. Disseminated VZV infection and asymptomatic VZV vasculopathy after steroid abuse. J Clin Virol. 2015;66:72–5. doi: 10.1016/j.jcv.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galetta SL, Balcer LJ, Lieberman AP, et al. Refractory giant cell arteritis with spinal cord infarction. Neurology. 1997;49:1720–3. doi: 10.1212/wnl.49.6.1720. [DOI] [PubMed] [Google Scholar]
- 44.Gilden D, White T, Galetta SL, et al. Widespread arterial infection by varicella zoster virus explains refractory giant cell arteritis. Neurol Neuroimmunol Neuroinflamm. 2015;2:3125. doi: 10.1212/NXI.00000000000000125.. [DOI] [PMC free article] [PubMed] [Google Scholar]