

SUMMARY
Skeletal muscle is a major site of postprandial glucose disposal. Inadequate insulin action in skeletal myocytes contributes to hyperglycemia in diabetes. While glucose is known to stimulate insulin secretion by β cells, whether it directly engages nutrient signaling pathways in skeletal muscle to maintain systemic glucose homeostasis remains largely unexplored. Here we identified the Baf60c-Deptor-AKT pathway as a target of muscle glucose sensing that augments insulin action in skeletal myocytes. Genetic activation of this pathway improved postprandial glucose disposal in mice, whereas its muscle-specific ablation impaired insulin action and led to postprandial glucose intolerance. Mechanistically, glucose triggers KATP channel-dependent calcium signaling that promotes HDAC5 phosphorylation and nuclear exclusion, leading to Baf60c induction and insulin-independent AKT activation. This pathway is engaged by the anti-diabetic drugs sulfonylureas to exert their full glucose-lowering effects. These findings uncover an unexpected mechanism of glucose sensing in skeletal myocytes that contributes to homeostasis and therapeutic action.
ETOC BLURB
Skeletal myocytes directly sense extracellular glucose concentrations and activate a signaling pathway to drive hormone-independent AKT activation. This glucose sensing pathway acts in concert with insulin to promote muscle glucose utilization and maintain systemic glucose homeostasis.
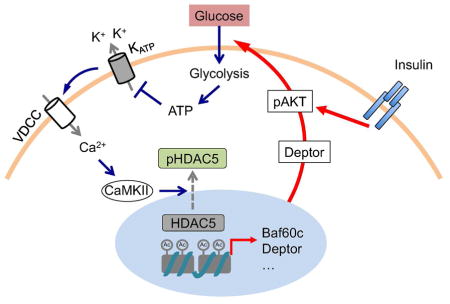
INTRODUCTION
Metabolic syndrome has become a global epidemic that markedly increases the risk for type 2 diabetes, cardiovascular disease, and non-alcoholic steatohepatitis. Impaired muscle insulin action is an early hallmark of obesity-associated metabolic disorders. Skeletal muscle insulin resistance has been associated with reduced mitochondrial oxidative function (Mootha et al., 2004; Patti et al., 2003; Petersen et al., 2003), an accumulation of intramyocellular lipids (Kelley et al., 2002; Samuel et al., 2010), and a shift from oxidative to glycolytic muscle fiber type in patients with type 2 diabetes (Lillioja et al., 1987; Simoneau et al., 1995; Simoneau and Kelley, 1997). These observations form the basis for the hypothesis that the oxidative to glycolytic shift of myofiber metabolism may be detrimental for muscle insulin sensitivity. However, the cause and effect relationship between skeletal myofiber energy metabolism and insulin sensitivity remains inconclusive (Muoio, 2010; Turner and Heilbronn, 2008). In fact, resistance training, which promotes the growth and function of fast-twitch glycolytic muscle, improves metabolic profiles in diabetic patients (Egan and Zierath, 2013; LeBrasseur et al., 2011). Further, genetic activation of glycolytic metabolism in skeletal muscle resulted in improved whole body glucose homeostasis in mice (Izumiya et al., 2008; Meng et al., 2013; Meng et al., 2014; Wilkes et al., 2009). As such, augmented glycolytic metabolism may instead represent an important aspect of the adaptive response in skeletal muscle when blood glucose levels are elevated.
Skeletal muscle is a major site of glucose disposal in the postprandial phase. The insulin-sensitive glucose transporter Glut4 is responsible for the bulk of glucose clearance from the circulation after a meal. Insulin triggers the activation of the PI3K-AKT signaling pathway, which mediates the pleiotropic effects of this hormone on nutrient metabolism, including insulin-stimulated glucose uptake (Leto and Saltiel, 2012; Taniguchi et al., 2006). A canonical cell type responsible for glucose sensing in the body is the pancreatic β cells. In response to rising blood glucose, the ATP-sensitive potassium (KATP) channel in β cells is inhibited, leading to membrane depolarization, elevation of cytosolic calcium, and insulin secretion (Newgard and McGarry, 1995; Nichols, 2006). Previous studies have shown that O-GlcNAc transferase (OGT) (Bond and Hanover, 2015; Erickson et al., 2013; Hardiville and Hart, 2014; Ruan et al., 2013), and ChREBP and MondoA together with their binding partner Mlx (Havula and Hietakangas, 2012; Postic et al., 2007; Uyeda and Repa, 2006) contribute to the transcriptional effects of glucose in diverse cell types, including skeletal muscle (Ahn et al., 2016; Stoltzman et al., 2008). However, whether glucose directly engages nutrient sensing pathways in skeletal myocytes to regulate glucose disposal remains largely unknown.
Skeletal myofibers are heterogeneous with regard to their contractile and metabolic properties. Recent work demonstrated that Baf60c, a subunit of the SWI/SNF chromatin-remodeling complex, as a core component of a regulatory network that drives glycolytic metabolism in skeletal myofibers (Meng et al., 2013; Meng et al., 2014). Baf60c recruits the SWI/SNF complexes to specific genomic loci and activates downstream target genes involved in glucose metabolism. Notably, Baf60c stimulates the expression of Deptor, an activator of AKT (Peterson et al., 2009), and promotes glycolysis in myocytes (Meng et al., 2014). Here, we discovered that glucose stimulates the Baf60c-Deptor-AKT signaling axis in skeletal myocytes through the KATP channel-dependent calcium signaling and the class IIa histone deacetylase HDAC5. Muscle-specific activation and ablation of this pathway in mice revealed a critical role of Baf60c-mediated glucose sensing in postprandial glucose metabolism and the extrapancreatic glucose-lowering effects of the anti-diabetic sulfonylurea drugs.
RESULTS
Glucose triggers the activation of the Baf60c-Deptor-AKT axis in skeletal myocytes
The Baf60 subunits endow the SWI/SNF chromatin-remodeling complexes with the ability to orchestrate context-dependent transcriptional responses. Baf60a coordinates the programs of hepatic fatty acid β-oxidation and bile acid synthesis (Li et al., 2008; Meng et al., 2015), whereas Baf60c regulates cardiac and skeletal myogenesis (Forcales et al., 2012; Lickert et al., 2004; Takeuchi and Bruneau, 2009), promotes glycolytic metabolism in skeletal muscle (Meng et al., 2013; Meng et al., 2014), and mediates nutritional regulation of lipogenesis (Wang et al., 2013). In skeletal muscle, Baf60c induces the expression of Deptor, a potent activator of AKT, and programs myocyte energy metabolism toward glycolysis (Meng et al., 2014). In mammals, peripheral tissues respond to nutritional and hormonal signals to fine-tune their metabolic functions and maintain homeostasis. Whether skeletal muscle engages the Baf60c-Deptor-AKT axis to sense nutrients in the circulation remains unknown.
To explore whether the Baf60c pathway is responsive to nutritional and hormonal cues, we analyzed mRNA and protein expression of Baf60c and Deptor in skeletal muscle from ad lib, fasted, and refed mice. As expected, AKT phosphorylation was reduced following overnight starvation (Figure 1A). The mRNA and protein levels of Baf60c were significantly decreased in quadriceps muscle from fasted mice and recovered to the pre-fasting levels following 16 h of refeeding. Deptor expression also exhibited a similar pattern of regulation in response to feeding status. In contrast, mRNA and protein expression of Glut4, an insulin-sensitive glucose transporter, remained largely unaltered by fasting and refeeding. To dissect the nutritional and hormonal signals involved, we treated differentiated C2C12 myotubes with factors known to fluctuate under different feeding conditions, including insulin, amino acids, fatty acids, and glucose. While insulin, amino acids, palmitic acid and oleic acid had modest effects on Baf60c expression, glucose treatment within its physiological range (4–10 mM) elicited a robust and dose-dependent increase in mRNA and protein levels of Baf60c and Deptor (Figure 1B–C). In contrast, Baf60a, Baf60b, Pgc-1α, Pgc-1β, and Glut1 mRNA expression remained largely unaltered by glucose treatment (Figure S1A). The induction of Baf60c and Deptor was accompanied by robust AKT activation in treated C2C12 myotubes, leading to increased phosphorylation of known AKT substrates, including FoxO1, AS160, and GSK3β (Figure 1C). Similarly, glucose also induced the expression of Baf60c and Deptor and stimulated AKT activation in cultured primary human myotubes (Figure 1D). In contrast, glucose treatment appeared to elicit modest impacts on Baf60c and Deptor expression and AKT phosphorylation in cultured mouse primary hepatocytes and differentiated brown adipocytes (Figure S1B–C). To further establish the relevance of Baf60c and Deptor in skeletal muscle glucose signaling, we treated ex vivo cultured EDL and plantaris muscles with different concentrations of glucose. Consistent with observations in cultured myotubes, glucose induced the mRNA expression of Baf60c and Deptor in a dose-dependent manner in EDL muscles (Figure 1E). Baf60c and Deptor protein levels and AKT phosphorylation were also markedly elevated in response to 10 mM glucose (Figure 1F). Glucose moderately increased Baf60c and Deptor mRNA levels in soleus muscles (Figure S1D), consistent with their enriched expression in glycolytic muscles (Meng et al., 2013). Together, these results demonstrate that the Baf60c-Deptor-AKT pathway is highly responsive to physiological concentrations of glucose in skeletal myocytes.
Figure 1. Glucose activates the Baf60c-Deptor-AKT axis in skeletal myocytes.

(A) qPCR analysis of gene expression (top) and immunoblots of total protein lysates (bottom) from quadriceps muscle. Data represent mean ± SEM (n=3–4). *p < 0.05, ***p < 0.001 Fasted vs. Fed; one-way ANOVA
(B) qPCR analysis of gene expression in C2C12 myotubes following treatments for 12 h. Veh, Vehicle; Ins, Insulin; AA, Amino Acids; FA, Fatty acids; Glc, Glucose; PA, Palmitic Acid; OA, Oleic Acid. Data represent mean ± SD. ***p < 0.001 vs. 1 mM glucose; one-way ANOVA.
(C, D) Immunoblots of total cell lysates from C2C12 myotubes (C) and primary human myotubes (D) treated with different concentrations of glucose for 12 h.
(E) qPCR analysis of gene expression in ex vivo cultured EDL muscle treated with different concentrations of glucose for 4 h. Data represent mean ± SEM (n=3). **p < 0.01, ***p < 0.001 vs. 1 mM glucose; one-way ANOVA
(F) Immunoblots of total protein lysates from ex vivo cultured plantaris muscle treated with glucose for 4 h.
See also Figure S1.
Baf60c-mediated nutrient sensing acts in concert with insulin to facilitate postprandial glucose disposal
We next examined whether glucose-induced AKT activation contributes to increased expression of Baf60c and Deptor in response to glucose. We treated differentiated C2C12 myotubes with 1 or 10 mM glucose in the absence or presence of 10 μM AKT1/2 inhibitor (AKTi). As expected, AKTi strongly inhibited AKT phosphorylation in treated myotubes (Figure 2A). Glucose-induced expression of Baf60c and Deptor remained largely unaffected by AKTi treatment. On the contrary, RNAi knockdown of Baf60c significantly impaired the induction of Deptor expression by glucose (Figure 2B). Glucose-induced AKT phosphorylation was also diminished in Baf60c knockdown myotubes (Figure 2C). These results demonstrate that AKT activation is downstream of Baf60c and Deptor following glucose stimulation.
Figure 2. Baf60c-mediated glucose sensing acts in concert with insulin to facilitate postprandial glucose disposal.

(A) Immunoblots of total cell lysates from C2C12 myotubes treated with 1 mM or 10 mM glucose in the absence or presence of 10 μM AKTi for 12 h.
(B) qPCR analysis of gene expression in control (siCtr) or Baf60c knockdown (siBaf) C2C12 myotubes treated with different concentrations of glucose for 12 h. Data represent mean ± SD. ***p < 0.001 siCtr vs. siBaf; two-way ANOVA.
(C) Immunoblots of total cell lysates from control and Baf60c knockdown C2C12 myotubes treated with different concentrations of glucose for 12 h.
(D) Glucose consumption and lactate production in C2C12 myotubes pretreated with different concentrations of glucose for 12 h, followed by switching to media containing 5 mM glucose for additional 12 h. Data represent mean ± SD. ***p < 0.001 vs. 1 mM; one-way ANOVA.
(E) Lactate production in ex vivo cultured muscles after chase with media containing 5 mM glucose. Data represent mean ± SEM (n=4). *p < 0.05, ***p < 0.001 vs. 1 mM; one-way ANOVA.
(F) Lactate production in ex vivo cultured muscles in the absence or presence of AKTi (10 μM). Data represent mean ± SEM (n=4). ***p < 0.001; two-way ANOVA.
(G) Clustering analysis of glucose-responsive genes in C2C12 myotubes. The red bar indicates a cluster of Baf60c-dependent glucose-responsive genes.
(H) Immunoblots of total cell lysates from C2C12 myotubes pretreated with 1 mM or 10 mM glucose for 12 h, followed by treatment with different concentrations of insulin for 30 min.
(I) Blood glucose levels in HFD-fed WT and Tg mice. Data represent mean ± SEM (n=5–7). **p < 0.01, ***p < 0.001; two-tailed unpaired Student’s t-test.
(J) Blood glucose and plasma insulin levels during OGTT. Data represent mean ± SEM (n=5–7). **p < 0.01; two-way ANOVA with multiple comparisons. Area under curve (AUC) was evaluated by two-tailed unpaired Student’s t-test.
(K) Blood glucose and plasma insulin levels in STZ-treated HFD-fed WT and Tg mice. Data represent mean ± SEM (n=5–7). **p < 0.01; two-tailed unpaired Student’s t-test.
(L) Postprandial blood glucose levels in mice as described in (K). Data represent mean ± SEM (n=5–7). **p < 0.01; two-way ANOVA with multiple comparisons. AUC was evaluated by two-tailed unpaired Student’s t-test.
See also Figure S1.
We next examined whether activation of myocyte glucose sensing pathway augments the glycolytic metabolism of C2C12 myotubes. Pre-treatment of C2C12 myotubes with glucose resulted in a dose-dependent increase in glucose consumption and lactate production after switching to 5 mM glucose during the chase period (Figure 2D). Further, glucose treatment strongly increased lactate production in ex vivo cultured plantaris, EDL, and soleus muscles in a dose-dependent manner (Figure 2E). The induction of lactate production by glucose was essentially abolished by AKTi, illustrating a critical role of AKT activation in mediating the stimulatory effects of glucose on glycolytic metabolism in muscle (Figure 2F). To test the possibility that Baf60c may play a broader role in myocyte glucose sensing, we performed microarray analysis on control (siCtr) and Baf60c (siBaf) knockdown myotubes following exposure to 1 or 10 mM glucose. We identified a cluster of glucose-responsive genes; 49 genes within this cluster required Baf60c to be fully induced by glucose (Figure 2G). As such, Baf60c appears to orchestrate the transcriptional response of a significant subset of glucose-regulated genes in skeletal myocytes.
Skeletal muscle is a major site of postprandial glucose disposal, a process mediated by insulin-stimulated glucose uptake. Insulin stimulates the PI3K-AKT signaling pathway and promotes the translocation of Glut4 to the plasma membrane of skeletal myocytes. To determine whether insulin and glucose cooperatively activate AKT phosphorylation, we treated differentiated C2C12 myotubes with 1 or 10 mM glucose in the presence of different concentrations of insulin. As shown in Figure 2H, insulin-stimulated AKT phosphorylation was significantly augmented under the high glucose conditions, suggesting that glucose may directly engage skeletal myocytes to promote AKT activation and augment postprandial glucose disposal. In support of this, high-fat diet (HFD)-fed muscle-specific Baf60c transgenic mice (Tg) had improved postprandial glycemic control following refeeding and during oral glucose tolerance test (OGTT), compared to wild type (WT) littermate controls (Figure 2I–J). Plasma insulin concentrations during OGTT remained similar. To dissect the relative contribution of Baf60c- and insulin-mediated AKT activation in glucose metabolism, we performed fasting/refeeding studies in WT and Tg mice following streptozotocin (STZ) treatments. Five consecutive injections of STZ (50 mg/kg) markedly reduced plasma insulin levels, leading to hyperglycemia in WT mice (Figure S1E). Similarly, while plasma insulin levels were low in STZ-treated WT and Tg mice, Tg group had significantly lower blood glucose levels under ad lib conditions and following overnight starvation (Figure 2K). Remarkably, Tg mice developed less pronounced postprandial hyperglycemia than WT mice following refeeding (Figure 2L). These results illustrate that insulin-independent AKT activation mediated by Baf60c is sufficient to drive postprandial glucose disposal and homeostasis.
Glucose triggers intramyocellular calcium response through a KATP channel-dependent pathway
Several downstream pathways have been identified to mediate the metabolic effects of glucose in peripheral tissues. Glucose regulates gene expression through post-translational modifications of transcriptional regulators, particularly O-linked glycosylation mediated by O-GlcNAc transferase (OGT) (Bond and Hanover, 2015; Erickson et al., 2013; Hardiville and Hart, 2014; Ruan et al., 2013). To test whether O-linked glycosylation plays a role in glucose-induced Baf60c expression and AKT activation in skeletal myocytes, we performed glucose treatment in C2C12 myotubes in the absence or presence of diazo-5-oxonorleucine (DON), an inhibitor of O-linked glycosylation, and PUGNAc and Thiamet-G, inhibitors of the O-GlcNAcase that removes O-GlcNAc from glycosylated proteins (Erickson et al., 2013; Li et al., 2013). The ability of glucose to induce Baf60c and Deptor gene expression and AKT phosphorylation was largely unaffected by these treatments (Figure S2A–E). Similarly, the induction of the Baf60c-Deptor-AKT pathway appeared to be independent of reactive oxygen species (Figure S2F), which contribute to the regulation of certain target genes in response to glucose (Inoguchi et al., 2000; Yu et al., 2011). In β cells, insulin secretion is stimulated in response to rising blood glucose through ATP-mediated closure of KATP channel and plasma membrane depolarization (Newgard and McGarry, 1995; Nichols, 2006). Interestingly, 2-deoxy glucose (2-DG), a glucose analog that cannot be metabolized through the glycolytic pathway, failed to induce Baf60c and Deptor expression and AKT phosphorylation in C2C12 myotubes (Figure 3A–B) and in ex vivo cultured EDL muscles (Figure 3C–D). Similarly, lactate and pyruvate, two metabolites of the glycolytic pathway, and galactose, a sugar that does not produce ATP through glycolytic metabolism, had modest effects on the expression of Baf60c and Deptor. These findings suggest that ATP production through glycolysis may play an important role in myocyte glucose sensing that leads to activation of the Baf60c-Deptor-AKT pathway.
Figure 3. Glucose elevates intramyocellular calcium levels through a KATP channel-dependent pathway.

(A) qPCR analysis of gene expression in C2C12 myotubes treated with vehicle (Veh) alone or 10 mM of indicated treatments for 12 h. Lac, lactate; Gal, galactose; 2-DG, 2-deoxyglucose; Pyr, pyruvate. Data represent mean ± SD. ***p < 0.001; one-way ANOVA.
(B) Immunoblots of total cell lysates from C2C12 myotubes as treated in (A).
(C) qPCR analysis of gene expression in ex vivo cultured EDL muscles with indicated treatments for 4 h. Data represent mean ± SEM (n=3). ***p < 0.001; one-way ANOVA.
(D) Immunoblots of total protein lysates from ex vivo cultured plantaris muscle as treated in (C).
(E) Top, calcium imaging traces in C2C12 myotubes in response to 10 mM glucose (left), 100 μM diazoxide (DZO) pretreatment followed by 10 mM glucose treatment (middle), or 10 mM 2-DG (right). Data represent mean ± SD. Bottom, representative fluorescent images in C2C12 myotubes treated with 10 mM glucose.
(F) ATP content in treated C2C12 myotubes. Data represent mean ± SD. ***p < 0.001; one-way ANOVA.
(G) Top, calcium imaging traces in primary mouse myotubes in response to 10 mM glucose (left), 100 μM diazoxide (DZO) pretreatment followed by 10 mM glucose treatment (middle), or 10 mM 2-DG (right). Data represent mean ± SD. Bottom, representative fluorescent images in primary mouse myotubes treated 10 mM with glucose.
(H) qPCR analysis of Baf60c and Deptor expression in C2C12 myotubes treated with 1 or 10 mM glucose in combination with Veh or DZO for 12 h. Data represent mean ± SD. *p < 0.05, ***p < 0.001; two-way ANOVA.
(I) Immunoblots of total lysates from C2C12 myotubes treated as in (H).
See also Figure S2 and S3.
Similar to previous findings (Flagg et al., 2010), analysis of a microarray dataset on mouse tissues and cell lines indicated that skeletal muscle expresses high levels of the KATP channel subunits Sur2 and Kir6.2 (Figure S3A). It is plausible that glucose metabolism may increase intracellular ATP levels in myocytes, leading to KATP channel closure, plasma membrane depolarization, and calcium influx through voltage-dependent calcium channels. To test this, we performed calcium imaging using Fluo-4 AM, a cell permeable and sensitive calcium indicator. Exposure of C2C12 myotubes to 10 mM glucose, but not 2-DG, resulted in a rapid and robust increase in intracellular calcium levels (Figure 3E). Consistantly, treatment with glucose, but not other sugars or metabolites, elevated intracellular ATP concentration in treated myotubes (Figure 3F). Similar calcium response was observed in primary mouse myotubes in response to glucose (Figure 3G). In contrast, galactose and pyruvate did not elicit significant changes in intracellular calcium concentrations (Figure S3B–C). Diazoxide (DZO), a potassium channel activator, abolished the calcium response to glucose in both C2C12 and primary mouse myotubes. Importantly, DZO dose-dependently blocked the induction of Baf60c and Deptor and phosphorylation of AKT and AS160 in response to glucose (Figure 3H–I), suggesting that glucose-induced KATP channel closure is required for activation of calcium response and downstream nutrient signaling.
HDAC5 is a nuclear target of myocyte glucose sensing
Calcium signaling plays an important role in myofiber development and metabolism. In skeletal muscle, several kinases, including AMP-activated protein kinase (AMPK), protein kinase C and D (PKC/PKD), mitogen-activated protein kinase (MAPK) and calcium/calmodulin-dependent protein kinase II (CaMKII) have been identified as downstream effectors of calcium signaling (Bassel-Duby and Olson, 2006). We next performed glucose treatment in the presence of specific inhibitors for these kinases to dissect the effector of glucose signaling in myocytes. Remarkably, the CaMKII-specific inhibitors KN-62 and KN-93, dose-dependently diminished the induction of Baf60c and Deptor mRNA and protein expression by glucose (Figure 4A–B and S4A). Glucose-induced AKT activation was also markedly reduced in the presence of KN-62. In contrast, specific inhibitors of PKC/PKD, MAPK, and AMPK had largely no effect on Baf60c and Deptor gene expression (Figure S4B–D). Further, adenoviral overexpression of constitutively active CaMKII, which exhibited calcium-independent kinase activation (Pfleiderer et al., 2004), stimulated Baf60c and Deptor expression in a dose-dependent manner (Figure 4C). As such, CaMKII likely serves as an important downstream mediator of glucose-induced calcium signaling in skeletal myocytes.
Figure 4. Glucose stimulates HDAC5 phosphorylation and nuclear exclusion in cultured myotubes.

(A) qPCR analysis of gene expression in C2C12 myotubes treated with 1 mM (open) or 10 mM (filled) glucose in the presence of indicated concentrations of CaMKII inhibitor KN-62 for 12 h. Data represent mean ± SD. ***p < 0.001; two-way ANOVA.
(B) Immunoblots of total cell lysates from C2C12 myotubes treated with indicated concentrations of glucose without or with 40 μM of KN-62 for 12 h.
(C) qPCR analysis of gene expression in C2C12 myotubes transduced with adenoviruses expressing GFP or constitutive active CaMKII (CA-CaMKII) at low (LD) or high (HD) doses. Data represent mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA.
(D) Immunoblots of total cell lysates from C2C12 myotubes with indicated treatment for 12 h.
(E) Time course response of HDAC5 phosphorylation in C2C12 myotubes treated with 10 mM glucose.
(F) Immunoblots of total protein lysates from quadriceps muscle.
(G) Immunoblots of cytoplasmic and nuclear fractions from C2C12 myotubes treated with 1 mM or 10 mM glucose for 12 h.
(H) Immunofluorescence images of endogenous HDAC5 (green) and nuclei (DAPI) in C2C12 myotubes treated as described in (G). Quantitation of myotubes exhibiting cytoplasmic only (C only) or both cytoplasmic and nuclear (C+N) localization of HDAC5 in response to 1 mM (n=21) or 10 mM (n=19) glucose is shown on the right. Scale bar: 25 μm.
(I) Immunoblots of total protein lysates from C2C12 myotubes as treated in (B).
(J) Immunoblots of nuclear extracts from C2C12 myotubes as treated in (B).
See also Figure S4.
The class IIa HDACs, particularly HDAC4, 5, and 7, have been implicated in linking CaMKII activation to the transcriptional response of muscle genes (Haberland et al., 2009). Interestingly, treatments of C2C12 myotubes with glucose, but not lactate and galactose, strongly induced phosphorylation of HDAC5-S498 and HDAC5-S259/HDAC7-S155, as detected by antibodies that recognize these phosphorylation sites (Figure 4D). Further immunoblotting analyses using phosphorylation site specific antibody ruled out HDAC7-S155 as the glucose-responsive phosphorylation site. Glucose induced a modest increase in HDAC4 phosphorylation on S246 residue. Phosphorylation of HDAC5 was induced within 5–10 min in response to glucose treatment, while total HDAC5 levels remained largely unchanged (Figure 4E). Importantly, HDAC5 phosphorylation exhibited a strikingly similar pattern of regulation by feeding status as Baf60c and Deptor. Phospho-HDAC5 levels were significantly reduced in response to starvation and recovered to high levels following refeeding (Figure 4F).
HDAC5 phosphorylation serves as an important switch for its subcellular localization (Haberland et al., 2009; Mihaylova et al., 2011; Parra and Verdin, 2010). Unphosphorylated HDAC5 is primarily localized in the nucleus and undergoes nuclear to cytosolic translocation upon phosphorylation. We next examined whether glucose-mediated HDAC5 phosphorylation promotes its export from the nucleus. As expected, HDAC5 is localized largely in the nuclear fraction and nearly undetectable in the cytosolic fraction under low glucose (1 mM) conditions (Figure 4G). Endogenous HDAC5, but not HDAC7, underwent a switch of subcellular localization from the nucleus to the cytosol in response to 10 mM glucose. This shift in HDAC5 localization was further confirmed using immunofluorescence staining of endogenous HDAC5 protein (Figure 4H). Further, KN-62 treatments diminished glucose-induced phosphorylation and nuclear exclusion of HDAC5 (Figure 4I–J). Importantly, abolishment of calcium response by the potassium channel activator DZO attenuated glucose-induced HDAC5 phosphorylation in a dose dependent manner (Figure 3I). Together, these results demonstrate that glucose regulates the subcelluar localization of HDAC5 in myocytes in a CaMKII-dependent manner downstream of KATP channel-mediated calcium response.
HDAC5 phosphorylation and nuclear exclusion are required for the induction of glucose-responsive genes
We next determined whether glucose-induced nuclear exclusion of HDAC5 is associated with a switch of chromatin structure to a transcriptional active state. Chromatin immunoprecipitation (ChIP) assays revealed that acetyl-histone H3 (Ace-H3) and trimethyl-H3K4 (H3K4m3), histone marks for transcriptionally active chromatin, were augmented at the proximal promoters of Baf60c and Deptor in response to glucose treatment (Figure 5A–B). Accordingly, glucose treatment significantly reduced the levels of dimethyl-H3K9 (H3K9m2), a histone mark frequently associated with repressive chromatin, near the transcriptional start sites of these two genes. Enrichment of Ace-H3 at distal sites on Baf60c and Deptor promoters remained unaffected by glucose treatment (Figure S5). To test whether nuclear exclusion of HDAC5 contributes to the induction of Baf60c and Deptor by glucose, we knocked down HDAC5 or HDAC7 in C2C12 myotubes and performed glucose treatment. As shown in Figure 5C, RNAi knockdown of HDAC7 had modest effects on the expression of Baf60c and Deptor, while HDAC5 depletion significantly augmented the induction of these genes in response to 2 mM glucose. This sensitizing effect of HDAC5 knockdown disappeared at high glucose concentrations as endogenous HDAC5 was excluded from the nucleus, masking the positive effects of HDAC5 depletion on gene expression (data not shown). These results suggest that glucose-regulated HDAC5 phosphorylation and translocation from the nucleus to cytosol may be critical for the induction of glucose-responsive genes. To test this, we transduced differentiated C2C12 myotubes with recombinant adenoviruses expressing GFP or a non-phosphorylatable HDAC5 mutant with serine residues at positions S259 and S498 mutated to alanine (GFP-HDAC5-Mut). GFP-HDAC5-Mut is constitutively localized in the nucleus and failed to undergo nucleus to cytosol translocation following glucose treatment (Figure 5D). Compared to control, the induction of Baf60c and Deptor by glucose was nearly abolished in myotubes expressing GFP-HDAC5-Mut (Figure 5E). Together, these results strongly suggest that HDAC5 phosphorylation and its nuclear export are required for the activation of glucose-responsive genes.
Figure 5. HDAC5 mediates the induction of glucose-responsive genes in cultured myotubes.

(A, B) ChIP assay in C2C12 myotubes treated with 1 mM (open) or 10 mM (filled) glucose using antibodies against Acetyl-Histone 3 (Ace-H3), trimethyl-H3K4 (H3K4m3) or dimethyl-H3K9 (H3K9m2). The locations of the qPCR primers relative to the transcription start site of Baf60c (A) and Deptor (B) are indicated. Data represent mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed unpaired Student’s t-test.
(C) qPCR analysis of gene expression in transduced C2C12 myotubes. Data represent mean ± SD. ***p < 0.001, two-way ANOVA.
(D) Immunofluorescence images of GFP-HDAC5-Mut and nuclei (DAPI) in C2C12 myotubes transduced with an adenovirus expressing GFP-HDAC5-Mut, and treated with 1 mM (top) or 10 mM (bottom) glucose. Scale bar: 25 μm.
(E) qPCR analysis of gene expression in C2C12 myotubes transduced with GFP or GFP-HDAC5-mut adenoviruses. Data represent mean ± SD. ***p < 0.001, two-way ANOVA.
See also Figure S5.
Skeletal muscle-specific inactivation of Baf60c impairs glucose homeostasis
Having established Baf60c as a key target of myocyte glucose sensing, a critical question remains whether this pathway is required for maintaining systemic glucose homeostasis. To address this, we generated muscle-specific Baf60c knockout (MKO) mice by crossing Baf60c flox/flox mice (flox/flox) with MLC-Cre transgenic mice. MKO mice were born at expected Mendelian ratio and appeared indistinguishable from control littermates. Compared to control (flox/flox), Baf60c mRNA and protein expression was nearly absent in MKO mouse muscles (Figure 6A and B). In contrast, Baf60c expression in other metabolic tissues, including liver, adipose tissues and heart, remained similar between two groups (Figure S6A). Baf60c deficiency in skeletal muscle had no obvious effects on body weight and muscle mass in chow-fed mice (Figure S6B). In addition, body weight and liver, adipose tissue and muscle weights were similar between two groups following HFD feeding (Figure S6C). Gene expression and histological analyses indicated that muscle development remains largely unaffected by Baf60c inactivation in mature myocytes (Figure S6D–E). In fact, endurance exercise capacity was similar between control and MKO mice (Figure S6F). The mRNA levels of Baf60a, Baf60b, and other transcriptional factors and cofactors that regulate muscle metabolism, including MondoA1, MondoA2, Pgc-1α, and Pgc-1β, remained largely unaltered by ablation of Baf60c in skeletal myocytes. Muscle-specific Baf60c deficiency resulted in a significant decrease of several genes involved in glucose metabolism, including Deptor, Glut4, Pgk1, Pgam2, and Gpd1, whereas mRNA levels of the major subunits of mitochondrial oxidative phosphorylation complexes were similar between control and MKO mice. Inactivation of Baf60c significantly impaired glucose-induced lactate production in plantaris, EDL, and soleus muscles cultured ex vivo (Figure 6C).
Figure 6. Skeletal muscle-specific Baf60c deficiency impairs glucose homeostasis.

(A) qPCR analysis of gene expression in EDL muscles from control (flox/flox, open) and MKO (blue) mice. Data represent mean ± SEM (n=5–7). *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed unpaired Student’s t-test.
(B) Immunoblots of total protein lysates from quadriceps muscles from flox/flox and MKO mice fasted for 24 h (Fasted) or fasted for 24 h followed by refeeding for 2 h (Refed).
(C) Lactate production in ex vivo cultured plantaris (PL), EDL and soleus (Sol) muscles from flox/flox and MKO mice pretreated with 4 or 10 mM glucose for 3 h followed by 3 h chase with media containing 5 mM glucose. Data represent mean ± SEM (n=4). ***p < 0.001; two-way ANOVA.
(D–E) ITT (E) and OGTT (E) in HFD-fed flox/flox and MKO mice. Data represent mean ± SEM (n=8–11). *p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA with multiple comparisons. AUC was evaluated by two-tailed unpaired Student’s t-test.
(F) Blood glucose levels in HFD-fed flox/flox and MKO mice fasted for 24 h followed by refeeding for 8 h. Data represent mean ± SEM (n=6–7). **p < 0.01; two-tailed unpaired Student’s t-test.
(G) Plasma insulin and blood glucose levels in STZ-treated HFD-fed flox/flox and MKO mice. Data represent mean ± SEM (n=6). ***p < 0.01; two-tailed unpaired Student’s t-test.
(H) ITT (left) and GTT (right) in mice as described in (G).
(I) Postprandial (fasted/refeeding) blood glucose levels in mice as described in (G). Data in (H–I) represent mean ± SEM (n=6). *p < 0.05, ***p < 0.001; two-way ANOVA with multiple comparisons. AUC was evaluated by two-tailed unpaired Student’s t-test.
(J) Immunoblots of total protein lysates from quadriceps muscles from STZ-treated HFD-fed flox/flox and MKO mice fasted for 24 h followed by refeeding for 2 h.
See also Figure S6.
We next determined whether Baf60c-mediated glucose sensing is required for postprandial AKT activation in skeletal muscle and systemic glucose homeostasis. As expected, AKT phosphorylation was markedly stimulated in WT muscle following the fasting/refeeding switch (Figure 6B). Baf60c deficiency greatly impaired feeding-induced AKT activation. ITT and OGTT studies indicated that HFD-fed MKO mice were more insulin resistant and glucose intolerant than control (Figure 6D–E). Plasma insulin concentrations were similar between two groups during OGTT. Consistently, MKO mice had elevated postprandial blood glucose after refeeding (Figure 6F). Gene expression analyses revealed that muscle-specific inactivation of Baf60c had a modest effect on metabolic gene expression in the liver and adipose tissues (Figure S6G), suggesting that defective skeletal muscle glucose sensing is likely responsible for impaired whole body glucose metabolism in MKO mice. We further examined systemic glucose homeostasis under the condition of insulin deficiency. STZ-treated MKO mice had significantly higher blood glucose levels than control despite similarly low levels of plasma insulin (Figure 6G). In addition, inactivation of Baf60c rendered MKO mice more insulin resistant and glucose intolerant (Figure 6H). Compared to control, MKO mice developed more severe postprandial hyperglycemia following refeeding (Figure 6I), as a result of diminished skeletal muscle AKT activation upon the fasting-refeeding switch (Figure 6J). Taken together, these results strongly implicate Baf60c as a core component of myocyte glucose sensing that is critical for muscle glucose disposal and whole body glucose homeostasis.
Sulfonylurea engages the Baf60c-Deptor-AKT pathway in skeletal muscle to lower blood glucose
Sulfonylureas are a class of widely used anti-diabetic drugs that function through inhibiting the KATP channel on the plasma membrane of pancreatic β cells, leading to augmented insulin secretion. Previous studies have demonstrated that sulfonylureas also exerted effects in extra-pancreatic tissues, particularly skeletal muscle, to lower blood glucose levels (Chutkow et al., 2001; Greenfield et al., 1982; Miki et al., 2002; Muller et al., 1995). Given that the KATP channel is indispensable for glucose-induced calcium response in myocytes, we postulated that sulfonylureas might engage the Baf60c-Deptor-AKT pathway to regulate skeletal muscle glucose metabolism. To test this, we performed calcium imaging studies in myocytes following exposure to sulfonylureas. Tolbutamide and glibenclamide, a first- and second-generation sulfonylurea, respectively, induced a rapid rise of intracellular calcium in C2C12 and primary mouse myotubes (Figure 7A–B and Figure S7). The mRNA and protein expression of Baf60c and Deptor was also significantly induced by glibenclamide treatment in ex vivo cultured EDL muscles, leading to AKT activation (Figure 7C–D). Importantly, glibenclamide-induced expression of Baf60c/Deptor and phosphorylation of AKT and its downstream substrates (FoxO1, AS160, and GSK3β) were significantly attenuated in cultured EDL muscles isolated from MKO mice (Figure 7E). To examine whether Baf60c is required for the full glucose-lowering effect of sulfonylurea, we treated HFD-fed STZ-treated control and MKO mice with glibenclamide through oral gavage and measured blood glucose and plasma insulin levels. The plasma insulin levels remained low and unaltered in response to glibenclamide treatment in both groups. While glibenclamide decreased blood glucose by approximately 30% in control mice, this glucose-lowering effect was significantly attenuated in MKO mice (Figure 7F). As such, the Baf60c-Deptor-AKT pathway serves as a molecular target downstream of sulfonylureas in skeletal muscle that is required for the full glucose-lowering effects of sulfonylurea drugs.
Figure 7. Sulfonylurea engages the Baf60c-Deptor-AKT pathway in skeletal muscle to lower blood glucose.

(A, B) Calcium imaging traces (left) and representative fluorescence images (right) in primary mouse myotubes treated with 100 μM tolbutamide (A) or 10 μM glibenclamide (B), as indicated by red arrows. Data represent mean ± SD.
(C) qPCR analysis of gene expression in ex vivo cultured EDL muscles treated with vehicle (Veh) or 10 μM glibenclamide (Glib) for 3 h. Data represent mean ± SEM (n=3). *p < 0.05, **p < 0.01; two-tailed unpaired Student’s t-test.
(D, E) Immunoblots of total protein lysates from ex vivo cultured plantaris muscles treated with Veh or 10 μM Glib for 3 h.
(F) Plasma insulin and blood glucose levels in STZ-treated HFD-fed flox/flox and MKO mice in response to glibenclamide treatment (5 mg/kg by gavage). Data represent mean ± SEM (n=5). **p < 0.01, flox/flox vs. MKO; two-way ANOVA.
See also Figure S7.
DISCUSSION
Skeletal muscle is a major site of postprandial glucose disposal through insulin-stimulated glucose uptake. Central to this dogma is that insulin activates the PI3K-AKT pathway, leading to Glut4 translocation and glucose uptake. Whether glucose itself is capable of triggering adaptive metabolic responses in skeletal myocytes to promote glucose clearance and contribute to systemic homeostasis is largely unknown. In this study, we identified the Baf60c-Deptor-AKT signaling pathway as a target of myocyte glucose sensing that augments muscle insulin action. Similar to β cells, skeletal myocytes actively respond to glucose by triggering a rapid rise of intracellular calcium, leading to activation of CaMKII, phosphorylation and nuclear exclusion of HDAC5, and induction of Baf60c and Deptor in a cell-autonomous manner. The engagement of this nutrient sensing pathway results in glucose-stimulated AKT activation that acts in concert with insulin to promote muscle glucose uptake. As such, muscle-specific transgenic activation of this pathway improved postprandial glucose clearance, whereas its deficiency impaired insulin action, leading to glucose intolerance. Our data provide evidence for the existence of a myocyte-intrinsic glucose sensing pathway that is coupled to the insulin signaling pathway to function as a rheostat in maintaining steady blood glucose levels.
In mammals, circulating glucose is sensed by diverse cell types in the body. A notable example is the pancreatic β cells, which increase insulin secretion in response to rising blood glucose levels. A subset of hypothalamic neurons also exhibits glucose-responsiveness and contributes to glucose homeostasis through modulating autonomic nervous activity (Chan and Sherwin, 2013; Kong et al., 2010; Steinbusch et al., 2015). Distinct molecular mechanisms have been implicated in linking extracellular glucose levels to biological responses in the cell (Efeyan et al., 2015; Erickson et al., 2013; Hardiville and Hart, 2014; Havula and Hietakangas, 2012; Ruan et al., 2013; Uyeda and Repa, 2006). An intriguing aspect of muscle glucose sensing is that the core molecular apparatus responsible for β cell glucose sensing is also engaged in myocytes. Glucose induced a rapid and robust increase in intracellular calcium levels in myotubes. In fact, pretreatments of myotubes with DZO, a KATP channel opener, completely abolished glucose-induced calcium response, suggesting that the KATP channel acts upstream of Baf60c. Remarkably, exposure of myotubes to tolbutamide and glibenclamide induced a strong calcium response and was sufficient to trigger the expression of Baf60c and Deptor, and AKT activation. The sulfonylurea drugs have been demonstrated to act on extrapancreatic tissues to exert the full blood glucose-lowering effects (Chutkow et al., 2001; Greenfield et al., 1982; Miki et al., 2002; Muller et al., 1995). Our findings demonstrated that the Baf60c-mediated glucose sensing pathway may serve as an important target of sulfonylureas in skeletal muscle.
Calcium plays an important role in orchestrating many aspects of muscle biology. A major target here is the activation of CaMKII, which is required for transducing the stimulatory effects of glucose on Baf60c and Deptor expression and AKT activation. CaMKII phosphorylates the class IIa histone deacetylase HDAC5 and promotes it translocation from nucleus to cytosol in response to glucose. This phosphorylation-dependent switch of the subcellular localization of HDAC5 has been previously implicated in skeletal muscle development and the regulation of hepatic metabolism (Haberland et al., 2009; McKinsey et al., 2000; Mihaylova and Shaw, 2013; Mihaylova et al., 2011). In the context of glucose sensing, we found that HDAC5 is exquisitely critical for transcriptional regulation of Baf60c and Deptor. RNAi knockdown of HDAC5 sensitizes the transcriptional response to glucose, whereas overexpression of a HDAC5 mutant lacking two CaMKII phosphorylation sites severely dampened the induction of glucose-responsive genes in myotubes. These studies established a novel molecular pathway that links extracellular glucose levels to the myocyte transcriptional program and AKT activation.
The expression of Baf60c and Deptor in skeletal muscle is elevated under fed conditions, suggesting that this pathway may be engaged during the postprandial phase to modulate glucose disposal. In support of this, muscle-specific Baf60c transgenic mice had improved glycemic control following the fasting/refeeding switch, even when insulin secretion was diminished by STZ treatments. In fact, this insulin deficiency model revealed a prominent role of Baf60c-mediated glucose sensing pathway in promoting postprandial glucose disposal. On the contrary, mice with muscle-specific deficiency of Baf60c had significantly reduced Deptor expression and lower AKT phosphorylation compared to control mice. MKO mice developed more severe insulin resistance and glucose intolerance following HFD-feeding, suggesting that the Baf60c-Deptor-AKT pathway is engaged in metabolic stress conditions to restore glucose homeostasis. Its deficiency compromised the ability of insulin to fully activate AKT and promote glucose uptake. Similarly, under the conditions of STZ-induced insulin deficiency, MKO mice developed more pronounced postprandial hyperglycemia. Finally, Baf60c appeared to be required for mediating the extrapancreatic action of sulfonylureas in lowering blood glucose. Taken together, this work illustrates a new nutrient sensing pathway underlying glucose-induced glucose disposal in skeletal muscle that serves a critical role in whole body glucose homeostasis.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jiandie Lin (jdlin@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal model
MCK-Baf60c mice have been described previously (Meng et al., 2013). For the generation of Baf60c muscle specific knockout (MKO) mice, a targeting vector that carries two loxP sites flanking exon 2 of mouse Baf60c gene was constructed using BAC recombineering. C57BL/6J ES cell line (Bruce 4) from Transgenic Animal Model Core at the University of Michigan was chosen for homologous recombination to facilitate breeding into C57BL/6J genetic background for metabolic studies. Transgenic mice expressing the Cre recombinase driven by the skeletal muscle specific myosin light chain promoter (MLC-Cre) (Bothe et al., 2000) were used to cross with Baf60c flox/flox mice to achieve muscle specific deletion of Baf60c. Mice were maintained in 12/12 h light/dark cycles and fed normal rodent chow or high-fat diet (D12492, Research Diets). Male mice of 2–8 months of age were used. Littermates were randomly assigned to experimental groups. All animal studies were performed according to procedures approved by the Institutional Animal Care and Use Committee at the University of Michigan.
Muscle cell culture, retroviral transduction and differentiation
C2C12 myoblasts were obtained from ATCC and cultured in DMEM containing 10% fetal bovine serum. Pooled primary human skeletal myoblasts isolated from three healthy adult donors were purchased from Zen-Bio (Research Triangle Park, NC) and maintained in skeletal muscle growth medium (SKM-M, Zen-Bio). Cells were cultured at 37°C in a humidified atmosph ere containing 95% air and 5% CO2. Both cells have been used in multiple previous publications and further confirmed by the differentiation assays. For the establishment of C2C12 stable cell lines, myoblasts were transduced with retroviruses expressing vector alone or Baf60c, or retroviruses expressing control shRNA or shRNAs targeting Baf60c (Meng et al., 2013), HDAC5 and HDAC7 (Mihaylova et al., 2011), and subjected to puromycin selection for 3 days. Myotube differentiation was initiated by switching confluent C2C12 or primary human myoblasts to DMEM containing 2% bovine growth serum or skeletal muscle differentiation medium (SKM-D, Zen-Bio), respectively. All studies were performed in differentiated myotubes.
METHOD DETAILS
Overall experimental design
For in vitro cell culture experiments, three or more independent experiments were performed and each with three or more replicates. For animal experiments, the gender and age of animals were matched. The sample size was determined based on generally expected variations of metabolic parameters and typical sample size for metabolic studies documented in the literature. No statistical method was used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment. All of the experiments were repeated three or more independent times with similar results. Images shown are representative of the images obtained from three independent experiments.
Mouse STZ treatment
For the STZ treatments, STZ (Cayman) was prepared freshly in sodium citric buffer (pH 4.2) and administrated to mice through intraperitoneal (IP) injection at 50 mg/kg body weight for consecutive five days.
ITT and GTT
ITT and GTT were performed as previously described (Meng et al., 2013; Wang et al., 2014). For GTT, mice were fasted overnight (16 h) and IP injected with a glucose solution (20% prepared in saline) at 2 g/kg body weight. Blood glucose concentrations were measured before and 15, 30, 60, and 120 min after glucose injection. For ITT, mice were fasted for 4 h and IP injected with insulin at 1 U/kg. Blood glucose levels were measured before and 15, 30, 60, and 120 min after insulin injection.
Glucose consumption and lactate production assays
Glucose consumption and lactate production assay in differentiated C2C12 myotubes were performed as previously described (Meng et al., 2013). Briefly, Cells grown in 6-well plates were glucose deprived in glucose free DMEM (Gibco, 11966) containing 0.1% BSA and 1 mM glucose for 12 h. Then, cells were treated with fresh glucose free DMEM plus 0.1% BSA and certain concentrations of glucose for 24 h. At the end of the treatment, culture medium was replaced with glucose free DMEM plus 0.1% BSA and 5 mM glucose. At 1 h, 2 h, 4 h, 8 h, 12 h and 24 h after incubation, 50 ul of culture medium was collected from each well and frozen in −80°C for fu ture lactate and glucose measurements using Lactic acid assay kit (K-LATE, Megazyme International Ireland Ltd.) and Glucose assay kit (GAGO-20, Sigma), respectively. For ex vivo culture, intact plantaris, soleus, and EDL muscles were dissected from male flox/flox and MKO mice, and subjected to different concentrations of gluocse treatment for 4 h. At the end of the treatment, culture medium was replaced with glucose free DMEM plus 0.1% BSA and 5 mM glucose. At 1 h, 2 h, 3 h after incubation, 50 ul of culture medium was collected for future lactate and glucose measurements.
Intracellular calcium imaging
C2C12 or primary mouse myoblasts were seeded and differentiated into myotubes in 35 mm glass bottom dishes (MatTek Corporation). Cells were pre-treated with 1 mM glucose for 12 h before loading with calcium indicator Fluo-4 AM (Thermo Fisher) and imaging with inverted fluorescent microscope. Images were acquired every other second to monitor the dynamic changes in intracellular calcium levels in response to different treatments.
ChIP assay
ChIP assay was performed according to the protocol developed by Upstate Biotechnology as described (Meng et al., 2013). Briefly, treated myotubes were fixed with 1% formaldehyde and sonicated to produce chromatin lysates. The lysates were pre-cleared with Protein-G agarose beads and immunoprecipitated overnight with antibodies against Ace-H3 (Millipore, 06–599), H3K4m3 (Millipore, 07–473), H3K9m2 (Abcam, ab1220), or control IgG in the presence of BSA and salmon sperm DNA. The next day, Protein-G agarose beads were added to each immunoprecipitation reaction for 1 h, followed by extensively wash and reverse crosslink. DNA was eluted from the beads and purified using a PCR Purification Kit (Invitrogen) and subsequently analyzed by qPCR using primers located on the proximal promoter regions of mouse Baf60c and Deptor genes (Table S1).
Gene expression analyses
TRIzol reagent (Life Technologies) was used to extract total RNA from tissues and cultured cells. Gene expression analyses were performed using SYBR Green reagent (Life Technologies). Relative abundance of mRNA was normalized to ribosomal protein 36B4. qPCR primers are listed in Table S2.
Western blot analyses
Total protein extracts were prepared using a lysis buffer containing 50 mM Tris-HCl (pH=7.5), 137 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 10 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4, and protease inhibitor cocktail (Roche). Cytosolic and nuclear fractions were prepared as previously described (Meng et al., 2014). Protein extracts were separated by SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane (Millipore), followed by immunoblotting with the following primary antibodies. Rabbit polyclonal antibody against Baf60c was generated with the recombinant GST fusion mouse Baf60c protein and affinity-purified. Antibodies against Phospho-AKT (Thr308) (1:1,000, #9275S), Phospho-AKT (Ser473S) (1:1,000, #4060), AKT (1:1,000, #4691S), Phospho-HDAC4 (Ser246)/HDAC5 (Ser259)/HDAC7 (Ser155) (D27B5) (1:1,000, #3443S), Phospho-HDAC4 (Ser632)/HDAC5 (Ser661S)/HDAC7 (Ser486) (1:1,000, #3424), and Lamin A/C (1:2,000, #2032), Phospho-FoxO1 (Ser256) (1:1,000, #9461), and FoxO1 (1:1,000, #2880) were from Cell Signaling. Antibodies against α-tubulin (1:2,000, T6199) and β-actin (1:2000, A4700) were from Sigma. Antibodies against HDAC4/5/7 (H-273) (1:1,000, sc-11421) and HA (1:2,000, sc-66181) were from Santa Cruz. Antibodies against Deptor (1:1,000, 09–463), and AS160 (1:1,000, 07–741) were from Millipore. Antibodies against Phospho-HDAC7 (Ser155) (1:1,000, ab111390), Phospho-HDAC5 (Ser498) (1:1,000, ab47283) and HDAC5 (1:1,000, ab1439) were from Abcam. Antibody against Phospho-AS160 (Thr642) (1:1,000, 44-1071G) was from Thermo Fisher.
Immunofluorescence
To study the translocation of endogenous HDAC5 in response to glucose treatment, C2C12 myotubes cultured and differentiated on glass coverslips in 6-well plates were pretreated with glucose free DMEM plus 1 mM glucose and 0.1% BSA for 12 hr, followed by treatment with 1 mM or 10 mM glucose for 2 hr. Myotubes were washed once with PBS, fixed in 4% paraformaldehyde on ice for 10min, permeabilized with 0.3% Triton X-100 for 10 min, and blocked with 5% BSA in TBST at room temperature for 1 hr, followed by incubation with rabbit anti HDAC5 antibody (1:200, Abcam ab1439) in 5% BSA in TBST at 4°C overnight. The next day, myotubes were wash three times with PBS, incubated with Alexa Fluor® 488 conjugated goat anti-rabbit IgG (H+L) secondary antibody (1:1,000, Invitrogen A11034) at room temperature for 1 hr, washed three times with PBS, and stained with DAPI for nuclei for 5 min. Coverslips were then mounted using VECTASHIELD Mounting Medium (Vector Laboratories), and images were captured using an Olympus confocal microscope. To study the intracellular localization of GFP-HDAC5 S259A/S498A mutant, C2C12 myotubes cultured and differentiated on glass coverslips in 6-well plates were transduced with recombinant adenovirus expressing GFP-HDAC5 S259A/S498A mutant. The following procedures were the same as described above except for the GFP-HDAC5 S259A/S498A mutant protein could be directly visualized by the confocal microscope. Therefore, after fixation with 4% paraformaldehyde, the myotubes on coverslips were washed three times with PBS and stained with DAPI for 5 min, followed by mounting and confocal imaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using GraphPad Prism 7. Statistical differences were evaluated using two-tailed unpaired Student’s t-test for comparisons between two groups, or analysis of variance (ANOVA) and appropriate post hoc analyses for comparisons of more than two groups. For ITT, GTT, OGTT, and fasting/refeeding blood glucose studies, two-way ANOVA with multiple comparisons was used for statistical analysis. Further, area under curve (AUC) was calculated for each mouse and evaluated by two-tailed unpaired Student’s t-test for statistical differences between two genotypes. A p value of less than 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001) was considered statistically significant. Statistical methods and corresponding p values for data shown in each panel were included in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Data resources
Raw data files for the microarray datasets reported in this paper have been deposited in the NCBI Gene Expression Omnibus under accession numbers GSE79925.
Supplementary Material
HIGHLIGHTS.
Skeletal myocytes engage in physiological glucose sensing via the KATP channel
Glucose stimulates insulin-independent AKT activation through HDAC5/Baf60c
Muscle glucose sensing is required for postprandial glucose homeostasis
Sulfonylureas lower blood glucose in part through the Baf60c-Deptor-AKT axis
Acknowledgments
We thank the staff at the University of Michigan Transgenic Animal Core for the generation of Baf60c flox/flox mice, Drs. Steven J. Burden (NYU) and Daniel Kelly (Sanford-Burnham Medical Research Institute) for sharing the MLC-Cre mice, and Dr. Harold Singer (Albany Medical College) and Dr. Lale Ozcan (Columbia University) for sharing CA-CaMKII and KD-CaMKII adenoviruses. This work was supported by NIH grants (DK112800 and DK102456 to J.D.L.), American Heart Association Scientist Development Grant (15SDG22970032 to Z.X.M.), and the Animal Phenotyping Core of the Michigan Diabetes Research Center (P30 DK020572), and the Michigan Nutrition and Obesity Center (P30 DK089503). The authors declare no competing financial interests.
Footnotes
AUTHOR CONTRIBUTIONS
J.D.L. and Z.X.M. conceived the project and designed research. Z.X.M., J.G., Z.C., J.S., Y.X., Y.L., J.L., X.Z.X. performed the studies. Z.X.M. and J.D.L. analyzed the data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn B, Soundarapandian MM, Sessions H, Peddibhotla S, Roth GP, Li JL, Sugarman E, Koo A, Malany S, Wang M, et al. MondoA coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J Clin Invest. 2016;126:3567–3579. doi: 10.1172/JCI87382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- Chan O, Sherwin R. Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol Metab. 2013;24:616–624. doi: 10.1016/j.tem.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Samuel V, Hansen PA, Pu J, Valdivia CR, Makielski JC, Burant CF. Disruption of Sur2-containing K(ATP) channels enhances insulin-stimulated glucose uptake in skeletal muscle. Proc Natl Acad Sci U S A. 2001;98:11760–11764. doi: 10.1073/pnas.201390398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302–310. doi: 10.1038/nature14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, Coutinho P, Saccone V, Consalvi S, Williams R, et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012;31:301–316. doi: 10.1038/emboj.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield MS, Doberne L, Rosenthal M, Schulz B, Widstrom A, Reaven GM. Effect of sulfonylurea treatment on in vivo insulin secretion and action in patients with non-insulin-dependent diabetes mellitus. Diabetes. 1982;31:307–312. doi: 10.2337/diab.31.4.307. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiville S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014;20:208–213. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havula E, Hietakangas V. Glucose sensing by ChREBP/MondoA-Mlx transcription factors. Semin Cell Dev Biol. 2012;23:640–647. doi: 10.1016/j.semcdb.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008;7:159–172. doi: 10.1016/j.cmet.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Goodpaster BH, Storlien L. Muscle triglyceride and insulin resistance. Annu Rev Nutr. 2002;22:325–346. doi: 10.1146/annurev.nutr.22.010402.102912. [DOI] [PubMed] [Google Scholar]
- Kong D, Vong L, Parton LE, Ye C, Tong Q, Hu X, Choi B, Bruning JC, Lowell BB. Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab. 2010;12:545–552. doi: 10.1016/j.cmet.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Walsh K, Arany Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am J Physiol Endocrinol Metab. 2011;300:E3–10. doi: 10.1152/ajpendo.00512.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Li MD, Ruan HB, Hughes ME, Lee JS, Singh JP, Jones SP, Nitabach MN, Yang X. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 2013;17:303–310. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008;8:105–117. doi: 10.1016/j.cmet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- Lillioja S, Young AA, Culter CL, Ivy JL, Abbott WG, Zawadzki JK, Yki-Jarvinen H, Christin L, Secomb TW, Bogardus C. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest. 1987;80:415–424. doi: 10.1172/JCI113088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZX, Li S, Wang L, Ko HJ, Lee Y, Jung DY, Okutsu M, Yan Z, Kim JK, Lin JD. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nat Med. 2013;19:640–645. doi: 10.1038/nm.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZX, Wang L, Chang L, Sun J, Bao J, Li Y, Chen YE, Lin JD. A Diet-Sensitive BAF60a-Mediated Pathway Links Hepatic Bile Acid Metabolism to Cholesterol Absorption and Atherosclerosis. Cell Rep. 2015;13:1658–1669. doi: 10.1016/j.celrep.2015.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZX, Wang L, Xiao Y, Lin JD. The Baf60c/Deptor pathway links skeletal muscle inflammation to glucose homeostasis in obesity. Diabetes. 2014;63:1533–1545. doi: 10.2337/db13-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol Metab. 2013;24:48–57. doi: 10.1016/j.tem.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Minami K, Zhang L, Morita M, Gonoi T, Shiuchi T, Minokoshi Y, Renaud JM, Seino S. ATP-sensitive potassium channels participate in glucose uptake in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2002;283:E1178–1184. doi: 10.1152/ajpendo.00313.2002. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller G, Satoh Y, Geisen K. Extrapancreatic effects of sulfonylureas--a comparison between glimepiride and conventional sulfonylureas. Diabetes Res Clin Pract. 1995;28(Suppl):S115–137. doi: 10.1016/0168-8227(95)01089-v. [DOI] [PubMed] [Google Scholar]
- Muoio DM. Intramuscular triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim Biophys Acta. 2010;1801:281–288. doi: 10.1016/j.bbalip.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Wong CC, Li G, Xu T, Pajvani U, Park SK, Wronska A, Chen BX, Marks AR, Fukamizu A, et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15:739–751. doi: 10.1016/j.cmet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. 2010;10:454–460. doi: 10.1016/j.coph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleiderer PJ, Lu KK, Crow MT, Keller RS, Singer HA. Modulation of vascular smooth muscle cell migration by calcium/calmodulin-dependent protein kinase II-delta 2. Am J Physiol Cell Physiol. 2004;286:C1238–1245. doi: 10.1152/ajpcell.00536.2003. [DOI] [PubMed] [Google Scholar]
- Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- Ruan HB, Singh JP, Li MD, Wu J, Yang X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab. 2013;24:301–309. doi: 10.1016/j.tem.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau JA, Colberg SR, Thaete FL, Kelley DE. Skeletal muscle glycolytic and oxidative enzyme capacities are determinants of insulin sensitivity and muscle composition in obese women. Faseb J. 1995;9:273–278. [PubMed] [Google Scholar]
- Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- Steinbusch L, Labouebe G, Thorens B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol Metab. 2015;26:455–466. doi: 10.1016/j.tem.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci U S A. 2008;105:6912–6917. doi: 10.1073/pnas.0712199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Turner N, Heilbronn LK. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol Metab. 2008;19:324–330. doi: 10.1016/j.tem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Wang GX, Zhao XY, Meng ZX, Kern M, Dietrich A, Chen Z, Cozacov Z, Zhou D, Okunade AL, Su X, et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med. 2014;20:1436–1443. doi: 10.1038/nm.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wong RH, Tang T, Hudak CS, Yang D, Duncan RE, Sul HS. Phosphorylation and recruitment of BAF60c in chromatin remodeling for lipogenesis in response to insulin. Mol Cell. 2013;49:283–297. doi: 10.1016/j.molcel.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes JJ, Lloyd DJ, Gekakis N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes. 2009;58:1133–1143. doi: 10.2337/db08-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.