

Abstract
Sex assignment at birth remains one of the most clinically challenging and controversial topics in 46,XY disorders of sexual development (DSD). This is particularly challenging in deficiency of 5-alpha reductase type 2 given that external genitalia are typically undervirilized at birth but most individuals virilize at puberty to a variable degree. Historically, most individuals with 5-alpha reductase deficiency were raised females. However, reports that over half of patients who underwent a virilizing puberty adopted an adult male gender identity have challenged this practice. Consensus guidelines on assignment of sex of rearing at birth are equivocal or favor male assignment in the most virilized cases. While a male sex of rearing assignment may avoid lifelong hormonal therapy and/or allow the potential for fertility, female sex assignment may be more consistent with external anatomy in the most severely undervirilized cases. Herein, we describe five patients with 46,XY DSD due 5-alpha-reductase type 2 deficiency, all with a severe phenotype. An inter-disciplinary DSD medical team at one of two academic centers evaluated each patient. This case series illustrates the complicated decision-making process of assignment of sex of rearing at birth in 5-alpha reductase type 2 deficiency and the challenges that arise when the interests of the child, parental wishes, recommendations of the medical team and state law collide.
Keywords: 5-alpha reductase deficiency, DSD, SRD5A2, 5aR2D, gender, sex assignment, ethics
INTRODUCTION
Deficiency of 5-alpha reductase type 2 (5α-R2D) is a rare, autosomal recessive 46,XY DSD that results in a reduced ability to convert testosterone to its more potent metabolite, dihydrotestosterone (DHT). Encoded by SRD5A2 (steroid-5-alpha reductase 2, MIM 607306), 5α-R2D results in undervirilized genitalia at birth and the possibility to virilize at puberty [Imperato-McGinley et al, 1974]. Initially recognized in large families in the Dominican Republic and Papua New Guinea, 5α-R2D also occurs outside of these ethnic groups. The overall incidence is unknown.
During embryogenesis, the fetus is exposed to normal levels of testosterone, the androgen responsible for normal development of male testes and ejaculatory ducts. In contrast, development of the prostate and typical male external genitalia is dependent on DHT; DHT deficiency results in undervirilization. Depending on the degree of 5-alpha reductase type 2 activity, affected individuals are exposed to varying amounts of DHT in utero. This results in phenotypic variability from hypospadias or micropenis in the most virilized individuals to external genitalia that appear normal female in the most severe cases [Imperato-McGinley J et al, 1974; Maimoun et al, 2011].
Sex of rearing is assigned based on diagnosis, genital appearance, surgical options, potential fertility and need for lifelong hormonal therapy [Hughes IA et al, 2006]. Though sex of rearing is assigned to address anatomic and hormonal needs, it also provides families with a social and cultural framework to raise their child before the child is able to express gender identity. Given the severity of the undervirilization in 5α-R2D at birth, affected individuals have historically been raised female [Kolesinska Z et al, 2014]. More recent studies showing that over half of patients who underwent a virilizing puberty because their gonads were not removed adopted a male gender identity in adulthood have challenged this practice [Cohen-Kettertis PT, 2013; Costa EMF et al, 2012; Maimoun L et al, 2011]. Improved phenotypic awareness and more comprehensive molecular diagnostics has led to earlier diagnosis and broadened the phenotypic understanding of several known DSDs, including 5α-R2D. This presents a challenging clinical and ethical dilemma for parents and medical providers when diagnosis occurs before the patient’s ability to express gender identity – and both treatment (gonadectomy) and the lack thereof (which can lead to a virilizing puberty) can lead to irrevocable changes [Flück CE et al, 2011].
The goals of sex assignment of rearing are to assign a sex with the greatest likelihood for concordant gender identity in adulthood. Gender identity develops over time. It is based on sex chromosomes, androgen exposure, psychosocial development, cultural expectations, family dynamics and social situation [Sandberg DE et al, 2012]. While young children can recognize gender, gender identity is likely not fixed until after puberty [Cohen-Kettenis PT, 2005]. The factors that contribute to gender identity are complex and difficult to report; there are few clinical and laboratory indicators of adult gender identity in 5α-R2D.
Herein, we describe five patients with 5α-R2D and severely undervirilized external genitalia, who presented over a 12-year period. All parents strongly desired to raise the child as female given external genital appearance, poor predicted surgical outcome and (when present) the patient’s expressed gender identity. Parents desired gonadectomy to reinforce sex of rearing assignment, prevent virilization at puberty and to alleviate parental distress. These cases illustrate the complicated decision-making process of sex of rearing assignment in the most severe phenotypic presentation of 5α-R2D and highlight the urgent need for clinical investigation into predictors of adult gender identity.
CLINICAL PRESENTATION
All patients described were spontaneously conceived by non-consanguineous couples. Cases two and three are sisters; no other patients had a family history of ambiguous genitalia or atypical sexual development. None of the patients described had an invasive procedure for karyotyping or non-invasive prenatal screening (NIPS) which may have suggested discordant biologic and phenotypic sex prenatally. Case 3, the sister of case 2, had ultrasound abnormalities detected at 36 weeks gestation; all other patients presented shortly after birth with palpable masses in the labioscrotal folds, prompting medical referral. Each patient was evaluated by an interdisciplinary DSD team that consisted of a medical geneticist, endocrinologist, urologist, gynecologist, psychologist and genetic counselor at one of two academic medical centers. At birth, all patients were severely undervirilized with no clitoromegaly. All patients had normal 46,XY chromosomal analysis and molecular diagnosis or confirmation of 5α-R2D (Table 1).
TABLE 1.
Laboratory values including chemical chemistry, cytogenetics and molecular results. Blank boxes indicate testing not performed or result is no longer available.
| CASE 1 | CASE 2 | CASE 3 | CASE 4 | CASE 5 | |
|---|---|---|---|---|---|
| Karyotype | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY |
| FISH | SRY+ | SRY+ | |||
| AR sequencing | Negative | Negative | Negative | Negative | |
| NR5A1 (SF-1) sequencing | Negative | ||||
| T/DHT after hCG stimulation | >50 days 3,5 & 6 (10 months of life) | 19.2 (day 6) (1 month of life) | |||
| Diagnostic method | SRD5A2 sequencing | T/DHT biochemical diagnosis | Clinical assessment, family history | SRD5A2 sequencing | SRD5A2 sequencing |
| Molecular diagnosis* | SRD5A2 p.Pro212Arg, homozygous | SRD5A2 p.Asn193Ser; p.His231Arg | SRD5A2 p.Asn193Ser; p.His231Arg | SRD5A2 p.Gln126Arg homozygous | SRD5A2 p.Glu57Gln; p.Gly183Ser |
| Age at definitive diagnosis (molecular or biochemical) | 2 ½ years old | 10 months | Suspected at 36 weeks gestation because of sister dx; molecular confirmation at 7 months | 4 years old | 9 years old; disclosed at 12 years old (due to persistent lack of follow-up) |
KEY: SRY: sex-determining region. AR: androgen receptor. NR5A1: Nuclear Receptor Subfamily 5, Group A, Member 1. T: testosterone. DHT: dihydrotestosterone. ng/dL: nanograms per deciliter. DSD: Disorder of sex development. SRD5A2: steroid 5 alpha-reductase 2. Pro: Proline. Arg: Arginine. Asn: Asparagine. Ser: Serine. His: Histidine. Gln: Glutamine. Glu: Glutamic acid.
Based on reference sequence NG_008365.1 for Cases 2, 3, and 5 and NW_927719.1 for Case 4.
The DSD teams at each academic center reached similar conclusions about sex assignment of rearing and medical management. For each patient, the DSD team considered the following options: 1) maintain female sex of rearing with early orchiectomy; 2) delay surgery to further assess longitudinal development of gender identity and allow for patient assent, with or without leuprolide therapy to delay onset of puberty; 3) re-assign to male sex of rearing; treat with testosterone supplementation and genital reconstruction. Option three was predicted to have a poor cosmetic and functional outcome in all patients and was uniformly rejected by the parents and inter-disciplinary medical DSD team. All parents clearly stated their decision to maintain a female sex of rearing and desired gonadectomy after counseling with the DSD team. However, due to a divergence in hospital policy and interpretation of state law, gonadectomy was only offered at one center.
CASE ONE
Case 1 was the third daughter born to a Mexican couple after an uncomplicated pregnancy and delivery. Physical examination at birth revealed phenotypically female genitalia, with bifid labioscrotal folds with minimal rugae, no hyperpigmentation and no labioscrotal fusion. She had a very small clitoris with a separate urethral opening and a posterior introitus (Figure 1). Masses were palpated in the labioscrotal folds bilaterally, prompting referral to our DSD team.
FIGURE 1.
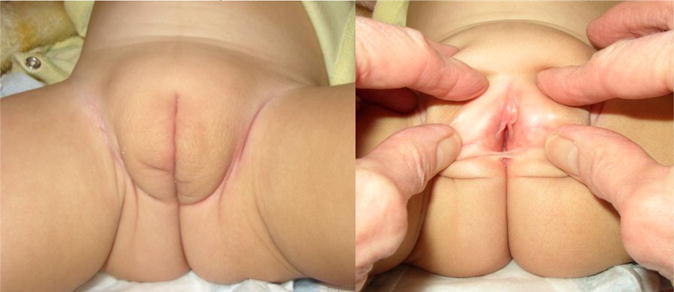
All patients had bifid labioscrotal folds with minimal rugae, little or no hyperpigmentation, separate urethra and introitus, no clitoromegaly and normal placement of the anus. Case 1 is shown below at 18 months of life. Testes have ascended into the inguinal canals.
After karyotype, molecular workup included FISH, which confirmed the presence of sex-determining region (SRY) and single-gene sequencing of the androgen receptor gene (AR), which was normal (Table 1). Ultrasound suggested the presence of a bicornuate uterus and masses in the labioscrotal folds were considered consistent with testes with visualized epididymis. Given the presumed presence of male and female structures, there was concern for dysplastic gonads and the recommendation was made for laparoscopic evaluation and gonadectomy.
However, at the time of laparoscopy at age 8 months, no Mullerian structures were identified. Vaginal vault measured 2.5 × 3 centimeters and no cervix was visualized. Masses in labial folds measured 1 × 0.4 × 0.7 cm (right) and 0.9 × 0.5 × 0.7 cm (left) and were consistent with normal testes, confirmed on surgical biopsy. Given these findings, testes were left in place.
When the patient returned to clinic at age two-and-a half, she was reevaluated by our DSD team, including our child psychologist. She fully identified as female. The family noted little difference in her behavior and interests compared to her two older sisters. Testes retracted into the inguinal canal around age 18 months and were apparent only by palpation. 5α-R2D was considered and SRD5A2 was sequenced. This identified homozygous pathogenic variants in SRD5A2 (p. Pro212Arg), which confirmed a diagnosis of 5α-R2D. This variant has previously been reported in families of Mexican descent [Vilchis F et al, 2010].
Given the appearance of her external genitalia and poor predicted surgical reconstruction outcome, the family strongly preferred a female sex of rearing, which the DSD team supported. The family did not feel the patient could have a satisfactory life as a male given her degree of undervirilization. The family strongly desired gonadectomy to reinforce their decision on sex of rearing, prevent virilization at puberty, and to alleviate the distress the parents felt due to the presence of the gonads and the ambiguity they represented. The family was unswayed by the potential for male fertility given her diagnosis of 5α-R2D, feeling this would be irrelevant if she were in a relationship as a female. While the DSD team supported the parents’ decision for gonadectomy, hospital policy and interpretation of Washington state law prohibits parents from providing informed consent for any procedure that removes the reproductive organs of a minor [Disability Rights Washington, 2012; Seattle Children’s Hospital Bioethics Policy, 2013]. Exceptions are allowed if they pose a health risk, such as the oncogenic risk posed by dysplastic gonads and/or if infertility is considered inevitable with standard treatment [Seattle Children’s Hospital Bioethics Policy, 2013]. A court order authorization must be obtained for any other exception. Given the knowledge available on 5α-R2D and the patient at the time, the medical team felt this policy precluded them from offering gonadectomy to the patient without a court order.
Case 1 is currently seven years old and fully identifies as female. The family did not have the means to independently pursue a court order for gonadectomy and the patient retains her testes. She is followed closely by our child psychologist and will follow-up with the full DSD team prior to puberty.
CASES TWO AND THREE
Case 2 was born at term, at an outside hospital, to a Caucasian couple. At birth, physical examination revealed a normal sized clitoris, separate vaginal and urethral openings, and labioscrotal folds containing bilateral palpable masses. She was seen by a local pediatric geneticist and clinically diagnosed with androgen insensitivity syndrome (AIS). Following discussions shortly after birth, a presumed diagnosis of AIS, and probable need for significant surgical reconstruction if raised as a male, the parents committed to raising the child as female. The infant was referred to Pediatric Endocrinology at our center for presumed AIS and seen at one month of age. At this time, pelvic ultrasound was not able to identify a uterus. Single-gene AR sequencing was normal, making a diagnosis of AIS unlikely (Table 1). An hCG stimulation test performed at 10 months of age revealed an elevated T/DHT ratio (>50 at Days 3, 5, and 6 following hCG injection), a level considered diagnostic of 5α-R2D. After diagnosis of 5α-R2D was achieved biochemically, the family elected to pursue gonadectomy due to concern for virilization at puberty. The patient underwent gonadectomy at 14 months. Testes, fully developed epididymis and appendix testis were removed through the inguinal canal. Pathological examination revealed asymmetrical immature testes; the smaller one contained focal scarring and calcification. There was no evidence of malignancy. Examination under anesthesia revealed a short, blind-ending vagina that was 1.5–2.2 cm in depth.
Case 3 is the sister of Case 2. Fetal genitalia appeared normal-female at a 20-week fetal anatomy ultrasound. However, bilateral masses were noted in the labioscrotal folds at a subsequent 36-week ultrasound. This finding was confirmed on post-natal physical examination following term delivery. External genital phenotype revealed a normal sized clitoris and separate urethral and vaginal openings. Postnatal pelvic ultrasound was unable to identify a uterus. Given her sister’s diagnosis, it was presumed that this child also had 5α-R2D and the parents decided to raise her as female. The parents chose to proceed with gonadectomy and felt comfortable with this decision due to her sister’s diagnosis and treatment. Gonadectomy was performed at age 7 months and revealed normal appearing, immature testicles with well-developed epididymis and vas deferens. Vaginoscopy performed in the operating room revealed a short, blind-ending vaginal pouch. SRD5A2 sequencing revealed biallelic pathogenic variants in exon 4, p. Asn193Ser and p. His231Arg. Targeted genetic testing in Case 2 confirmed the same two pathogenic variants.
Cases 2 and 3 are currently 7 and 3 years old, respectively, and otherwise healthy. Both patients identify with female gender. The 7-year-old is treated by child psychiatry for behavioral problems, including aggressive behavior. The family is seen every 2–3 years for DSD follow-up and will be introduced to our adolescent gynecologist when the patients reach pubertal age.
CASE FOUR
Case 4 was the third child born at term to a Caucasian couple following an unremarkable pregnancy. A 20-week ultrasound suggested female external genitalia. After birth, the neonate was noted to have palpable masses in the labioscrotal folds bilaterally and was transferred to our center for further evaluation and management. The external genitalia showed smooth labioscrotal folds with palpable gonads, no clitoromegaly, and a single opening below the clitoris. Pelvic ultrasound was unable to identify a uterus. After karyotype, sequential single-gene sequencing of NR5A1 and AR genes was normal. The T/DHT ratio was slightly high at 13 hours of life at 15.2. An hCG stimulation test at age 1 month revealed a T/DHT ratio of 19.2 at day 6 following hCG injection, which was high, but not considered diagnostic based on the knowledge at that time (Table 1).
Given the external genital phenotype, the parents strongly preferred to raise the child with a female sex of rearing. Gonadectomy was requested to reinforce the female sex of rearing assignment and alleviate parental anxiety regarding the presence of palpable gonads in the labioscrotal folds. Gonadectomy was performed at age 18 months. Intraoperative findings included a 2-centimeter vaginal canal and a urogenital sinus with a normal-appearing bladder. Normal-appearing testes with epididymis and vas deferens were removed through the inguinal region. Pathological analysis confirmed age-appropriate immature testicular tissue with no evidence of malignancy. Following gonadectomy, the possibility of 5α-R2D was reintroduced given the borderline high T/DHT ratio. Sequencing of SRD5A2 revealed homozygous, pathogenic missense variants in exon 2 resulting in the substitution of an arginine for a glutamine at position 126 (p. Gln126Arg). This variant has been previously observed in individuals with 5α-R2D of French and Spanish ancestry. Parental testing confirmed both were carriers.
The child is currently 9 years old and otherwise healthy. She continues to identify as female. The plan is to return to DSD clinic for care around age 10, prior to puberty, to initiate discussions about hormone replacement therapy and to establish care with adolescent gynecology with the anticipated need for vaginal management in the future.
CASE FIVE
Case 5 was born prematurely to an African-American couple. Pregnancy was complicated by maternal drug use and prematurity. At birth, palpable gonads were present in labioscrotal folds bilaterally. There was no clitoromegaly and the separate introital opening was unremarkable. Testosterone, DHT and 17-hydroxyprogesterone were all elevated by outside report. Pelvic ultrasound was unable to identify a uterus and the patient was given a working clinical diagnosis of androgen insensitivity syndrome. The family and guardians committed to a female sex of rearing assignment. After weighing hormonal benefits versus risk of gonadal malignancy and further virilization, biological mother and grandmother opted for bilateral gonadectomy, which was performed at 19 months of life. Exam under anesthesia revealed a slightly enlarged clitoris with 3–4mm diameter but no phallic length, a 1 cm common urogenital sinus, a short, 1cm deep blind-ending vagina, and mildly rugated labioscrotal folds. Pathologic exam of the gonads demonstrated normal immature testicular tubules and epididymal tissues. Based on these masculinizing features, clinical diagnosis was modified to partial androgen insensitivity syndrome.
Post-operatively, the patient was lost to follow-up until age 9 when there was a change in her caregiver; biological parents were no longer involved. Neither the new caregiver nor the patient had any knowledge about the patient’s prior medical care or evaluation, so disclosure and education was a focus of the visit. Estrogen therapy was initiated and molecular evaluation of AR and SRD5A2 was pursued (Table 1). This revealed two known pathogenic variants in SRD5A2 (p. Glu57Gln, p. Gly183Ser) and the patient was diagnosed with 5α-R2D. AR sequencing was negative.
The patient was again lost to follow-up from ages 10 to 12. Estrogen therapy was self-discontinued from ages 11 to 12, and recently restarted. The patient is currently 12 years old and identifies as female. Her guardian reports the patient had a more “tomboyish” and “aggressive” demeanor after estrogen therapy was suspended. The patient will continue to follow with the DSD team. She was referred to adolescent gynecology to establish care for eventual vaginal management.
DISCUSSION
These cases illustrate the complexities of diagnosis, sex assignment and management of patients with 5α-R2D and highlight the urgent need for investigation into clinical markers of adult gender identity. Improved recognition and molecular diagnostics have allowed for earlier diagnosis, broadened the phenotypic understanding and increased awareness that 5α-R2D exists outside of specific populations. Previously, these patients frequently received a clinical diagnosis of partial or complete androgen insensitivity syndrome (PAIS, CAIS) or did not come to medical attention until virilization at puberty [Berra M et al; Maimoun et al, 2011; Imperato-McGinley J et al, 1974; Costa EMF et al, 2012; Cohen-Kettenis PT, 2005]. We anticipate early diagnosis will continue to expand with the widespread adoption of non-invasive prenatal screening, which reports a sex chromosome complement and may highlight genotype-phenotype discordance in severely affected individuals at birth. While early diagnosis can assist with medical management, it also presents unique challenges.
Parents (and later patients) often experience a great deal of stress with a diagnosis of 46,XY DSD [Suorsa KI et al, 2015; Sandburg DE et al, 2012; Hughes IA et al, 2006; Maimoun L et al, 2011; Thyen U et al, 2014; Herdt GH et al, 1988]. Sex assignment in 46,XY DSD due to defects of androgen biosynthesis is particularly challenging given the potential for virilization at puberty. Exposure to varying amounts of androgens in utero and at puberty may affect adult gender identity, though this is not easily measured or assessed [Wilson JD, 2010]. Both male and female adult gender identities have been reported in patients who have undergone a virilizing puberty [Berra M et al, 2011; Maimoun L et al, 2011; Cohen-Kettentis PT, 2005; Houk CP et al, 2005]. Systematic review of adult gender identity in patients raised as females with early gender-enforcing surgery is lacking; it is not known if early surgery leads to higher rates of concordant female adult gender identity. Consensus guidelines for sex assignment in 5α-R2D are non-directive and rely on considerable clinical judgment [Hughes IA et al, 2006; Douglas G et al, 2010]. Therefore, it is critical that patients are evaluated in a specialized center with expertise in evaluating and managing patients with DSD.
Overall, 5α-R2D is rare and has a broad phenotypic spectrum. The severe phenotype we describe makes up less than 10% of reported cases [Maimoun L et al, 2011]. The literature on 5α-R2D is largely reflective of the most common, characteristic phenotypic presentations and may not be generalizable to patients with a severe phenotype. Genotype-phenotype correlations have been shown for some recurrent variants, though intra-familial differences in phenotype and sex assignment continue to make definitive genotype-phenotype correlations challenging [Vilchis F et al, 2010]. The patients in this case series were evaluated over a 12-year period and reflect clinical knowledge and molecular diagnostics available at the time. Today, we consider 5α-R2D as part of the differential diagnosis of CAIS or PAIS for any patient with 46,XY chromosomal analysis, normal to high testosterone and ambiguous or under-virilized genitalia. Current molecular workup would include karyotype with FISH for SRY or microarray followed by a panel testing selected by phenotype.
The practice of early genital surgery and sex assignment has shifted in the past decade. Prior to 1990, 14% of patients with 46,XY DSD due to androgen biosynthesis defect were assigned male, compared to 54% after 1999 [Kolesinska Z et al, 2014]. Various factors contribute to this trend, including shifting cultural and societal views, improved surgical reconstruction techniques, and better understanding about the potential fertility, oncogenic risk and adult gender identity in this cohort [Kolesinska Z et al, 2014; Hughes IA et al, 2006]. Though evidence is lacking, it was commonly believed in practice that cosmetic genital surgery performed prior to the child’s first birthday reduced parental anxiety and improved parent-child bonding [Hughes IA et al, 2006]. Though based on a small number of patients, oncogenic risk in patients with 5α-R2D who do not undergo orchiectomy is considered low and may be even lower in patients with biopsy-proven normal testes and corrected cryptorchidism [Hughes IA et al, 2006; Sasaki G et al, 2003]. In the cases reported here, external genital appearance played a large role in parental and clinical decision-making. All parents reported feeling highly distressed by the presence of gonads, even after gonads ascended into the inguinal canal and were minimally visible. Parents advocated for gonadectomy to reinforce their decision to raise the child as female, prevent virilization and reduce parental anxiety.
Fertility was a crucial consideration regarding gonadectomy for Case 1. Given the advanced reproductive technologies (ART) available today, the connotation of fertility continues to evolve. Fertility is defined as the ability to conceive a child – and does not note whether interventions are required to achieve that end. Most patients affected with 5α-R2D have oligospermia, arrested spermatogenesis or anatomic limitations that would require ART for conception [Kang HJ et al, 2014]. In patients without 46,XY DSD, fertility is improved with corrected cryptorchidism. Spontaneous conception has been reported in a single patient with 5α-R2D, though his phenotype was much less severe than those presented here [Nordenskjöld A et al, 1998]. Many states have laws regarding sterilization. In most states, state law and hospital policy do not explicitly address genital surgery and/or the removal of reproductive organs in minors with DSD. In Washington State, a policy was explicitly defined after a public controversy arose following the removal of reproductive organs and hormonal treatment in a child with severe developmental delay [Disability Rights Washington, 2012; Seattle Children’s Hospital Bioethics Policy, 2013]. Though the (male) fertility of Case 1 may be compromised by her uncorrected cryptorchidism, female phenotypic sex and gender identity, the medical team could not state infertility was inevitable – meaning she did not qualify for a policy exception to allow gonadectomy. Though this hospital policy and interpretation of state law may be unique, recommendations for when gonadectomy is an appropriate course of medical management, with an emphasis on involving expert clinical teams, would help clarify the need for complex clinical decision-making to ensure the intention of the procedure is not misinterpreted. We anticipate that it will continue to be necessary to consider each patient individually and that guidelines based only on diagnosis will continue to present challenges when phenotype is severe.
The 2006 Consensus statement concluded that patients with 46,XY due to androgen biosynthesis defects who are raised females should undergo gonadectomy prior to puberty to prevent virilization [Hughes IA et al, 2006]. The increased use of LHRH analogs to prevent pubertal progression in children with gender dysphoria suggest that this therapy can also be used in patients with 5α-R2D who are raised female in order to give them more time to participate in decision making regarding gonadectomy. Deferring surgery until puberty allows the child to express free will and provide assent. However, the ability of a child to recognize gender identity and make an irreversible decision about sexuality at an age when psychosocial aspects of sexual maturity and sexual identity are just emerging is unclear [Houk CP et al, 2005; Mendez JP et al, 1994; Costa EM et al, 2012].
Individuals with 5α-R2D will require lifelong, ongoing support and education. Regular DSD clinic visits can provide ongoing age-appropriate support and education. Caregivers often question their decisions when the child exhibits behaviors not typical to the sex of rearing; it is beneficial to have knowledgeable providers available to explore gender identity concerns. Families often need guidance on and assistance with disclosing this information to their child and other friends and relatives. Children and their families may need psychosocial support surrounding any physical differences, infertility, and knowledge that they have a lifelong medical condition. As patients get older, continual care with a DSD program allows for easy transition into new care needs such as hormone replacement and vaginal management.
CONCLUSIONS
In conclusion, these five patients highlight the complexity of diagnosis and management for patients with 5α-R2D and severe undervirilization, particularly regarding the decision for early gonadectomy. These cases illustrate the importance of considering the broad phenotypic spectrum of 5α-R2D in larger studies and consensus statements, as recommendations may not be generalizable to patients at the extremes of the phenotypic spectrum. Ongoing care from an experienced interdisciplinary DSD team is critical and should evolve to meet the changing needs of the patient and family. Given the lifelong consequences of irreversible surgery and virilization at puberty, investigation into outcomes and predictors of gender identity are urgently needed to help guide decisions about sex of rearing in patients diagnosed with 5α-R2D.
Acknowledgments
We would like to thank the patients and their families for participating in this report and Mr. Edward Goldman for the interesting and helpful legal discussions. Heather Byers is supported by NIH5T32GM007454. We acknowledge the Disorders of Sex Development Translational Research Network, supported by grant R01 HD068138 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Biographies
Heather M. Byers, M.D. is a Senior Fellow in the Division of Medical Genetics at the University of Washington in Seattle, WA. She is trained as a clinical geneticist and has a special interest in the evaluation, diagnosis and management of children and adults with DSD.
Lauren Mohnach, MS, CGC is a Certified Genetic Counselor at the University of Michigan in Ann Arbor, MI. She is the clinical genetic counselor and coordinator of the University of Michigan Interdisciplinary Disorders/Differences of Sex Development program.
Patricia Y. Fechner, M.D., an Associate Professor of Pediatrics in the Division of Endocrinology at the University of Washington in Seattle, WA is a board certified pediatric endocrinologist with a special interest in the diagnosis, management, and etiology of DSD. She is the Medical Director of the Multidisciplinary Differences of Sex Development (DSD) Clinic at Seattle Children’s Hospital.
Ming Chen, M.D., Ph.D. is an Associate Professor of Pediatric Endocrinology at the University of Michigan in Ann Arbor, MI. He is the pediatric endocrinologist of the University of Michigan Interdisciplinary Disorders/Differences of Sex Development program. He has an interest in advancing diagnosis and management for individuals with DSD to optimize their care.
Inas H. Thomas, M.D. is an Assistant Professor of Pediatric Endocrinology at the University of Michigan in Ann Arbor, MI. She is a pediatric endocrinologist who works with the University of Michigan Interdisciplinary Disorders/Differences of Sex Development Program. As the Co-director of the Pediatric Endocrine Fellowship, she is interested in improving education among other clinicians as well as treatment for individuals with DSD.
Linda Ramsdell, MS, LGC is a licensed genetic counselor in the Division of Genetic Medicine at Seattle Children’s Hospital. She has provided genetic counseling services for patients and their families seen in the Multidisciplinary Differences in Sex Development Clinic for the past 28 years.
Margarett Shnorhavorian, MD, MPH, FAAP, FACS is an Associate Professor of Urology at the University of Washington in Seattle, WA. She is a pediatric urologist who is the Surgical Director of the Multidisciplinary Differences of Sex Development (DSD) Clinic at Seattle Children’s Hospital and has a special interest in the diagnosis and management of DSD.
Elizabeth McCauley, PHD, ABPP, is a Professor in the Department of Psychiatry and Behavioral Medicine at the University of Washington. She is a child psychologist and a member of the Multidisciplinary DSD Clinic at Seattle Children’s Hospital with a particular interest in promoting positive psychosocial functioning for the youth and their families dealing with a DSD.
Anne-Marie Amies Oelschlager, M.D. is an Associate Professor of Obstetrics and Gynecology at the University of Washington in Seattle, WA. She is the chief of Pediatric and Adolescent Gynecology and a member of the multidisciplinary Differences of Sex Development (DSD) Clinic at Seattle Children’s Hospital. She has clinical expertise in the evaluation, diagnosis, and management of DSD conditions.
John M. Park, MD is a Professor of Pediatric Urology and the Director of pediatric urology at the University of Michigan. He is a member of the University of Michigan Disorders of Sex development Program.
David E. Sandberg, Ph.D. is Professor of Pediatrics at the University of Michigan in Ann Arbor, MI. He is Director of the Division of Pediatric Psychology and Faculty Investigator in the Child Health Evaluation and Research Center. Dr. Sandberg is a member of the University of Michigan Interdisciplinary Disorders/Differences of Sex Development program and serves as the psychosocial lead. He is also a principal investigator of the NIH-funded DSD – Translational Research Network.
Margaret P. Adam, M.D. is a Professor of Pediatrics in the Division of Genetic Medicine at the University of Washington in Seattle, WA. She is a clinical geneticist who works in the Multidisciplinary Differences of Sex Development (DSD) Clinic at Seattle Children’s Hospital and has a special interest in the diagnosis, management, and etiology of DSD.
Catherine E. Keegan, M.D., Ph.D. is an Associate Professor of Pediatrics and Human Genetics at the University of Michigan in Ann Arbor, MI. She is trained as a medical geneticist and is the Director of the University of Michigan Interdisciplinary Disorders/Differences of Sex Development program. She has an interest in advancing genetic diagnosis for individuals with DSD to optimize interdisciplinary care.
Footnotes
The authors declare no conflicts of interest.
References
- Berra M, Williams EL, Muroni B, Creighton SM, Honour JW, Rumsby G, Conway GS. Recognition of 5α-reductase-2 deficiency in an adult female 46XY DSD clinic. Eur J Endocrinol. 2011;164(6):1019–25. doi: 10.1530/EJE-10-0930. [DOI] [PubMed] [Google Scholar]
- Cohen-Kettenis PT. Gender change in 46,XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005;34(4):399–410. doi: 10.1007/s10508-005-4339-4. [DOI] [PubMed] [Google Scholar]
- Costa EM, Domenice S, Sircili MH, Inacio M, Mendonca BB. DSD due to 5α-reductase 2 deficiency - from diagnosis to long-term outcome. Semin Reprod Med. 2012;30(5):427–31. doi: 10.1055/s-0032-1324727. [DOI] [PubMed] [Google Scholar]
- Disability Rights Washington. Devaluing people with disabilities: medical procedures that violate civil rights – 2012. Retrieved from: http://www.disabilityrightswa.org/devaluing-people-disabilities.
- Douglas G, Axelrad ME, Brandt ML, Crabtree E, Dietrich JE, French S, Gunn S, Karaviti L, Lopez ME, Macias CG, McCullough LB, Suresh D, Sutton VR. Consensus in Guidelines for Evaluation of DSD by the Texas Children’s Hospital Multidisciplinary Gender Medicine Team. Int J Pediatr Endocrinol. 2010;2010:919707. doi: 10.1155/2010/919707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flück CE, Meyer-Böni M, Pandey AV, Kempná P, Miller WL, Schoenle EJ, Biason-Lauber A. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am J Hum Genet. 2011;89(2):201–18. doi: 10.1016/j.ajhg.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdt GH, Davidson J. The Sambia “turnim-man”: sociocultural and clinical aspects of gender formation in male pseudohermaphrodites with 5-alpha-reductase deficiency in Papua New Guinea. Arch Sex Behav. 1988;17(1):33–56. doi: 10.1007/BF01542051. [DOI] [PubMed] [Google Scholar]
- Houk CP, Damiani D, Lee PA. Choice of gender in 5alpha-reductase deficiency: a moving target. J Pediatr Endocrinol Metab. 2005;18(4):339–45. doi: 10.1515/jpem.2005.18.4.339. Review. [DOI] [PubMed] [Google Scholar]
- Hughes IA, Houk C, Ahmed SF, Lee PA, Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148–62. doi: 10.1016/j.jpurol.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5α -reductase deficiency in man: An inherited form of male pseudohermaphroditism. Science. 1974;186:1213–1216. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Imperato-McGinley J, Zhu YS, Rosenwaks Z. The effect of 5α-reductase-2 deficiency on human fertility. Fertil Steril. 2014;101(2):310–6. doi: 10.1016/j.fertnstert.2013.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesinska Z, Ahmed SF, Niedziela M, Bryce J, Molinska-Glura M, Rodie M, Jiang J, Sinnott RO, Hughes IA, Darendeliler F, Hiort O, van der Zwan Y, Cools M, Guran T, Holterhus PM, Bertelloni S, Lisa L, Arlt W, Krone N, Ellaithi M, Balsamo A, Mazen I, Nordenstrom A, Lachlan K, Alkhawari M, Chatelain P, Weintrob N. Changes over time in sex assignment for disorders of sex development. Pediatrics. 2014;134(3):e710–5. doi: 10.1542/peds.2014-1088. [DOI] [PubMed] [Google Scholar]
- Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, Fenichel P, Cartigny M, Pienkowski C, Polak M, Skordis N, Mazen I, Ocal G, Berberoglu M, Reynaud R, Baumann C, Cabrol S, Simon D, Kayemba-Kay’s K, De Kerdanet M, Kurtz F, Leheup B, Heinrichs C, Tenoutasse S, Van Vliet G, Grüters A, Eunice M, Ammini AC, Hafez M, Hochberg Z, Einaudi S, Al Mawlawi H, Nuñez CJ, Servant N, Lumbroso S, Paris F, Sultan C. Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: an extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011;96(2):296–307. doi: 10.1210/jc.2010-1024. [DOI] [PubMed] [Google Scholar]
- Nordenskjöld A, Ivarsson SA. Molecular characterization of 5 alpha-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metab. 1998;83(9):3236–8. doi: 10.1210/jcem.83.9.5125. [DOI] [PubMed] [Google Scholar]
- Sandberg DE, Gardner M, Cohen-Kettenis PT. Psychological aspects of the treatment of patients with disorders of sex development. Semin Reprod Med. 2012;30(5):443–52. doi: 10.1055/s-0032-1324729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki G, Nakagawa K, Hashiguchi A, Hasegawa T, Ogata T, Murai M. Giant seminoma in a patient with 5 alpha-reductase type 2 deficiency. J Urol. 2003;169(3):1080–1. doi: 10.1097/01.ju.0000047621.66463.29. [DOI] [PubMed] [Google Scholar]
- Suorsa KI, Mullins AJ, Tackett AP, Scott Reyes KJ, Austin P, Baskin L, Bernabé K, Cheng E, Fried A, Frimberger D, Galan D, Gonzalez L, Greenfield S, Kropp B, Meyer S, Meyer T, Nokoff N, Palmer B, Poppas D, Paradis A, Yerkes E, Wisniewski AB, Mullins LL. Characterizing Early Psychosocial Functioning of Parents of Children with Moderate to Severe Genital Ambiguity due to Disorders of Sex Development. J Urol. 2015;194(6):1737–42. doi: 10.1016/j.juro.2015.06.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyen U, Lux A, Jürgensen M, Hiort O, Köhler B. Utilization of Health Care Services and Satisfaction with Care in Adults Affected by Disorders of Sex Development (DSD) Journal of General Internal Medicine. 2014;29(Suppl 3):752–759. doi: 10.1007/s11606-014-2917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchis F, Ramos L, Méndez JP, Benavides S, Canto P, Chávez B. Molecular analysis of the SRD5A2 in 46,XY subjects with incomplete virilization: the P212R substitution of the steroid 5alpha-reductase 2 may constitute an ancestral founder mutation in Mexican patients. J Androl. 2010;31(4):358–64. doi: 10.2164/jandrol.109.009407. [DOI] [PubMed] [Google Scholar]
- Wilson JD. Androgens, androgen receptors, and male gender role behavior. Horm Behav. 2001;40(2):358–66. doi: 10.1006/hbeh.2001.1684. [DOI] [PubMed] [Google Scholar]