

Abstract
ACDC (arterial calcification due to deficiency of CD73) is an autosomal recessive disease resulting from loss-of-function mutations in NT5E, which encodes CD73, a 5′-ectonucleotidase that converts extracellular adenosine monophosphate to adenosine. ACDC patients display progressive calcification of lower extremity arteries, causing limb ischemia. Tissue-nonspecific alkaline phosphatase (TNAP), which converts pyrophosphate (PPi) to inorganic phosphate (Pi), and extracellular purine metabolism play important roles in other inherited forms of vascular calcification. Compared to cells from healthy subjects, induced pluripotent stem cell–derived mesenchymal stromal cells (iMSCs) from ACDC patients displayed accelerated calcification and increased TNAP activity when cultured under conditions that promote osteogenesis. TNAP activity generated adenosine in iMSCs derived from ACDC patients but not in iMSCs from control subjects, which have CD73. In response to osteogenic stimulation, ACDC patient–derived iMSCs had decreased amounts of the TNAP substrate PPi, an inhibitor of extracellular matrix calcification, and exhibited increased activation of AKT, mechanistic target of rapamycin (mTOR), and the 70-kDa ribosomal protein S6 kinase (p70S6K), a pathway that promotes calcification. In vivo, teratomas derived from ACDC patient cells showed extensive calcification and increased TNAP activity. Treating mice bearing these teratomas with an A2b adenosine receptor agonist, the mTOR inhibitor rapamycin, or the bisphosphonate etidronate reduced calcification. These results show that an increase of TNAP activity in ACDC contributes to ectopic calcification by disrupting the extracellular balance of PPi and Pi and identify potential therapeutic targets for ACDC.
INTRODUCTION
Vascular calcification (VC) is a deleterious result of aging and disease. It alters vascular hemodynamics, contributes to stenosis and ischemia, and predicts cardiovascular morbidity and mortality (1). VC is associated with various common and complex diseases. For example, intimal layer calcification is frequently present in atherosclerotic plaques, and medial layer calcification is associated with maladies such as diabetes, end-stage renal disease, and rheumatoid arthritis (2). VC was once considered as a degenerative, passive biochemical process, but current evidence suggests that this pathology mimics active bone remodeling and is highly regulated (3).
We identified ACDC (arterial calcification due to deficiency of CD73) as an autosomal recessive disease caused by loss-of-function mutations in 5′-ectonucleotidase (NT5E), which encodes CD73, the major enzyme that converts extracellular adenosine monophosphate (AMP) to adenosine (4). ACDC patients exhibit disabling intermittent pain due to obstructive peripheral artery disease stemming from extensive calcification in the medial layer of their lower extremity arteries, as well as calcification in joint capsules of their hands and feet (4, 5). We previously showed that skin fibroblasts from ACDC patients have increased activity of tissue-nonspecific alkaline phosphatase (TNAP) and exhibited calcification in vitro, which can be inhibited with exogenous adenosine (4). Here, we address the mechanism by which CD73 deficiency leads to calcification in ACDC.
TNAP, an important enzyme in bone and tooth mineralization, metabolizes pyrophosphate (PPi), a potent endogenous inhibitor of VC, to inorganic phosphate (Pi) (6). Reduced amounts of PPi are in patients with generalized arterial calcifications of infancy (GACI), a rare recessive disease characterized by extensive medial arterial calcification in infants who also frequently develop joint calcifications. GACI is caused by inactivating mutations in ectonucleotide pyrophosphatase phosphodiesterase 1 (ENPP1). ENPP1 is a component of the purine metabolic pathway upstream of CD73 and converts extracellular adenosine 5′-triphosphate (ATP) to AMP and PPi. In genetic murine models, inhibition of TNAP rescues VC caused by functional inactivation of ENPP1 (7), and TNAP overexpression in vascular smooth muscle cells (VSMCs) or endothelial cells (ECs) causes extensive VC through dysregulation of PPi metabolism (8, 9). TNAP is also important in extracellular purine metabolism for hydrolyzing ATP and AMP, although it does so at lower metabolic rates compared to ENPP1 and CD73, respectively. Crosstalk between TNAP and CD73 has been described in both in vivo and in vitro mouse models (10–12). Adenosine is an important regulator of vascular homeostasis, exerting its effects through the P1 purinergic receptors A1, A2a, A2b, and A3. A1 and A3 adenosine receptors (ARs) couple with Gi proteins and inhibit the cyclic AMP (cAMP) pathway; A2a and A2b ARs couple with Gα proteins and activate cAMP pathways (13). In addition to these canonical AR signaling pathways, noncanonical signaling downstream of ARs includes activation of phosphatidylinositol 3-kinase (PI3K) and AKT.
Determining the role of AR signaling in regulating TNAP will be key to understanding the disease mechanism that leads to VC in ACDC patients. Mice with CD73 deficiency, however, cannot be used to study the ACDC disease mechanism because they do not mimic the human vascular phenotype (14). Therefore, we have established patient-specific in vitro and in vivo disease models using induced pluripotent stem cell (iPSC) technology to determine the mechanisms by which CD73 deficiency leads to VC in ACDC patients. Generating iPSCs from patients with genetic disorders offers enormous promise for understanding the pathogenesis of human diseases. In particular, disease-specific iPSCs and iPSC-derived cells open new possibilities for generating continuous supply of diseased human cells for mechanistic studies and drug screening (15, 16).
Here, we show accelerated and severe calcification in ACDC patient iPSC–derived mesenchymal stromal cells (iMSCs) in culture and in ACDC patient iPSC–derived teratomas in mice. We found that cells from ACDC patients had increased abundance of TNAP relative to cells from control subjects and that this increased TNAP reduced the amount of PPi, which promotes calcification. We also found that the increased abundance of TNAP and increased calcification in ACDC patients are mediated by A2bAR, AKT, and the mechanistic target of rapamycin complex (mTORC). We evaluated several treatment strategies and demonstrated that the bisphosphonate etidronate, the A2bAR agonist BAY 60-6583, and the mTOR inhibitor rapamycin each inhibit calcification in ACDC patient–specific in vitro and in vivo models. Our findings provide proof of concept for the use of these patient-specific iPSC-based disease models for identifying new disease mechanisms and therapeutic strategies for patients with VC.
RESULTS
Cells from ACDC patients can be used to derive iPSCs
We generated iPSCs from dermal skin fibroblasts isolated from healthy control volunteers and patients with ACDC using a lentiviral delivery system for the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) under feeder-free and chemically defined conditions. We selected colonies that expressed the pluripotency marker Tra-1-60 (tumor rejection antigene 1 to 60) and expanded them for further characterization. We obtained similar numbers of Tra-1-60–positive colonies from control and ACDC fibroblasts (76.67 ± 21.9 and 67.75 ± 14.66 colonies per 1 × 105 fibroblasts, respectively; fig. S1), indicating that CD73 is not required for reprogramming fibroblasts into iPSCs. All iPSC lines from five affected individuals in two different families were propagated and expressed the endogenous pluripotency markers Nanog, Oct4, Tra-1-60, and SSEA-4 (Fig. 1A). We verified the pluripotency of these iPSC lines by demonstrating that they could differentiate into cells of each of the three germ layers. After 5 days of monolayer differentiation, iPSCs exhibited reduced abundance of the endogenous pluripotency markers Nanog, Oct4, and Sox2 (Fig. 1B) and a concomitant increase in germ layer differentiation markers for the mesoderm (RUNX1), endoderm (AFP), and ectoderm (Nestin) (Fig. 1C). All the ACDC iPSC lines that we generated retained their original homozygous (c.662C→A, p.S221X) or compound heterozygous (S221X and c.1609dupA, p.V537fsX7) mutations in NT5E (Fig. 1D) and exhibited no chromosomal abnormalities (fig. S2). Control iPSCs and ACDC iPSCs also displayed the capacity to generate the three developmental germ layers in teratomas after injection into the hindlimb of NOD/SCID/IL2rγnull (nonobese diabetic/severe combined immunodeficient/interleukin-2 receptor γ null) (NSG) mice (Fig. 1, E and F). These data illustrate that iPSCs can be generated from ACDC dermal fibroblasts and that these cells are functionally similar to iPSCs generated from healthy controls.
Fig. 1. Characterization of iPSCs from control subjects and ACDC patients.
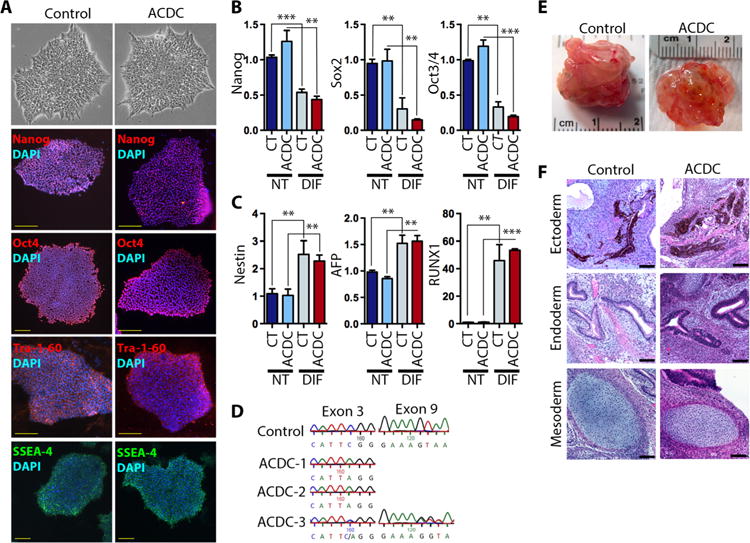
Six iPSC lines from three control and three ACDC patients were characterized. (A) Control and ACDC iPSC colonies stained for pluripotency transcription factors Nanog and Oct4 and cell surface markers Tra-1-60 and SSEA-4. Scale bars, 100 μm. Representative images from six clones are shown. DAPI, 4′,6-diamidino-2-phenylindole. (B and C) Expression of endogenous pluripotency markers (B) and differentiation markers (C) in control (CT) and ACDC iPSCs after monolayer differentiation. Gene expression was normalized to 18S; data are means ± SD from six iPSC clones for pluripotent (NT) and differentiated stage (DIF). P values in (B) and (C) were determined using unpaired two-tailed Student’s t test, n = 6. **P < 0.01; ***P < 0.001. (D) Representative genomic DNA sequencing results of established control and three ACDC iPSC lines. (E) Representative teratomas of control and ACDC iPSCs at 6 to 8 weeks after injection. (F) Control and ACDC iPSC teratoma histology. Scale bars, 100 μm.
ACDC iPSC teratomas develop calcification
In our initial histological evaluation of teratomas, we noted an increase of macroscopic calcifications in teratomas derived from ACDC iPSCs in comparison to teratomas derived from control iPSCs. Microcomputed tomography (microCT) analysis revealed that teratomas from ACDC iPSCs, unlike those from control iPSCs, developed extensive and distinct nodules of calcification, and restoring CD73 expression in ACDC iPSCs rescued this phenotype (Fig. 2A and fig. S3). In line with the microCT analysis, von Kossa histological staining, which indicates mineralization, showed that sections of teratomas derived from ACDC iPSCs were significantly more calcified than sections of teratomas derived from control iPSCs and that these mineralized structures displayed typical features of ectopic calcification (Fig. 2B). Alizarin Red S staining confirmed the presence of calcium in the calcified areas, and the osteoblast-specific transcription factor osterix and the structural protein osteopontin were also present in calcified areas (Fig. 2C).
Fig. 2. ACDC iPSC teratomas exhibit extensive calcification.
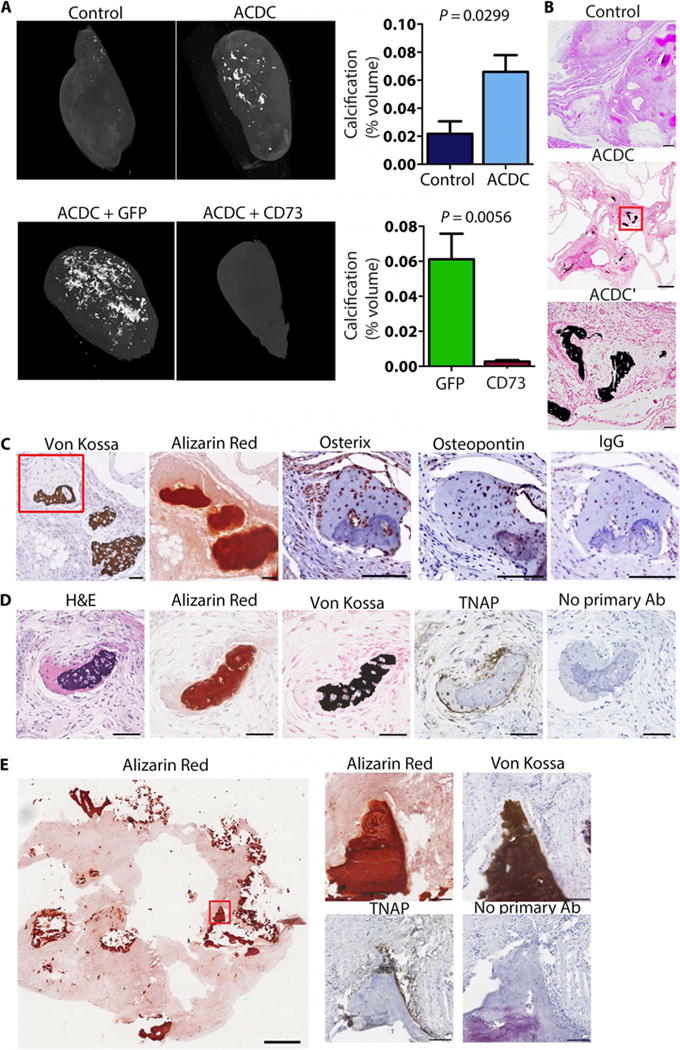
(A) Representative microCT images of teratomas of control iPSCs, ACDC iPSCs, and ACDC iPSCs that overexpressed enhanced green fluorescent protein (GFP) or CD73. Data are means ± SEM of n = 11 controls from seven patient lines, n = 27 ACDC from five patient lines, and n = 6 GFP teratomas and n = 5 CD73 rescue teratomas from one patient line. P values were determined using unpaired two-tailed Student’s t test. (B) Histological analysis of teratomas with von Kossa calcium stain (dark brown). Scale bars, 250 μm (control and ACDC) and 50 μm (ACDC′). (C and D) Serial sections from ACDC iPSC teratomas demonstrating colocalization of osterix, osteopontin, and TNAP with calcification (von Kosa and Alizarin Red). The red box indicates the area shown in the osterix, osteopontin, and immunoglobulin G (IgG) images. Both IgG and no primary antibody (Ab) stainings were used as negative controls. Scale bars, 100 μm. Representative images from three teratomas are shown. H&E, hematoxylin and eosin. (E) Tissue scraped from the inner wall of the popliteal artery of an ACDC patient undergoing embolectomy and patch reconstruction showing the presence of TNAP at sites of calcification.
TNAP is more abundant in dermal fibroblasts from ACDC patients compared to dermal fibroblasts from control subjects and increases further after osteogenic stimulation (4). Similarly, ACDC iPSC teratomas exhibited increased TNAP staining in the vicinity of calcified nodules (Fig. 2D). Tissue scraped from the inner wall of the popliteal artery of an ACDC patient undergoing an embolectomy and patch reconstruction further confirmed up-regulation of TNAP at sites of calcification in vivo (Fig. 2E), whereas a control femoral artery from a patient who did not have ACDC showed no presence of TNAP (fig. S4). These results confirm the link between CD73 and ectopic calcification in the in vivo iPSC teratoma assay and affirm that TNAP is a key component in this calcification process and in that occurring in the vessels of ACDC patients.
ACDC iPSCs display an accelerated calcium precipitation potential
The bone-forming cells (osteoblasts and osteocytes) that stimulate calcium precipitation arise from the MSC lineage. Because ACDC iPSC teratomas exhibit calcification, we hypothesized that CD73-deficient stem or progenitor cells may more readily induce calcium precipitations. To test this, we developed a protocol to differentiate control and ACDC iPSCs into iMSCs (fig. S5). MSCs are characterized by the presence of CD105, CD73, and CD90 and the absence of CD31 and CD45 (17). After differentiation, we confirmed by flow cytometry that our control and ACDC iMSCs had this well-established MSC signature (Fig. 3, A and B). CD73 is a cell surface marker used to identify MSC populations, but the genetic mutations in ACDC patients (4) render the CD73 protein inactive. Control iMSC lines exhibited robust production of Pi when given exogenous AMP, whereas ACDC iMSC lines did not (Fig. 3C). Stable overexpression of CD73 rescued this effect (fig. S6). MSCs are defined by their ability to give rise to adipocyte-, osteoblast-, and chondrocyte-like lineages (18). Both control and ACDC iMSCs exhibited similar adipocyte morphology with accumulation of lipid droplets following an adipogenic stimulation protocol (Fig. 3D). After undergoing a chondrogenic stimulation protocol, both control and ACDC iMSCs showed characteristics of chondrogenic cells, as illustrated by Alcian Blue staining and the presence of the cartilage marker aggrecan (Fig. 3E). After undergoing osteogenic stimulation, we assessed calcification using Alizarin Red S staining, as described previously (4). All tested cells were calcified in vitro at the 21-day time point; however, we observed robust calcium deposition in ACDC iMSCs at earlier time points than for control iMSCs or commercially available hMSC lines (Fig. 3F). These data indicate that a lack of enzymatically functional CD73 enhances calcification potential.
Fig. 3. Enhanced osteogenesis in ACDC iMSCs.
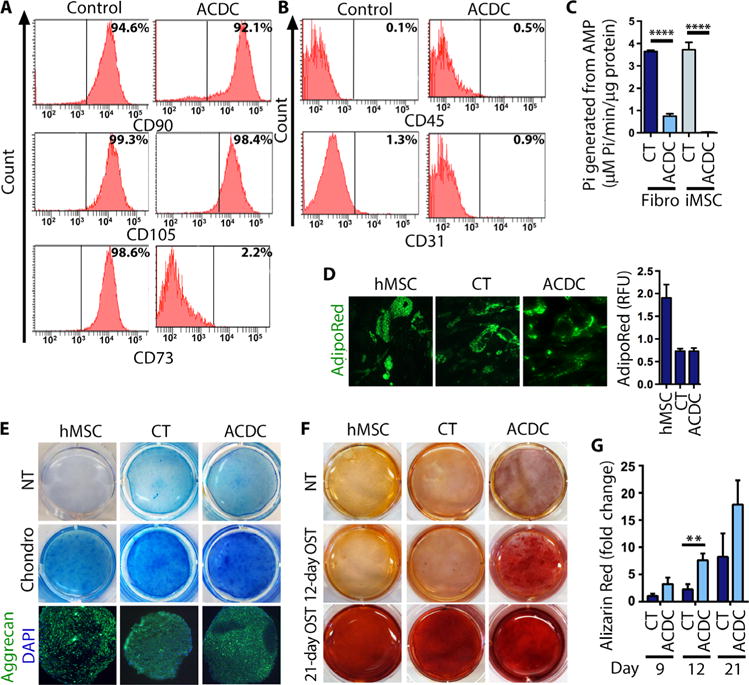
(A and B) Fluorescence-activated cell sorting (FACS) analysis of control and ACDC iMSCs at day 14 of differentiation detecting the MSC-specific surface markers as indicated. Representative analysis from three independent experiments is shown. (C) Production of Pi from exogenous AMP measured in control and ACDC iMSCs and parent fibroblast (Fibro) cell lines. Data are means ± SD: CT fibroblasts, CT iMSCs, and ACDC iMSCs had two cell lines; n = 6; ACDC fibroblast had four cell lines, n = 12. P value was determined using unpaired two-tailed Student’s t test. ****P < 0.0001. (D) Accumulation of lipid droplets in human MSCs (hMSCs) and CT and ACDC iMSCs after adipogenic stimulation. The bar graph shows the quantifications of intracellular lipid accumulation. Representative images are shown. Data are means ± SD, n = 5 (hMSC), n = 11 (CT), and n = 8 (ACDC). RFU, relative fluorescence units. (E) After chondrogenic (Chondro) stimulation of hMSCs, CT iMSCs, and ACDC iMSCs, cells were stained with Alcian Blue to show cartilage formation (representative images are shown). Cell pellets were cultured under the same conditions and subjected to immunostaining for aggrecan. (F) hMSCs, CT iMSCs, and ACDC iMSCs were cultured under osteogenic conditions (OST) and stained after 12 or 21 days with Alizarin Red S for calcium deposition (representative images are shown). (G) Quantification of calcification. Data are means ± SEM; day 9, n = 3 each group; day 12, n = 5 in CT and n = 6 in ACDC; day 21, n = 4 each group. **P = 0.009, unpaired two-tailed Student’s t test.
Increased TNAP activity generates adenosine and decreases PPi in ACDC cells
We previously showed that lack of CD73 function is directly linked to the enhanced calcification phenotype in fibroblasts from ACDC patients (4), and we observed increased TNAP activity in patient fibroblasts (4). However, the functional link between these two closely related glycosylphosphatidylinositol (GPI)–anchored ectoenyzmes, both of which can metabolize AMP, although at different affinities, is not understood (10–12), nor is their impact on the generation of extracellular adenosine during osteogenic stimulation.
Similar to primary fibroblasts from ACDC patients, ACDC iMSCs exhibited a significant increase in TNAP activity compared to control iMSCs when exposed to osteogenic stimulation medium (Fig. 4A). Stable overexpression of CD73 in ACDC iMSCs rescued this effect (fig. S7). ACDC-associated mutations ablate CD73 enzymatic functions (4), so there was significantly less adenosine in the medium of ACDC iMSCs under osteogenic stimulation compared to control iMSCs; nevertheless, there was no concomitant increase in AMP (Fig. 4B). When provided with an excess of the CD73 substrate AMP, control iMSCs produced robust quantities of adenosine (Fig. 4C, upper left). With an excess of AMP, ACDC iMSCs were also able to generate adenosine, albeit to a much lesser extent than control iMSCs (1.438 ± 0.23 versus 131.8 ± 26.15 SEM, respectively; Fig. 4C, upper right). This occurred despite the absence of CD73 function, indicating that another enzyme is generating adenosine from the excess AMP. Under these conditions, control iMSCs did not accumulate AMP because it is rapidly converted to adenosine by CD73 (Fig. 4C, lower left), but ACDC iMSCs did accumulate AMP (Fig. 4C, lower right). Because TNAP can also hydrolyze AMP to produce adenosine (6) and is abundant in cells from ACDC patients, we tested whether the TNAP inhibitor levamisole reduced adenosine production in the presence of excess AMP. In ACDC iMSCs, levamisole alone significantly decreased adenosine (Fig. 4C, upper right), whereas there was no effect in control iMSCs (Fig. 4C, upper left). Furthermore, in the presence of excess AMP, control iMSCs showed no difference in adenosine production from AMP when cells were pretreated with levamisole (Fig. 4C, upper left). However, in ACDC iMSCs, levamisole significantly reduced adenosine production to the degree seen under conditions of no exogenous AMP treatment (Fig. 4C, upper right). This suggests that TNAP can produce adenosine in the absence of CD73 activity but likely does not produce enough adenosine to stimulate ARs (19).
Fig. 4. TNAP activity in iMSCs.
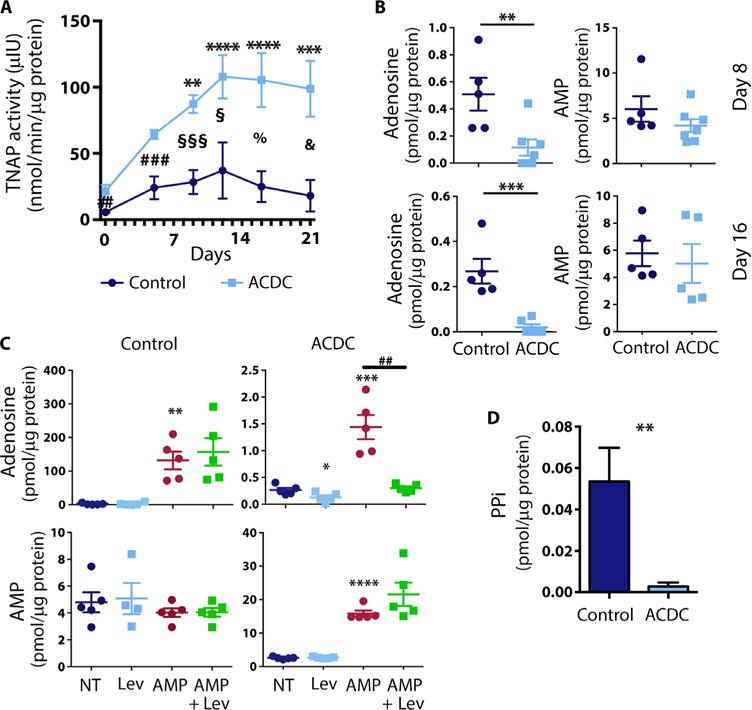
(A) TNAP activity time course in control and ACDC iMSCs treated with osteogenic medium. Data are means ± SEM; n = 6 to 9 (ACDC), n = 5 to 10 (CT). Two-way analysis of variance (ANOVA) multiple comparison analysis was performed for CT and ACDC lines using Dunnett’s multiple comparison test to day 0 in CT (not significant) and ACDC cell lines (**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001); unpaired two-tailed Student’s t test compares data at each day (##P = 0.005, ###P = 0.0009, §P = 0.0228, §§§P = 0.0002, %P = 0.0121, &P = 0.045). (B) Quantification of adenosine and AMP by liquid chromatography–mass spectrometry (LC-MS) measurement in CT iMSCs and ACDC iMSCs at day 8 (top panel) and day 16 (bottom panel). Unpaired two-tailed t test was used for statistical analysis: day 8: **P = 0.0096, n = 5 for CT iMSC, n = 7 for ACDC iMSC; day 16: ***P = 0.0010, n = 5 for CT iMSCs, n = 6 for ACDC iMSCs. (C) Quantification of adenosine (top panel) and AMP (bottom panel) after treatment with exogenous AMP, with and without the TNAP inhibitor levamisole (Lev) after day 8 of osteogenic treatment. Unpaired two-tailed t test was used for statistical analysis; data are means ± SEM. For adenosine measurements: CT iMSC, n = 5, **P = 0.001; ACDC iMSCs, n = 5, *P = 0.0382, ***P = 0.0009, ##P = 0.0011. For AMP measurement: CT iMSCs, n = 5; ACDC iMSCs, n = 5, ****P < 0.0001, n = 5. For (B) and (C), all data were normalized against total protein. (D) PPi quantification in CT iMSCs and ACDC iMSCs after 9 days of osteogenic stimulation. Unpaired two-tailed t test was used for statistical analysis; data are means ± SEM. CT iMSCs, n = 3; ACDC iMSCs, n = 5. **P = 0.0059.
The main substrate for TNAP, PPi, is a potent endogenous inhibitor of ectopic calcification. We hypothesized that increased TNAP activity would accelerate PPi degradation during osteogenic stimulation in ACDC iMSCs compared to control iMSCs, leading to ectopic calcification. In support of this hypothesis, the concentration of PPi was significantly reduced in supernatants from ACDC iMSCs compared to control iMSCs (Fig. 4D). These findings suggest that whereas increased TNAP activity in ACDC iMSCs partially compensates for the lack of CD73-generated adenosine, this increased activity also drastically decreases PPi in ACDC cells, which potentiates calcification and overwhelms thecompensatory effect of the small amount (100-fold less; Fig. 4C) of adenosine generated by TNAP.
Activation of the A2b AR reduces calcification in vivo
Adenosine generated from the breakdown of AMP by CD73 can be converted to inosine by adenosine deaminase (ADA). Alternatively, adenosine can act as a signaling molecule by binding to one of the four ARs—A1, A2a, A2b, and A3 (20, 21). ACDC cells produced significantly less adenosine than did control cells (Fig. 4), and we hypothesized that the consequent reduction in AR signaling leads to a concomitant increase in TNAP, which degrades PPi, thus promoting calcification. To evaluate the effects of AR activation on calcification in vivo, we used teratoma formation as a model. Mice were injected with ACDC iPSCs with concurrent administration of AR agonists or vehicle control. Treatment with A2aAR (CGS-21680) and A3AR (IB-MECA) agonists caused a significant increase in calcification volume, whereas the A1AR agonist (CCPA) produced no significant change in calcification volume (fig. S8) compared to the vehicle control. Treatment with the A2bAR agonist (BAY 60-6583) significantly reduced the calcification volume in ACDC iPSC–derived teratomas (Fig. 5A). None of the treatments altered the total volume of teratomas (fig. S9). In vitro administration of BAY 60-6583 during osteogenic stimulation reduced calcification in ACDC iMSCs (Fig. 5B) and further decreased TNAP activity (Fig. 5C), confirming the in vivo data.
Fig. 5. A2b AR agonist inhibits calcification.
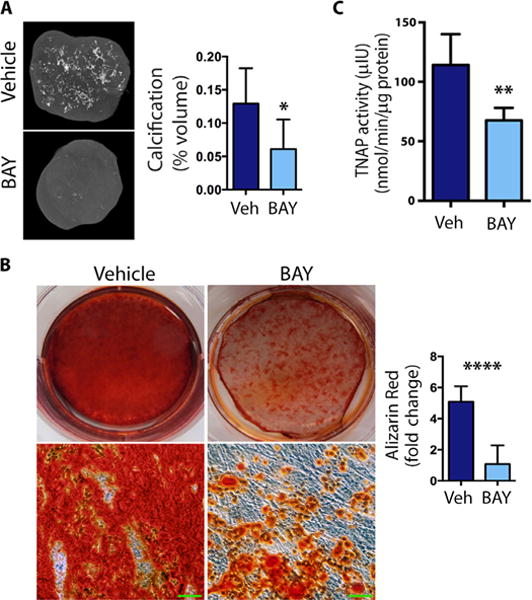
(A) The microCT analysis in ACDC teratomas with A2bAR agonist BAY 60-6583 (BAY) treatment or dimethyl sulfoxide (DMSO) (Veh) control. Calcification as a percentage of teratoma volume was assessed using CT Analyzer software. Representative images are shown. Bright spots showed dense mineralized areas. Data are means ± SEM; vehicle, n = 12 teratomas; BAY, n = 6 teratomas; *P = 0.0157, unpaired two-tailed Student’s t test. (B) Vehicle or BAY was added every other day to ACDC iMSCs under osteogenic conditions, and calcification was detected and quantified by Alizarin Red staining on day 21. Representative images are shown. The bottom images are high-magnification views of images above them. Scale bars, 100 μm. Quantification shows means ± SEM, n = 6 each group; ****P < 0.0001, unpaired two-tailed Student’s t test. (C) ACDC iMSCs were treated with osteogenic medium, and BAY or vehicle were given every other day for 9 days before TNAP activity was measured. Data are means ± SEM; n = 8 each group; **P = 0.0039, two-tailed paired Student’s t test.
Treating primary fibroblasts from ACDC patients with the extracellular signal–regulated kinase 1 and 2 (Erk1/2) inhibitor U0126, the PI3K inhibitor LY249002, or the mTOR inhibitor rapamycin decreased TNAP activity (fig. S10). These inhibitors also decreased TNAP activity and calcification of primary hMSCs (figs. S10 and S11). We next investigated A2bAR downstream signaling in ACDC and control iMSCs during osteogenic stimulation. A2bAR activation by BAY 60-6583 significantly decreased AKT and p70S6K phosphorylation in ACDC iMSCs during osteogenic stimulation to quantities seen in the control iMSCs, indicating activation of the mTOR pathway in ACDC iMSCs (Fig. 6A). On the basis of these in vitro findings, we tested the effects of the mTOR inhibitor rapamycin in the in vivo teratoma model. Mice harboring ACDC iPSCs were given daily intraperitoneal injections of rapamycin or vehicle control. Similar to in vivo BAY 60-6583 treatment, rapamycin significantly reduced calcification in ACDC iPSC–derived teratomas in vivo (Fig. 6B) but did not significantly alter the size of teratomas compared to vehicle controls (fig. S12). These findings indicate that, in cells deficient in CD73, inhibiting the mTOR pathway significantly reduces osteogenesis, which highlights a potential therapeutic treatment regime.
Fig. 6. Activation of A2bAR attenuates mTOR pathway activation.
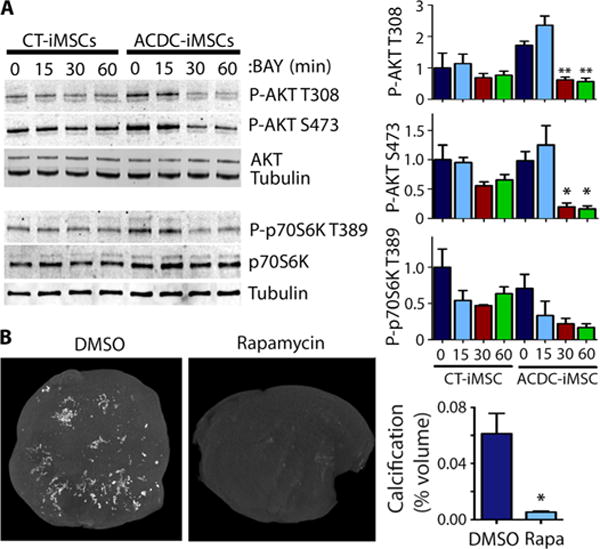
(A) A2b agonist BAY 60-6583 (BAY) attenuates AKT and p70S6K activity. Western blot analysis of AKT phosphorylated on Thr308 (P-AKT T308) or Ser473 (P-AKT S473) and phosphorylated p70S6K (P-p70S6K) relative to total AKT and total p70S6K, respectively. Vehicle or BAY was added at day 9 to control and ACDC iMSCs under osteogenic conditions for 15, 30, or 60 min. Representative images are shown. Densitometry data (right) are means ± SEM, one-way ANOVA with Dunnett’s comparison to time 0, n = 3 independent experiments. *P < 0.05, **P < 0.001. (B) MicroCT analysis in ACDC teratomas with rapamycin (Rapa) treatment or DMSO (Veh) control. Calcification percentage was valued in CT Analyzer software. Data are means ± SEM; DMSO, n = 6 teratomas; rapa-mycin, n = 3 teratomas; *P = 0.0354, unpaired two-tailed Student’s t test.
Etidronate inhibits calcification in the in vivo and in vitro disease models
The lower extremity arteries of ACDC patients present with medial calcifications along the internal elastic lamina and myointimal proliferation (5), thus histologically resemble the disease GACI. Because the bisphosphonate etidronate has been used successfully for long-term treatment of infants with GACI (22–24), we tested the effect of this drug on ACDC cells in vitro and in vivo. ACDC iMSCs exhibited a striking decrease in calcification when etidronate was administered during the 21-day osteogenic stimulation assay (Fig. 7A). Bisphosphonates can inhibit mineral deposition by competing with hydroxyapatite formation (25). To test this function in our biological system, we differentiated hMSCs under osteogenic conditions for 21 days, which causes the accumulation of calcified deposits in the culture and were then given a bolus of 1 mM etidronate. After seven additional days, calcification was resolved (Fig. 7B), suggesting that this drug has potential for treating calcifications that have already developed. To explore these effects in vivo, we injected mice with ACDC iPSCs, followed by a clinically relevant dosage of 30 mg/kg subcutaneous injection of etidronate or vehicle control three times per week starting either the day of or 10 days after iPSC injection and continuing until teratomas reached 20 mm or 8 weeks after injection. MicroCT analysis showed that both treatment regimens resulted in significantly reduced calcification of ACDC iPSC teratomas (Fig. 7C). These data from our in vivo ACDC disease model show that etidronate is a potential treatment for this ectopic mineralizing disorder. Furthermore, the patient-specific iPSC teratoma model may be used as a drug screening tool in cases where the teratoma itself exhibits features similar to human disease pathology.
Fig. 7. Etidronate inhibits calcification in vitro and in vivo.
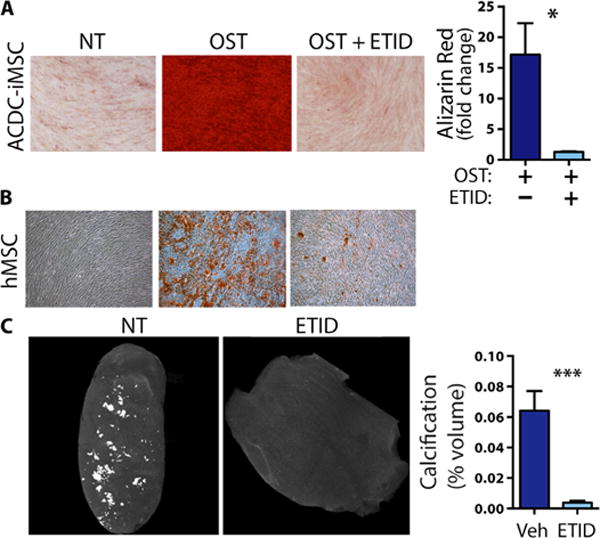
(A) Alizarin Red staining for calcification in ACDC iMSCs under osteogenic conditions and treated with etidronate (ETID) for 21 days. Representative images are shown. Data are means ± SEM, n = 3 independent experiments, *P = 0.0356, unpaired two-tailed Student’s t test. (B) Etidronate treatment was administered to hMSCs at day 21 of osteogenic stimulation, and the cells were stained for calcium with Alizarin Red at day 28. Representative images are shown from three independent experiments. (C) iPSC lines generated from two ACDC patients were injected intramuscularly into the hindlimbs of NSG mice. Control animals received an injection of an equal volume of vehicle (Veh). After 6 weeks, teratomas were harvested and subjected to microCT analysis. Calcification percentage was valued in CT Analyzer software. Data are means ± SEM; ACDC etidronate, n = 7 teratomas; ACDC control, n = 6 teratomas; ***P = 0.0004, unpaired two-tailed Student’s t test.
DISCUSSION
We developed a patient-specific in vitro and in vivo disease modeling system using iPSC technology to identify the signaling mechanisms underlying the calcification process in ACDC patients. The findings will not only enhance our understanding of this detrimental pathology, but the mechanisms identified may be operative in more common disorders characterized by ectopic VC. Injection of primary MSCs has been widely used to study bone formation and ectopic calcification in vivo (26, 27). The biology of MSCs is complex, and MSCs display different characteristics depending on their origin (28). It is well established that iMSCs exhibit a trilineage (chondrogenic, adipogenic, and osteogenic) differentiation potential similar to that observed in primary MSCs (29, 30). We successfully generated iMSCs from iPSCs derived from both control and ACDC patients. Our in vitro studies focused on ectopic calcification using standard osteogenic stimulation medium because this provides a source of Pi only in the presence of TNAP, which is increased in ACDC cells at baseline. However, we did not perform ectopic bone formation assays in mice because although ACDC patients develop extensive ectopic calcification, they do not develop ectopic bone formation and do not have impaired formation of hard tissue such as bone and teeth. iPSC-derived teratomas (31) have previously been used for in vivo disease modeling, and now, we demonstrate that patient-specific iPSC-derived teratomas can model ectopic calcification and are useful to test new treatment strategies for VC.
Here, we show accelerated calcification and increased TNAP abundance and activity in ACDC iMSCs and high abundance adjacent to sites of calcification in a sample of a femoral artery isolated from an ACDC patient. In ACDC but not control iMSCs, TNAP activity generates low amounts of adenosine to compensate for the loss of CD73 function but also leads to a concomitant decrease in the amount of PPi, resulting in calcification. These findings, which highlight the importance of extracellular purine metabolism in the development of VCs, are consistent with the disease mechanisms proposed for other monogenetic diseases involving ectopic calcification, such as GACI (32, 33). GACI, for example, is a rare, recessive, severe infantile form of medial VC disease characterized by rapidly progressing medial calcification caused by loss-of-function mutations in ENPP1, which is upstream of CD73 and converts extracellular ATP to AMP and PPi.
Treatment strategies in GACI are focusing on inhibition of TNAP and downstream processes involved in initiation and propagation of calcification. Bisphosphonates have been used successfully in small patient cohorts, on the basis of their functions as inhibitors of mineralization. Further evidence of the important role of TNAP as a key enzyme in mineralization stems from the rare inherited systemic bone disease called hypophosphatasia (HPP), which is caused by loss-of-function mutations in ALPL, the gene that encodes TNAP, and leads to high amounts of PPi in the serum and hypomineralization of hard tissues in affected patients (34).
Furthermore, crosses between TNAP and ENPP1 knockout mice rescue the calcification phenotypes, thus emphasizing the importance of the TNAP to PPi ratio in murine models of VC (35). Two murine models overexpressing TNAP in either VSMCs (9) or ECs (8) exhibit extensive medial arterial calcification, indicating that increased TNAP function alone is sufficient to induce extensive VCs. Last, PPi administration in chronic kidney disease rodent models prevents VC (36). These studies support our findings and also indicate the importance of these metabolic pathways in other rare or common forms of inherited medial VC.
The increased abundance of TNAP in ACDC iMSCs and the finding that TNAP can generate adenosine in our in vitro calcification assays were unexpected because gain of TNAP activity results from the loss of CD73-generated adenosine signaling. However, crosstalk between CD73-TNAP and bilateral modulation of AMP and adenosine metabolism has been described. Similar to CD73, TNAP is a GPI-anchored 5′-ectonucleotidase and is important in purine metabolism, including the generation of adenosine from AMP, depending on the organ system. Although CD73 is generally regarded as the major source for extracellularly generated adenosine, TNAP plays a role in generating adenosine, especially when CD73 function is genetically or pharmacologically diminished (9, 10, 12). In our model, TNAP can produce adenosine where there is loss of CD73 function, but TNAP-dependent generation of adenosine is not sufficient to prevent ACDC-specific pathologies. Inhibition of TNAP does not affect adenosine generation in the setting of control cells that have normal CD73 function. However, when CD73 activity is absent, inhibiting TNAP blocks the remaining extracellularly generated adenosine, which has the potential to exacerbate the disease pathology.
The precise mechanism by which diminished abundance of adenosine triggers increasing TNAP activity requires further investigation. Treating ACDC skin fibroblasts with adenosine inhibits TNAP activity and calcification (4), suggesting that downstream targets of AR signaling are key factors in preventing VC. In hMSCs and human primary osteoblast cells, expression of the different ARs changes during the course of osteogenic differentiation (37). A2bAR is a low-affinity receptor for adenosine, but A2bAR expression is increased by stressors such as inflammation (13) and oxidative stress (38). Although A2bAR is the dominant AR receptor (39) in hMSCs and rat MSCs, the mechanism by which AR signaling regulates calcification in humans is not clear. Here, we discovered that the increased TNAP activity and calcification observed in ACDC iMSCs and ACDC teratomas can be inhibited by the adenosine A2b receptor–specific agonist BAY 60-6583 but not by A1AR, A2aAR, and A3AR receptor agonists. This further confirms A2bAR as the predominant receptor regulating TNAP activity and calcification in ACDC patients.
A2bAR is a Gαs and Gαq protein–coupled receptor. Activation of A2bAR increases cAMP production by Gαs-activating adenylyl cyclase. A2bAR also couples to Gαq to activate the PLC (phospholipase C) and PKC (protein kinase C) pathways (40). The PI3K-AKT-mTOR signaling pathway promotes VC, and AKT and activation of downstream targets induce calcification in mouse models (41). We found increased activation of the Akt, mTOR, and p70S6K in ACDC iMSCs during osteogenic stimulation, and treatment with the A2bAR agonist or the mTOR inhibitor rapamycin significantly reduced calcification in vitro and in vivo, suggesting that A2bAR signaling negatively intersects with mTOR signaling.
In contrast to GACI and HPP mouse models, CD73 knockout mice do not develop VC, although they do develop joint capsule calcification, another hallmark feature of ACDC (14, 42). Mice are resistant to VC, and the lack of VC in CD73 knockout mice may be exacerbated due to differences in the adenosine metabolism between mice and humans: The half-life of adenosine in human blood is less than 15 s, but in mouse blood, the half-life is 2 min (43). Although this difference could be affected by factors such as heart rate, a substantial component of this difference could be the lack of ADA2 in the murine genome. ADA2 plays a key role in vascular homeostasis, and humans with ADA2 deficiency develop extensive vascular and inflammatory phenotypes (44).
The central finding of our study is that, in the absence of CD73 function, a compensatory increase in TNAP abundance results in decreased PPi, leading to the ectopic calcification of ACDC. Bisphosphonate drugs are widely used to interfere with bone metabolism and are structurally similar to PPi. The bisphosphonate etidronate has been used successfully to treat some GACI patients (45). Here, we demonstrated that etidronate is a potent inhibitor of in vitro and in vivo calcification in ACDC patient–specific disease models. Although these in vivo iPSC teratomas and in vitro iMSC models can be used to study the molecular basis of this disease, they are not the cell types affected in ACDC patients; thus, further confirmation of these findings should be conducted in an in vivo mammalian system. On the basis of these findings, we developed an ongoing clinical study to examine the effectiveness of etidronate on VC in ACDC patients (NCT01585402).
Our study emphasizes the key role of iPSCs for in vitro and in vivo disease modeling and for translating findings to the clinic. Using an iPSC-based system, we uncovered a compensatory mechanism by which CD73 deficiency leads to an increase in TNAP activity, resulting in calcification. We also identified potential therapeutic targets for preventing this compensatory mechanism in ACDC and perhaps other ectopic calcification disorders.
MATERIALS AND METHODS
Derivation of patient-specific fibroblasts and generation and culture of human iPSCs
All patient samples were collected from patients enrolled in a study that had been approved by the respective institutional review board. Control and ACDC-derived human dermal fibroblasts (HDFs) were generated from explants of 4-mm punch biopsy skin specimen and grown in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (FCS) and 1% penicillin-streptomycin as previously described (46). After 1 to 2 weeks, fibroblast outgrowths from the explants were passaged. HDFs were transduced with Human STEMCCA Cre-Excisable Constitutive Polycistronic (OKSM) Lentivirus Reprogramming Kit [Millipore; (47)]. Cells were harvested 3 to 4 days after transduction and replated on plates coated with Matrigel (BD Biosciences). On the following day, E8 medium without transforming growth factor–β (TGF-β) supplemented with 1 μM hydrocortisone and 100 μM butyrate was added to cells and replaced every other day. After 2 weeks of transduction, those cells were fed in full E8 medium. The Tra-1-60–positive iPSC colonies were collected 18 days after transduction and maintained in full E8 medium (STEMCELL Technologies) and passaged with 0.5 mM EDTA (K&D Medical). Twenty-five iPSC lines from five individuals (five lines for each patient) in two families and seven nonrelated control subjects were generated. Two ACDC lines were used in the TALEN rescue experiments.
Karyotyping and DNA sequencing
Chromosome karyotyping was performed by WiCell Research Institute (WiCell Cytogenetics Laboratory). All iPSC lines derived from ACDC patients (ACDC-iPSCs) and controls (CT-iPSCs) were tested. For each cell line, 20 total metaphases were examined. For DNA sequencing, genomic DNA was isolated using the DNeasy Kit (Qiagen) and sequenced as previously described (4).
Pluripotency markers and germ layer markers analysis
Formaldehyde-fixed iPSCs were assessed for pluripotency markers Tra-1-60, SSEA-4, Oct4, and Nanog by immunohistochemical staining as previously described (48). Trilineage differentiation potential of iPSCs was tested in vitro by monolayer differentiation assays for 5 days and in vivo by teratoma formation assays using established protocols (49). Total RNA was collected, and complementary DNA was produced as previously described (4). Expression of markers of undifferentiated cells (Oct3, Oct4, Nanog, and Sox2) and three germ layer markers (AFP, RUNX1, and Nestin) was assayed by quantitative reverse transcription polymerase chain reaction using standard TaqMan primer sets.
Overexpression of CD73 in ACDC iPSCs
The plasmids of AAVS1 (adeno-associated virus integration site 1) TALENs (pZT-AAVS1-L1 and pZT-AAVS1-R1) and AAVS1-CAG-CD73 or AAVS1-CAG-EGFP were constructed as shown in (50). To establish CD73 overexpression of ACDC iPSC clones, we seeded the cells at a density of 50 to 70% confluency in a six-well plate (Falcon) precoated with Matrigel. Five micrograms of each AAVS1 TALENs (pZT-AAVS1-L1 and pZT-AAVS1-R1) and AAVS1-CAG-CD73 or AAVS1-CAG-EGFP donor was transfected into cells the next day after seeding using the Lipofectamine 3000 reagent (Invitrogen) following the manufacturer’s protocol. After 48 hours of transfection, the iPSCs were passed at a 1:2 ratio into six-well plates coated with DR4 mouse embryonic fibroblasts. Puromycin (0.5 to 1 μg/ml) was administered on the day after plating. Single clones from each well were isolated and expanded for evaluation of CD73 expression by FACS.
Generation of iMSCs from iPSCs
iPSCs (1 × 105) were seeded onto Matrigel-coated six-well plates and cultured in E8 medium containing the Rho-associated protein kinase inhibitor Y-27623. The next day, the medium was replaced with mesoderm differentiation medium supplement of human recombi-nant bone morphogenetic protein 4 (10 ng/ml), human recombinant vascular endothelial growth factor (10 ng/ml), and human recombinant basic fibroblast growth factor (bFGF) (10 ng/ml). At day 5, the medium was replaced with MSC medium [80% α–minimal essential medium (α-MEM) without ribonucleic and deoxyribonucleic acids and 20% fetal bovine serum] and bFGF (4 ng/ml) for additional 5 days. Subsequently, the cells were passaged with 0.25% TrypLE (Life Technologies) and replated on new culture plates with fresh MSC medium. The medium was changed every 2 to 3 days.
Characterization of iMSCs
hMSCs were purchased from Lonza and cultured according to the manufacturer’s protocols. Differentiation of iPSC-derived iMSCs and control hMSCs (Lonza) along osteoblastic, adipogenic, and chondrogenic lineages was performed. For osteoblastic differentiation, cells were plated (3.1 × 103 MSC/cm2) and cultured in MSC medium. When the cells were confluent, the medium was replaced with osteoblast-inducing medium containing 90% α-MEM supplemented with 10% FCS and 1% penicillin-streptomycin, 0.1 μM dexamethasone, 50 μM ascorbic acid–2-phosphate, and 10 mM β-glycerophosphate. The medium was changed every 4 to 5 days. At day 21, mineralization was quantified using the Alizarin Red S staining. For adipogenic differentiation, cells were plated (2.1 × 104 MSC/cm2) and cultured in MSC medium. At confluence, cells were exposed to three cycles (7 days per cycles; 21 days in total) of induction medium (4 days), followed by maintenance medium (3 days) to induce adipogenic differentiation. At the end of the three cycles plus one additional week, adipogenic cells were quantified using AdipoRed Assay Reagent (Lonza). For chondrogenic differentiation, cells were cultured as pellets (2.5 × 105 iMSCs) in 15-ml tubes, and the medium was changed every 2 to 3 days using complete chondrogenic medium (Lonza). Cells are cultured in medium for three cycles of 3 days with TGF-β3 supplementation and 3 days without TGF-β3 supplementation. After culturing, pellets were harvested, fixed in formalin, and embedded in paraffin for histological processing. Additionally, cells were cultured in standard wells for 14 to 21 days and then stained using Alcian Blue to visualize chrondrogenic differentiation (Acian Blue/periodic acid–Schiff Stain Kit, Leica Biosystems). Cell lysates were collected for mRNA analysis using the Qiagen RNeasy Kit (Qiagen).
Production of Pi from exogenous AMP (CD73 activity), TNAP activity, and Alizarin Red staining
Production of Pi from exogenous AMP (CD73 activity), TNAP activity, and Alizarin Red staining were performed as previously described (4). For TNAP coloration, treatment with osteogenic medium was stopped after 10 days, and wells were stained for the presence of alkaline phosphatase (ALP) using SIGMAFAST BCIP/NBT kit (Sigma-Aldrich) following the manufacturer’s instructions.
Measurement of TNAP activity in iMSCs
TNAP activity was measured in iMSC protein extracts using the StemTAG Alkaline Phosphatase Activity Assay Kit (Cell Biolabs Inc.) following the manufacturer’s instructions. Protein concentration in the extracts was measured by bicinchoninic acid protein assay (Fisher Thermo Scientific). ALP activity was expressed in μIU (nmol/min per microgram of protein), which is the amount of p-nitrophenylphosphate (pNPP) converted to p-nitrophenol by 1 μg of the protein extracts in 1 min.
Teratoma formation
All animal experiments were approved by the National Heart, Lung, and Blood Institute (NHLBI) Animal Care and Use Committee. For in vivo teratoma formation, 3 × 106 to 5 × 106 iPSCs were suspended in 30% Basement Membrane Matrix High Concentration, lactose dehydrogenase elevating virus–free solution (BD Biosciences) and injected intramuscularly into the hindlimbs of NSG mice. Teratomas formed within 6 to 8 weeks and were excised for analysis.
Immunohistological detection of TNAP in paraffin sections of teratomas and human arteries
Teratomas and human arteries were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Sections were cut and mounted on positively charged slides. Sections were deparaffinized with xylene and rehydrated in a graded ethanol series. Endogenous peroxidase was quenched by placing the slides in 3% hydrogen peroxide in methanol for 10 min. Heat-induced epitope retrieval was performed by boiling the slides for 6 min in citrate buffer solution (pH 6.0) (Invitrogen). Slides were washed with phosphate-buffered saline and then stained with the monoclonal antibody MAB2909, which recognizes alkaline phosphatase (R&D Systems), using the Histostain-SP Kit (Invitrogen) according to the manufacturer’s instructions. In brief, after exposure to serum blocking solution, the sections were incubated successively with primary antibody, biotinylated secondary antibody, and streptavidin–horseradish peroxidase (HRP) conjugate. Visualization was performed with 3,3′-diaminobenzidine (Molecular Probes), which generates a brown-colored oxidation product upon reaction with the HRP-labeled streptavidin. Nuclei were stained with hematoxylin. For images, slides were scanned by NanoZoomer 2.0-RS slide scanner (Hamamatsu Photonics).
Immunoblot analysis
For Western blotting, cell lysates were prepared using CHAPS buffer, separated by SDS–polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes. Images were captured using the Odyssey system (LI-COR). The following antibodies were used: phospho-AKT T308 (Cell Signaling Technology), phospho-AKT S473 (Cell Signaling Technology), AKT1 (Cell Signaling Technology), phospho-p70 S6 kinase Thr389 (Cell Signaling Technology), p70 S6 kinase (Cell Signaling Technology), and α-tubulin mouse monoclonal (LI-COR).
MicroCT acquisition and analysis
Teratoma were harvested, weighed, and then fixed in 4% PFA 6 to 8 weeks after injection of iPSCs. MicroCT data were then acquired on the SkyScan1172 microCT scanner (Bruker microCT) with the x-ray source (focal spot size, 4 μm; energy range, 20 to 100 kV) biased at 60 kV and 67 μA. No filtering was used, allowing for visibility of lower CT densities. Each teratoma had 490 projections acquired every 0.4° while rotating about 220°, with a pixel size of 17.16 μm. Each projection consisted of four frame averages with each frame exposed for 590 ms. Scan duration was about 34 min for most teratomas, with some requiring two scans that automatically stitched together.
Tomographic images were reconstructed using the vendor-supplied software NRecon (Bruker microCT) based on the Feldkamp cone-beam algorithm. An appropriate dynamic range was applied to allow for proper air attenuation and prevent any image saturation.
Reconstructed images were then analyzed using the vendor-supplied software CT Analyzer (Bruker microCT). Binary masks of both teratoma tissue and their denser components (calcifications) were created to allow for quantification and morphometric analysis of their volumes. The percentage of calcified tissue (areas significantly denser than the surrounding tissue) relative to the total teratoma volume was calculated. Three-dimensional renderings of teratoma volumes were created, showing the denser calcified regions within the lighter density cast of the teratoma volume.
Rescue with etidronate, AR agonists, and rapamycin
The hindlimbs of NSG mice (51) were injected with iPSCs as previously described. Etidronate, AR antagonists, and rapamycin were dissolved in an appropriate solution and drawn through a 0.22-μm filter. Subcutaneous injections of etidronate (30 mg/kg) (Sigma) were performed three times per week, starting either 10 days after iPSC injection (group 1; three mice and six teratomas) or the same day as iPSC injection (group 2; three mice and six teratomas). AR antagonists (1 mg/kg per day) and rapamycin (5 mg/kg per day) were injected every day, starting the same day as iPSC injection. After 6 to 8 weeks or when teratomas reached 20 mm in diameter, teratomas were harvested, fixed in 4% PFA, and imaged as described above.
HPLC-MS for nucleosides
Culture media from iMSCs were collected for extracellular adenosine and AMP measurements. Cells were pretreated with or without levamisole (1 mM) overnight and then treated with or without AMP. Samples were prepared from 0.25 ml of cultured medium by deproteinization with 0.25 ml of ice-cold 20% trichloroacetic acid for 20 min on ice. After maximum-speed centrifugation, supernatants were collected for analysis. Extracted nucleosides were applied into a reversed-phase high-performance LC (HPLC) (Agilent 1100 Series HPLC, Agilent Technologies) with a mono C18 reversed-phase column (238EV5405, 4.6 mm × 50 mm, 5 μm, Vydac) and introduced into the mass spectrometer as described (52). Positive-ion electrospray ionization (ESI) mass spectra for target compounds were obtained with an Agilent G1956B MSD (mass selective detector) equipped with an ESI interface (Agilent Technologies). Sensitivity optimization and calibration of MSD were performed by injection of adenosine standards (fragmentor voltage, 80; gain, 10; drying gas flow, 13 liter/min, nebulizer pressure, 20 per square inch gauge; drying gas temperature, 350°C; and capillary voltage, 4.5 kV). Target compounds were quantified in selected ion monitoring mode using mass/charge ratio (M + H)+ 268.10 (adenosine) and 348.07 (AMP). Mass spectra were analyzed using the software ChemStation version B.04.03 (Agilent Technologies). Sample nucleoside concentration was calculated by adenosine and AMP calibration curves and normalized by total protein concentration.
Supplementary Material
Fig. S1. Reprogramming efficiency in ACDC fibroblasts.
Fig. S2. Representative karyotypes of ACDC iPSCs.
Fig. S3. Generation and characterization of ACDC iPSC lines overexpressing CD73.
Fig. S4. TNAP is not present in control human artery that is free from calcification.
Fig. S5. Schematic of differentiation of iPSCs into iMSCs.
Fig. S6. CD73 overexpression rescues osteogenic defects in ACDC iMSCs.
Fig. S7. Overexpression of CD73 decreases TNAP in ACDC iPSCs.
Fig. S8. Effect of AR agonists on calcification in teratomas.
Fig. S9. AR agonists do not affect teratoma size.
Fig. S10. TNAP activity in fibroblasts from control and ACDC patients.
Fig. S11. TNAP activity and calcification in hMSCs.
Fig. S12. Rapamycin treatment does not affect teratoma size.
Acknowledgments
We thank and acknowledge professional skills, advice, and help of the staff of the National Heart, Lung, and Blood Institute (NHLBI) Core Facilities; J. Zou at Induced Pluripotent Stem Cell Core for help with CD73 overexpression construct; C. Liu at Transgenic Core for help with mouse teratoma assays; Z.-X. Yu at Pathology Core for help with histology; and C. Combs at Light Microscopy Core for help with microscopy. We thank Y. Yang (NHLBI) for the statistical review and advice. We thank A. Brofferio for clinical evaluation of the patients and R. Huffstutler for assistance with clinical samples collection. We are grateful to E. Ladich at CVPath Institute Inc., Gaithersburg, MD, for paraffin blocks of control femoral arteries and to M. Siegenthaler at Suburban Hospital, Bethesda, MD, for surgical samples of calcified arteries. We also thank ACDC patients and their families for participating in the study.
Funding: This study was supported by the Intramural Research Program of the NHLBI (M.B.) and K22 HL117917 (C.S.H.), Ministry of Science and Technology of China (2016ZY05002341, 2011CB964800, and 2013CB966902), and the National Natural Science Foundation of China (81421002).
Footnotes
SUPPLEMENTARY MATERIALS
Author contributions: M.B. initiated the study, designed experiments, and analyzed and interpreted the data. G.C. led the protocol development and creation of patient-specific iPSC and iMSC cell lines. H.J., C.S.H., Y.H., G.C., D.Y., N.I.D., and A.N. designed and performed experiments and analyzed and interpreted the data. Y.L., C.S.H., J.-M.D., J.C., K.S.H., and D.-Y.L. performed experiments. A.W. and Z.Y. participated in the creation of patient-specific cell lines. D.D. and C.S.H. analyzed teratoma calcifications. R.S. managed the mouse colony. T.C. and W.G. advised on study design. M.B., C.S.H., D.Y., A.N., G.C., and N.I.D. wrote the manuscript.
Competing interests: M.B, C.S.H., and W.G. filed a patent regarding the use of AR agonists for the treatment of vascular or joint capsule calcification disorders (U.S. patent application no. US13/582,035). M.B. and G.C. filed a patent regarding mesoderm differentiation. The other authors declare that they have no competing interests.
Data and materials availability: All human cell lines must be obtained through a material transfer agreement according to NIH protocols.
REFERENCES AND NOTES
- 1.Shaw LJ, Raggi P, Schisterman E, Berman DS, Callister TQ. Prognostic value of cardiac risk factors and coronary artery calcium screening for all-cause mortality. Radiology. 2003;228:826–833. doi: 10.1148/radiol.2283021006. [DOI] [PubMed] [Google Scholar]
- 2.Persy V, D’Haese P. Vascular calcification and bone disease: The calcification paradox. Trends Mol Med. 2009;15:405–416. doi: 10.1016/j.molmed.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Thompson B, Towler DA. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nat Rev Endocrinol. 2012;8:529–543. doi: 10.1038/nrendo.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Krishna Chaganti R, Nussbaum RL, Kleta R, Gahl WA, Boehm M. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011;103:44–50. doi: 10.1016/j.ymgme.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picher M, Burch LH, Hirsh AJ, Spychala J, Boucher RC. Ecto 5′-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem. 2003;278:13468–13479. doi: 10.1074/jbc.M300569200. [DOI] [PubMed] [Google Scholar]
- 7.Hessle L, Johnson KA, Clarke Anderson H, Narisawa S, Sali A, Goding JW, Terkeltaub R, Luis Millán J. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savinov AY, Salehi M, Yadav MC, Radichev I, Luis Millán J, Savinova OV. Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc. 2015;4:e002499. doi: 10.1161/JAHA.115.002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Nicole Chhea T, Sergienko EA, Kapoor K, Jackson MR, Hoylaerts MF, Pinkerton AB, Charles O’Neill W, Luis Millán J. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res. 2015;30:824–836. doi: 10.1002/jbmr.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson EK, Cheng D, Verrier JD, Janesko-Feldman K, Kochanek PM. Interactive roles of CD73 and tissue nonspecific alkaline phosphatase in the renal vascular metabolism of 5′-AMP. Am J Physiol Renal Physiol. 2014;307:F680–F685. doi: 10.1152/ajprenal.00312.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gan XT, Taniai S, Zhao G, Huang CX, Velenosi TJ, Xue J, Urquhart BL, Karmazyn M. CD73-TNAP crosstalk regulates the hypertrophic response and cardiomyocyte calcification due to α1 adrenoceptor activation. Mol Cell Biochem. 2014;394:237–246. doi: 10.1007/s11010-014-2100-9. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Xiong W, Chu S, Sun C, Albensi BC, Parkinson FE. Inhibition of hippocampal synaptic activity by ATP, hypoxia or oxygen-glucose deprivation does not require CD73. PLOS ONE. 2012;7:e39772. doi: 10.1371/journal.pone.0039772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire Cynthia, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Investig. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Price TP, Sundberg JP, Uitto J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: Similarities to patients with NT5E mutations. Cell Cycle. 2014;13:2609–2615. doi: 10.4161/15384101.2014.943567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Stepniewski J, Kachamakova-Trojanowska N, Ogrocki D, Szopa M, Matlok M, Beilharz M, Dyduch G, Malecki MT, Jozkowicz A, Dulak J. Induced pluripotent stem cells as a model for diabetes investigation. Sci Rep. 2015;5:8597. doi: 10.1038/srep08597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–2749. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 18.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 19.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 20.St Hilaire C, Carroll SH, Chen H, Ravid K. Mechanisms of induction of adenosine receptor genes and its functional significance. J Cell Physiol. 2009;218:35–44. doi: 10.1002/jcp.21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets—What are the challenges? Nat Rev Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramjan KA, Roscioli T, Rutsch F, Sillence D, Munns CFJ. Generalized arterial calcification of infancy: Treatment with bisphosphonates. Nat Clin Pract Endocrinol Metab. 2009;5:167–172. doi: 10.1038/ncpendmet1067. [DOI] [PubMed] [Google Scholar]
- 23.Meradji M, de Villeneuve VH, Huber J, de Bruijn WC, Pearse RG. Idiopathic infantile arterial calcification in siblings: Radiologic diagnosis and successful treatment. J Pediatr. 1978;92:401–405. doi: 10.1016/s0022-3476(78)80427-2. [DOI] [PubMed] [Google Scholar]
- 24.van der Sluis IM, Boot AM, Vernooij M, Meradji M, Kroon AA. Idiopathic infantile arterial calcification: Clinical presentation, therapy and long-term follow-up. Eur J Pediatr. 2006;165:590–593. doi: 10.1007/s00431-006-0146-8. [DOI] [PubMed] [Google Scholar]
- 25.Graham R, Russell G, Smith R. Diphosphonates. Experimental and clinical aspects. J Bone Joint Surg Br. 1973;55:66–86. [PubMed] [Google Scholar]
- 26.Lian Q, Zhang Y, Zhang J, Kun Zhang H, Wu X, Zhang Y, Fu-Yuen Lam F, Kang S, Chuan Xia J, Lai W-H, Au K-W, Yen Chow Y, Siu C-W, Lee C-N, Tse H-F. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010;121:1113–1123. doi: 10.1161/CIRCULATIONAHA.109.898312. [DOI] [PubMed] [Google Scholar]
- 27.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini FC, Krause DS, Deans RJ, Keating A, Prockop DJ, Horwitz EM. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 28.Efthymiou AG, Chen G, Rao M, Chen G, Boehm M. Self-renewal and cell lineage differentiation strategies in human embryonic stem cells and induced pluripotent stem cells. Expert Opin Biol Ther. 2014;14:1333–1344. doi: 10.1517/14712598.2014.922533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lepage SI, Nagy K, Sung HK, Kandel RA, Nagy A, Koch TG. Generation, characterization, and multilineage potency of mesenchymal-like progenitors derived from equine induced pluripotent stem cells. Stem Cells Dev. 2016;25:80–89. doi: 10.1089/scd.2014.0409. [DOI] [PubMed] [Google Scholar]
- 30.Zomer HD, Vidane AS, Goncalves NN, Ambrósio CE. Mesenchymal and induced pluripotent stem cells: General insights and clinical perspectives. Stem Cells Cloning. 2015;8:125–134. doi: 10.2147/SCCAA.S88036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham JJ, Ulbright TM, Pera MF, Looijenga LHJ. Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotechnol. 2012;30:849–857. doi: 10.1038/nbt.2329. [DOI] [PubMed] [Google Scholar]
- 32.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nürnberg P. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 33.Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier M-F, Chassaing N, Roche O, Feenstra I, Loechner K, Deshpande C, Garber SJ, Chikarmane R, Steinmann B, Shahinyan T, Martorell L, Davies J, Smith WE, Kahler SG, McCulloch M, Wraige E, Loidi L, Höhne W, Martin L, Hadj-Rabia S, Terkeltaub R, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenberg CR, Evans JA, McKendry-Smith S, Redekopp S, Haworth JC, Mulivor R, Chodirker BN. Infantile hypophosphatasia: Localization within chromosome region 1p36.1-34 and prenatal diagnosis using linked DNA markers. Am J Hum Genet. 1990;46:286–292. [PMC free article] [PubMed] [Google Scholar]
- 35.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Luis Millán J. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: An integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riser BL, Carvalho Barreto F, Rezg R, Valaitis PW, Cook CS, White JA, Gass JH, Maizel J, Louvet L, Drueke TB, Holmes CJ, Massy ZA. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol Dial Transplant. 2011;26:3349–3357. doi: 10.1093/ndt/gfr039. [DOI] [PubMed] [Google Scholar]
- 37.Adelina Costa M, Barbosa A, Neto E, Sá-e-Sousa A, Freitas R, Neves JM, Magalhães-Cardoso T, Ferreirinha F, Correia-de-Sá P. On the role of subtype selective adenosine receptor agonists during proliferation and osteogenic differentiation of human primary bone marrow stromal cells. J Cell Physiol. 2011;226:1353–1366. doi: 10.1002/jcp.22458. [DOI] [PubMed] [Google Scholar]
- 38.St Hilaire C, Koupenova M, Carroll SH, Smith BD, Ravid K. TNF-α upregulates the A2B adenosine receptor gene: The role of NAD(P)H oxidase 4. Biochem Biophys Res Commun. 2008;375:292–296. doi: 10.1016/j.bbrc.2008.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gharibi B, Abraham AA, Ham J, Evans BA. Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res. 2011;26:2112–2124. doi: 10.1002/jbmr.424. [DOI] [PubMed] [Google Scholar]
- 40.Lasley RD. Adenosine receptors and membrane microdomains. Biochim Biophys Acta. 2011;1808:1284–1289. doi: 10.1016/j.bbamem.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell’Italia LJ, Chatham JC, Wu H, Chen Y. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. 2014;114:1094–1102. doi: 10.1161/CIRCRESAHA.114.302968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, Kingman J, Uitto J. Mineral content of the maternal diet influences ectopic mineralization in offspring of Abcc6−/− mice. Cell Cycle. 2015;14:3184–3189. doi: 10.1080/15384101.2015.1068473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Söderbäck U, Sollevi A, Fredholm BB. The disappearance of adenosine from blood and platelet suspension in relation to the platelet cyclic AMP content. Acta Physiol Scand. 1987;129:189–194. doi: 10.1111/j.1748-1716.1987.tb08058.x. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Jin Chae J, Rosenzweig SD, Bishop K, Barron KS, Sun Kuehn H, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee C-CR, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–920. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edouard T, Chabot G, Miro J, Christina Buhas D, Nitschke Y, Lapierre C, Rutsch F, Alos N. Efficacy and safety of 2-year etidronate treatment in a child with generalized arterial calcification of infancy. Eur J Pediatr. 2011;170:1585–1590. doi: 10.1007/s00431-011-1572-9. [DOI] [PubMed] [Google Scholar]
- 46.Normand J, Karasek MA. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev Biol Anim. 1995;31:447–455. doi: 10.1007/BF02634257. [DOI] [PubMed] [Google Scholar]
- 47.Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mali P, Ye Z, Chou BK, Yen J, Cheng L. An improved method for generating and identifying human induced pluripotent stem cells. Methods Mol Biol. 2010;636:191–205. doi: 10.1007/978-1-60761-691-7_12. [DOI] [PubMed] [Google Scholar]
- 49.Gordeeva OF, Nikonova TM. Development of experimental tumors formed by mouse and human embryonic stem and teratocarcinoma cells after subcutaneous and intraperitoneal transplantations into immunodeficient and immunocompetent mice. Cell Transplant. 2013;22:1901–1914. doi: 10.3727/096368912X657837. [DOI] [PubMed] [Google Scholar]
- 50.Cerbini T, Luo Y, Rao MS, Zou J. Transfection, selection, and colony-picking of human induced pluripotent stem cells TALEN-targeted with a GFP gene into the AAVS1 safe harbor. J Vis Exp. 2015 doi: 10.3791/52504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 52.Apffel A, Fischer S, Goldberg G, Goodley PC, Kuhlmann FE. Enhanced sensitivity for peptide-mapping with electrospray liquid-chromatography mass-spectrometry in the presence of signal suppression due to trifluoroacetic acid-containing mobile phases. J Chromatogr A. 1995;712:177–190. doi: 10.1016/0021-9673(95)00175-m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Reprogramming efficiency in ACDC fibroblasts.
Fig. S2. Representative karyotypes of ACDC iPSCs.
Fig. S3. Generation and characterization of ACDC iPSC lines overexpressing CD73.
Fig. S4. TNAP is not present in control human artery that is free from calcification.
Fig. S5. Schematic of differentiation of iPSCs into iMSCs.
Fig. S6. CD73 overexpression rescues osteogenic defects in ACDC iMSCs.
Fig. S7. Overexpression of CD73 decreases TNAP in ACDC iPSCs.
Fig. S8. Effect of AR agonists on calcification in teratomas.
Fig. S9. AR agonists do not affect teratoma size.
Fig. S10. TNAP activity in fibroblasts from control and ACDC patients.
Fig. S11. TNAP activity and calcification in hMSCs.
Fig. S12. Rapamycin treatment does not affect teratoma size.