

Abstract
8-Hydroxy-2-deoxyguanosine (8-OHdG), a marker of oxidative stress, has been recently rediscovered to inhibit Rac1 in neutrophils and macrophages, thereby inhibiting Rac1-linked functions of these cells, including reactive oxygen species production through NADPH oxidase activation, phagocytosis, chemotaxis, and cytokine release. In vascular smooth muscle cells (VSMCs), reactive oxygen species also induce abnormal proliferation and migration leading to progression of atherosclerosis. Based upon the involvement of reactive oxygen species in phagocytic cells and VSMCs during the atherosclerotic process, we hypothesized that 8-OHdG could have antiatherosclerotic action and tested this hypothesis in an experimentally induced atherosclerosis in mice. Partially ligated ApoE knockout mice, a more physiologically relevant model of low and oscillatory flow, developed an advanced lesion in 2 weeks, and orally administered 8-OHdG significantly reduced plaque formation along with reduced superoxide formation, monocyte/macrophage infiltration, and extracellular matrix (ECM) accumulation. The effects of 8-OHdG observed in primary VSMCs were consistent with the in vivo effects of 8-OHdG and were inhibitory to angiotensin II or platelet-derived growth factor-induced production of reactive oxygen species, proliferation, migration, and ECM production. Also, angiotensin II-induced Rac1 activity in VSMCs was significantly inhibited by 8-OHdG, and transfection of constitutively active Rac1 reversed the inhibitory effect of 8-OHdG on VSMC activation. Molecular docking study showed that 8-OHdG stabilizes Rac1–GEF complex, indicating the physical contact of 8-OHdG with Rac1. These findings highly suggest that the antiatherosclerotic effect of 8-OHdG is mediated by inhibition of Rac1 activity. In conclusion, our results show a novel action of orally active 8-OHdG in suppressing atherosclerotic plaque formation in vivo and VSMC activation in vitro through inhibition of Rac1, which emphasizes a new therapeutic avenue to benefit atherosclerosis.
Keywords: 8-Hydroxy-2-deoxyguanosine, Partial ligation, Atherosclerosis, Rac1, Vascular smooth muscle cell, Free radicals
Introduction
Atherosclerosis, an inflammatory disease of the blood vessel wall, is the primary cause of cardiovascular disease [1–3], and reactive oxygen species (ROS) are known to play important roles as signaling molecules in vascular physiology [4]. Vascular smooth muscle cells (VSMCs) form the medial layer of blood vessels, and abnormal proliferation of VSMCs with subsequent formation of intimal thickening is thought to play a role in the development of atherosclerotic lesions. Accumulating evidence implies that pathophysiological responses in VSMCs including migration, proliferation, and inflammation are mediated through ROS signal transduction pathways [5]. VSMCs contain several sources of ROS, among which the NADPH oxidases are predominant [6]. To date, efforts have been devoted to developing NADPH oxidase inhibitors that may have therapeutic potential in the treatment of human vascular diseases [7]. However, the results have been unsuccessful and therefore an effective therapeutic molecule still remains elusive.
Guanine in DNA is easily changed into 8-hydroxyguanine (8-oxo-Gua), which is removed from the DNA into body fluids by a repair process as a nucleoside, 8-hydroxy-2-deoxyguanosine (8-OHdG) [8,9]. 8-OHdG is usually detected in the urine or sera of patients and, therefore, elevated levels of 8-OHdG have been generally acknowledged as a biomarker for oxidative damage in diverse disease conditions, including atherosclerosis [10] and diabetes mellitus [11,12]. However, the exact biological significance of 8-OHdG has not been described. Recently, 8-OHdG has been rediscovered as an inhibitor of Rac1 in neutrophils and macrophages [13–15], thereby inhibiting Rac1-linked functions of these cells, including ROS production through NADPH oxidase activation, phagocytosis, chemotaxis, and cytokine release. The small G protein Rac1 is required in both phagocytes and non-phagocytic cells to activate NADPH oxidases [16]. Also, studies have shown that Rac1 plays an important role in angiotensin II (Ang II)- and platelet-derived growth factor (PDGF)-mediated NADPH oxidase activation in VSMCs [17,18]. Based on these ideas, we hypothesized that 8-OHdG could have potential effects in the prevention of VSMC activation and, furthermore, in the attenuation of atherosclerosis.
To test this hypothesis, we used a partial carotid ligation model using the ApoE knockout (KO) mouse, which is a newly developed and more physiologically relevant model compared to complete ligation of the common carotid artery or the perivascular cuff model [19]. In this model, three of the four caudal branches of the left common carotid arteries (LCA) are ligated to induce a substantial flow reduction in the LCA and the mice are fed a high-fat diet to accelerate the development of atherosclerotic lesions. Antiatherosclerotic action of 8-OHdG was evaluated in terms of its effects on the lesion development including superoxide formation, monocyte/macrophage infiltration, and extra-cellular matrix (ECM) accumulation. To observe the effects of 8-OHdG on nonphagocytic cells, primary VSMCs were selected to examine the effects of 8-OHdG on ROS liberation, VSMC proliferation, and ECM secretion. Also, the involvement of Rac1 was examined using Rac1 binding assay, transfection of constitutively active Rac1, and molecular docking study.
Materials and methods
Experimental animals and 8-OHdG treatment
Male ApoE KO mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and ligated at 8 weeks of age. All mice were fed a chow diet and water ad libitum until partial ligation. Partial ligation of the LCA was carried out as previously described [19]. Briefly, three of four caudal branches of the LCA (left external carotid, internal carotid, and occipital artery) were ligated with 6-0 silk suture and the superior thyroid artery was left intact. The incision was then closed with Tissue-Mend (Veterinary Product Laboratories, Phoenix, AZ, USA). After surgery, the mice were monitored until recovery in a chamber on a heating pad. A single subcutaneous injection of buprenorphine (0.1 mg/kg) was given 1 h after partial ligation for additional pain relief. Mice were fed the Paigen high-fat diet [20] (Science Diets, Topeka, KS, USA) immediately after partial ligation and sacrificed after 14 day. 8-OHdG (30 mg/kg/day) was dissolved in water and administered orally once a day starting at 2 day before ligation. The superoxide dismutase analog tempol (100 mg/kg/day) was used for comparison. The doses of 8-OHdG and tempol were decided through our preliminary study and previous studies [21,22]. Six mice were used for each group. Plasma triglycerides, cholesterol, and free fatty acids were analyzed using commercially available kits (Callegari, Spain, and BioAssay Systems, Hayward, CA, USA) according to the manufacturer’s instructions. Plasma aspartate aminotransferase (AST) levels were assayed at the Green Cross Reference Lab (Yongin-si, Kyunggi-do, Korea). All animal studies were carried out using procedures approved by the Emory University Institutional Animal Care and Use Committee.
Histology and immunohistochemical staining
Mice were sacrificed and pressure perfused with saline containing heparin. The LCA and the right common carotid artery (RCA) were collected en bloc with the trachea and esophagus. For frozen sections, tissue was embedded in Optimum Cutting Temperature medium (Sakura Finetek, Torrance, CA, USA), frozen on dry ice, and stored at −70 °C until used. Lesion (intimal) area was calculated as the difference between the internal elastic lamina area and the luminal area as described previously [19,23]. Micrographs were taken with a light microscope (Carl Zeiss, Thornwood, NY, USA). Images were analyzed with NIH ImageJ software to quantify lesion size in each animal.
For immunohistochemistry, MOMA-2 (anti-monocyte+macrophage antibody, 1:100; BMA Biomedicals, Switzerland) antibody was used. Oil red O staining was carried out using frozen sections as described previously [24]. For collagen staining, Masson’s trichrome staining kit (Sigma, St. Louis, MO, USA) was used according to the manufacturer’s instructions. Stained sections were counterstained with Mayer’s hematoxylin solution.
Dihydroethidium staining
Thick frozen sections (30 μm) obtained from the LCA of partially ligated mice were stained in 2 μm dihydroethidium (DHE) in phosphate-buffered saline for 30 min at 37 °C. Mounted cells were examined by a confocal microscope (LSM 510 META; Carl Zeiss).
Cell culture
VSMCs were obtained from the thoracic aorta of Sprague–Dawley rats by enzymatic digestion [25]. Briefly, the aortas were removed, cleaned of connective tissue and fat, and digested with collagenase and elastase to remove adventitia and to dissociate the VSMCs. Isolated cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. VSMCs from passages 3 to 7 were used for experiments.
Measurement of intracellular and extracellular ROS
VSMCs were seeded in 96-well plates or 8-well Lab-Tek chamber slides (Nunc, Rochester, NY, USA) at a density of 5 × 103 per well. Subconfluent cells were serum starved for 48 h, pretreated with 8-OHdG (100 μg/ml) for 1 h, and stimulated with Ang II (1 μm; Sigma), PDGF-BB (10 ng/ml; Sigma), or H2O2 (1 mM; Sigma) for 30 min and then loaded with the peroxide-sensitive fluorophore 2′,7′-dichlorofluorescin diacetate (DCF-DA; 5 μM; Molecular Probes, Carlsbad, CA, USA) for 20 min. Then, the cells were examined either by confocal microscopy or by fluorimeter (Molecular Devices, Sunnydale, CA, USA) at 485 nm excitation and 530 nm emission. In some experiments, N-acetylcysteine (5 mM) was used as a positive control. Hydrogen peroxide released from VSMCs was detected using an Amplex Red Hydrogen Peroxide Assay Kit (50 μM; Molecular Probes) according to the manufacturer’s instructions. Briefly, after 1 mM H2O2 stimulation for 30 min, the cells were washed and further incubated at 37 °C in fresh medium with Amplex red to detect the secretion of endogenous H2O2 levels. After 30 min incubation, the absorbance was measured at 560 nm. The fluorescence and absorbance were measured in triplicate to avoid well-to-well variation.
Gene expression analysis
Total RNA was extracted from VSMCs using Trizol (Invitrogen, Carlsbad, CA, USA) according to the standard protocol. mRNA expression was measured by real-time PCR using an ABI7300 (Applied Biosystems, Carlsbad, CA, USA) with 20 μl reaction volume consisting of cDNA transcripts, primer pairs, and SYBR Green PCR Master Mix (Applied Biosystems). Quantifications were normalized to 18S in each reaction. The sequences for the primers were as follows: 18S forward, 5′-AGGAATTGACGGAAGGGCAC-3′, reverse, 5′-GTGCAGCCCCGGACATCTAAG-3′; collagen type I forward, 5′-TCTAAGACATCCCTGGTCAC-3′, reverse, 5′-GTCCTTCCAGAAGAAACCTT-3′; fibronectin forward, 5′-CATGAAGGGGGTCAGTCCTA-3′, reverse, 5′-GTCCATTCCCCTTTTCCATT-3′; intracellular adhesion molecule-1 (ICAM-1) forward, 5′-AAACGGGAGATGAATGGTACCTAC-3′, reverse, 5′-TGCA CGTCCCTGGTGATACTC-3′; monocyte chemotactic protein-1 (MCP-1) forward, 5′-CTGTAGCATCCACGTGCTGT-3′, reverse, 5′-CCGACTCATT GGGATCATCT-3′; Nox1 forward, 5′-TCCTAAGAGGCTCCAGACCTCCA-3′, reverse, 5′-TGGCCAAGGCCAAGGCAGTT-3′; vascular cell adhesion molecule-1 (VCAM-1) forward, 5′-TGCACGGTCCCTAATGTGTA-3′, reverse, 5′-TGCCAATTTCCTCCCTTAAA.
Cell proliferation assay
Cell viability was determined using the TACS MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay kit (Trevigen, Gaithersburg, MD, USA). Briefly, cells (5 × 103 cells/well) were seeded into 96-well plates and incubated until 60–70% confluent. After 48 h of serum starvation, cells were stimulated with 15% FBS and 10 ng/ml PDGF-BB with or without 1 h 100 μg/ml 8-OHdG pretreatment. After 48 h, the tetrazolium compound MTT was added to the wells and the cells were incubated for an additional 4 h. MTT is reduced by metabolically active cells to insoluble purple formazan dye crystals. Detergent was then added to the wells to solubilize the crystals for 2 h at 37 °C. Absorbance was read at 570 nm using an ELISA plate reader (Molecular Devices). MTT assay was also performed to examine the cell viability in 8-OHdG (0–500 μg/ml)-treated VSMCs.
Aortic ring assay
Ex vivo proliferation and migration of VSMCs were measured by an aortic ring assay with some modifications [26]. The endothelium and adventitium of the aorta from Sprague–Dawley rats were removed enzymatically, and then the vessels were cut into rings (1 mm). The rings were placed and embedded in 48-well plates coated with Matrigel (BD Bioscience, San Diego, CA, USA). Embedded rings were incubated in 10% FBS DMEM for 2 day and then stimulated with PDGF-BB with or without 8-OHdG. After 48 h, the rings were photographed and the lengths of sprouts were analyzed using Scion Image software.
Western blot analysis
Fibronectin and collagen type I secretion was determined in the culture medium from treated VSMCs. Medium equivalent to 20 μg of cell lysates was loaded, separated by SDS–PAGE, and transferred to polyvinylidene difluoride membrane for 60 min and probed with antibodies (β-tubulin, 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA; fibronectin, 1:1000, Dako, Copenhagen, Denmark; collagen type I, 1:1000, Southern Biotech, Birmingham, AL, USA). The blots were detected using LAS-3000 (Fuji photo film; Tokyo, Japan).
DNA transfection
Plasmids encoding myc-tagged wild-type Rac1 (Rac1-WT) and the constitutively active form of Rac1 (Rac1-V12) were kindly provided by Professor Yun Soo Bae at Ewha Women’s University (Seoul, Korea). Both constructs were in the mammalian expression vector pEXV. For transient transfection of these vectors, the Amaxa Basic Nucleofector Kit (Lonza, Germany) for primary smooth muscle cells was used. Briefly, trypsinized VSMCs were mixed with the vectors (1 μg) and Nucleofector solution and then electroporated (program D-33). Cells were immediately seeded in six-well plates and grown for 72 h before experiment. Transfection efficiency of myc-tagged constructs was detected with anti-myc antibody (1:1000; Sigma) by Western blot analysis.
Rac1 activity assay
Active Rac1 was pulled down by interacting with GST-PAK-PBD (p21 activated kinase-protein binding domain) using the Rac1 activation assay kit (Upstate, Billerica, MA, USA), according to the manufacturer’s instructions. The kit provides better reactivity for mouse cells and therefore VSMCs obtained from C57BL/6 mice were used. Briefly, mouse VSMCs were pretreated with 8-OHdG for 1 h and then activated with 100 nM Ang II for 5 min. The cell lysates (2 mg) were incubated with GST-PAK-PBD beads for 2 h at 4 °C. The beads were washed, suspended in 2× Laemmli sample buffer, and separated by SDS–PAGE as described above. GTP-bound Rac1 levels were determined using monoclonal mouse anti-Rac1 antibody.
Molecular modeling study
Three-dimensional structures for 8-OHdG and its tautomer 8-oxo-2-deoxyguanosine (8-oxodG) were generated with Concord and energy minimized using the MMFF94s force field and MMFF94 charge until the rms of the Powell gradient was 0.05 kcal mol−1 A−1 in SYBYL-X 1.2 (Tripos International, St. Louis, MO, USA). Among the reported X-ray crystal structures of the Rac1–GEF complex, the Rac1–Vav1 structure was selected (PDB ID: 2VRW) because Vav1 is one of the GEFs involved in superoxide generation [27,28]. The X-ray crystal structure was prepared with the Biopolymer Structure Preparation tool in SYBYL, and the flexible docking study was performed using GOLD version 5.0.1 (Cambridge Crystallographic Data Centre, Cambridge, UK). To define the ligand binding site, the structure of the Rac1–GDP–RhoGDI complex (PDB ID: 1HH4) was aligned to that of the Rac1–Vav1 complex. The binding site was defined as 8 Å around the coordinates of the aligned GDP molecule. We used the protein H-bonding constraints in GOLD with a weighting factor of 10 for the respective carboxy oxygen of Asp118 and the backbone amide hydrogen of Ala159 to keep the key hydrogen bonds of the guanine base part of the GDP and GTP cocrystallized in Rac1 (PDB ID: 1HH4 [29], 1E96 [30]). Other parameters were set at their default values, and 30 docking runs per ligand were performed with the GoldScore scoring function. The docking results showed that both 8-OHdG and 8-oxodG bind to Rac1 in similar binding modes. The molecular graphic figures were generated with PyMOL software (http://www.pymol.org). All computation calculations were undertaken on an Intel Xeon Quad-Core 2.5-GHz workstation with Linux Cent OS release 5.5.
Statistics
Data are presented as the means±SE. Mean values obtained from each group were compared using ANOVA with subsequent Fisher’s significant difference method. Nonparametric analyses were also used where appropriate. A P value of <0.05 was used as the criterion for a statistically significant difference.
Results
8-OHdG inhibits plaque formation in partially ligated ApoE KO mice
To examine the effect of 8-OHdG treatment on lesion formation, ApoE KO mice were partially ligated. By 2 weeks, ligated LCA developed robust lipid lesion with reduced lumen area and increased vessel size compared to nonligated control RCA, which did not show any intima formation (Fig. 1 and Supplementary Fig. 1). The lesion area, measured as the difference between the internal elastic lamina area and the luminal area, was larger distal to the heart (distal section, nearer to the ligation site) with nearly completely blocked aortas. As shown in Fig. 1, 8-OHdG (30 mg/kg/day) treatment significantly reduced the lesion area (Fig. 1B), leading to prevention of vessel lumen occlusion (Fig. 1C). Tempol, which is a superoxide dismutase mimetic, was recently reported to ameliorate aortic ROS production in type 2 diabetes [22] and was used as a comparison in our animal model. As a result, tempol treatment (100 mg/kg) also effectively reduced lesion formation in LCA, comparable to 8-OHdG treatment. Interestingly, the effects of 8-OHdG and tempol were more evident at the distal section, which is closer to the site of ligation. Lipid profiles were only marginally different, with a small but significant reduction in plasma triglycerides in 8-OHdG-treated mice (Supplementary Table 1). The body weights were not different among groups before ligation and 2 weeks after ligation (before sacrifice). Plasma AST levels stayed in the normal range in all groups and at up to 120 mg/kg 8-OHdG treatment (48.84±9.39 U/L).
Fig. 1.
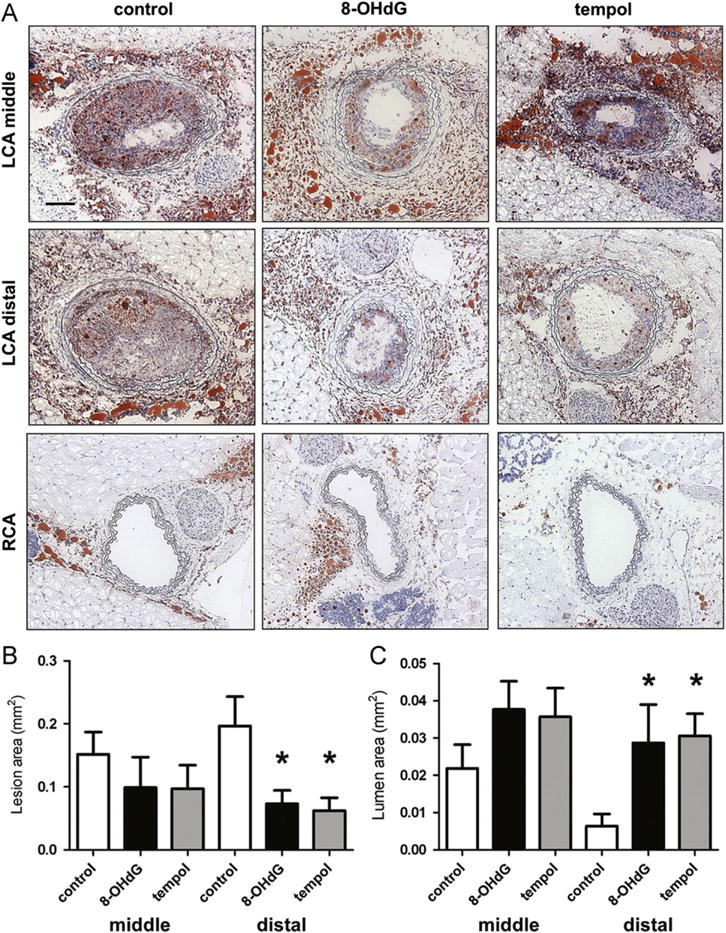
8-OHdG inhibits plaque formation in partially ligated ApoE KO mice. (A) ApoE KO mice were partially ligated and fed high-fat diet for 2 weeks. Frozen sections were stained with Oil Red O and hematoxylin. Shown are representative images of left and right carotid arteries. Scale bar; 100 μm. (B) Lesion area was calculated as the difference between internal elastic lamina area and luminal area and was quantified using Image J software. (C) Lumen area was also quantified. Values are means±SE of 6 mice per group. *P<0.05 vs. control distal left carotid artery (LCA). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
8-OHdG inhibits ROS formation in ApoE KO mice and cultured VSMCs
Because ROS are a well-known factor in lesion development, superoxide levels were measured by DHE staining and visualized by confocal microscopy (Fig. 2A). Excessive superoxide production was observed in the ligated vessel of control ApoE KO mice and was significantly reduced in 8-OHdG- and tempol-treated LCA. In cultured VSMCs, Ang II-, PDGF-, and H2O2-mediated intracellular ROS levels detected by DCF-DA were also effectively inhibited by 8-OHdG pretreatment (Fig. 2B and C), which was similar to pretreatment with N-acetylcysteine, a well-known antioxidant (Fig. 2B). To examine the effects of 8-OHdG on ROS secretion, Amplex red was used. As shown in Fig. 2D, secretion of ROS in the medium was increased by H2O2 stimulation, and 8-OHdG pretreatment significantly ameliorated the endogenous H2O2 secretion. Nox1 is a subunit of NADPH oxidase that is confined to VSMCs and expressed at low levels in the physiological state but upregulated by proatherogenic stimuli [31]. In our study, PDGF-induced Nox1 mRNA expression in VSMCs was significantly reduced by 8-OHdG treatment (Fig. 2E). These results suggest that 8-OHdG ameliorates plaque formation by suppressing oxidative stress particularly in VSMCs.
Fig. 2.
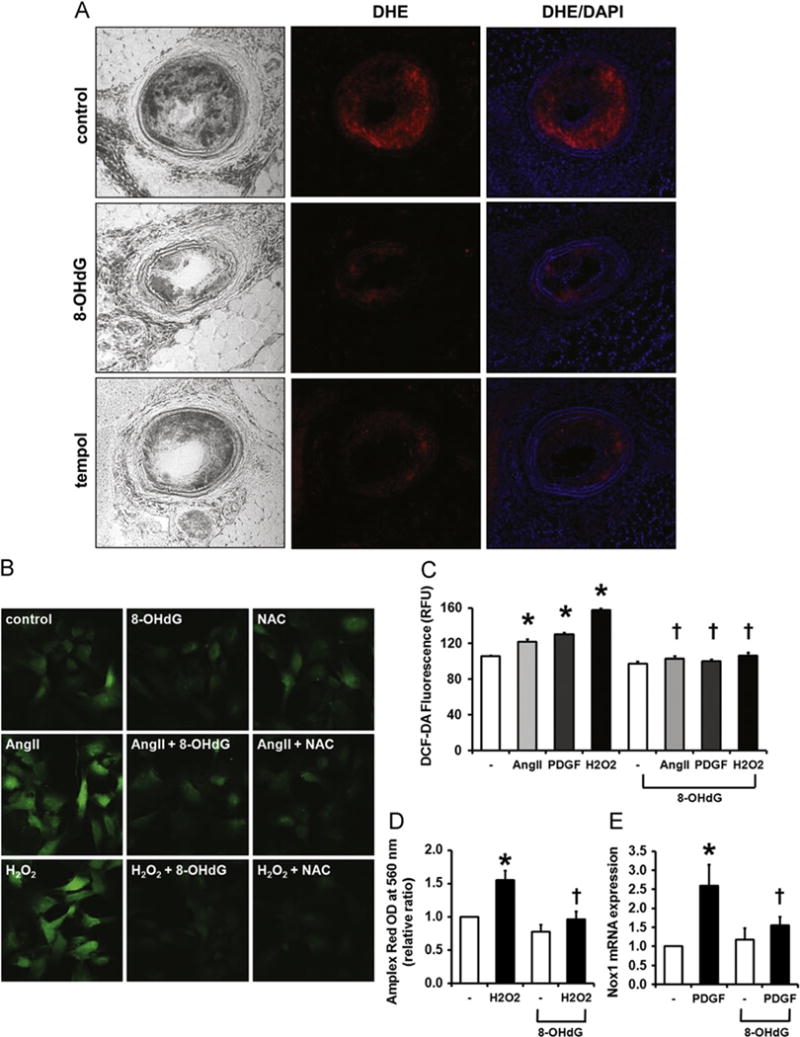
8-OHdG inhibits ROS formation in ApoE KO mice and cultured VSMCs. (A) Frozen sections of LCAs stained with DHE show decreased superoxide production in 8-OHdG treated aortas. DAPI was used to visualize nuclei. (B) Representative image of DCF-DA stained VSMCs. VSMCs were pretreated with 8-OHdG (100 μg/mL) or N-acetyl cysteine (NAC, 5 mM) for 1 h and stimulated with Ang II (1 μm) or H2O2 (1 mM) for 30 min. After stimulation, DCF-DA (5 μm) was loaded for 20 min, then fixed and mounted. (C) DCF-DA fluorescence was quantified by fluorometer in Ang II (1 μm), PDGF-BB (100 ng/mL), or H2O2 (1 mM) treated VSMCs. 8-OHdG (100 μg/mL) was pretreated for 1 h. (D) Secreted ROS was detected using Amplex Red. VSMCs were pretreated with 8-OHdG (100 μg/mL) for 1 h and stimulated with H2O2 (1 mM) 20 min. To remove the H2O2 stimulated, the cells were changed to fresh media and incubated for 30 min. Amplex Red (50 μm) was added to the medium the absorbance was detected at 560 nm. (E) Relative mRNA expression of Nox 1 in PDGF-BB treated VSMCs. 8-OHdG (100 μg/mL) was pretreated for 1 h and stimulated with PDGF-BB (10 ng/mL) for 24 h. Values are means±SE of 4 experiments. *P<0.05 vs. control, †P<0.05 vs. Ang II or PDGF-BB treated cells. DHE; dihydroethidium. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
8-OHdG inhibits PDGF- or serum-induced proliferation and migration of VSMCs
ROS induced by various stimuli results in VSMC activation [5]. To examine whether 8-OHdG modulates VSMC proliferation and migration, a rat aortic ring assay was performed. As shown in Fig. 3A and B, 8-OHdG treatment significantly prevented PDGF-induced sprouting of VSMCs in a dose-dependent manner. The inhibitory effect of 8-OHdG on proliferation was further confirmed by MTT assay in cultured VSMCs (Fig. 3C). VSMC proliferation increased by either 15% FBS or 10 ng/ml PDGF stimulation was significantly reduced by 8-OHdG treatment. Notably, MTT assay using VSMCs treated with 8-OHdG alone revealed that 8-OHdG has no toxicity with up to 500 μg/ml treatment (Supplementary Fig. 2).
Fig. 3.
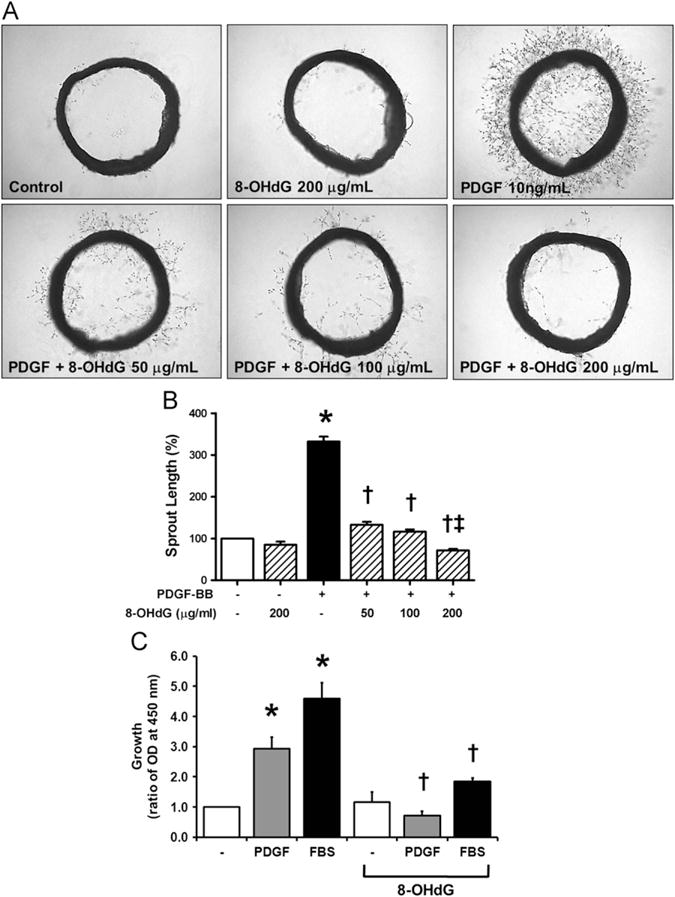
8-OHdG inhibits PDGF- or serum-induced proliferation and migration of VSMCs. (A) Representative images of rat aortic ring assay. (B) Sprout length was assessed by Scion Image Software. (C) Proliferation of VSMCs was examined by MTT assay. VSMCs were pretreated with 8-OHdG (100 μg/mL) for 1 h and stimulated with PDGF-BB (10 ng/mL) or FBS (15%) for 48 h. Values are means±SE of 3–4 experiments. *P<0.05 vs. control, †P<0.05 vs. PDGF-BB or FBS treated cells, ‡P<0.05 vs. PDGF-BB+8-OHdG (100 μg/mL).
8-OHdG inhibits monocyte/macrophage infiltration in ApoE KO mouse aortas and adhesion molecule expression in cultured VSMCs
Fatty streaks, which consist of lipid-laden monocytes and macrophages, are linked with migrated VSMCs in the progression to advanced lesion. Because 8-OHdG was previously reported to inhibit the activation of macrophages [13], we anticipated seeing differences in monocyte/macrophage infiltration in 8-OHdG-treated ApoE KO mice. Aortic sections stained with MOMA-2 showed that partial ligation resulted in massive accumulation of inflammatory cells inside the plaque (Fig. 4A). The MOMA-2 staining correlated with the oil red O-stained areas, which implies the development of foam cells. In contrast, monocyte/macrophage infiltration was completely ameliorated by 8-OHdG treatment. Along with activation of leukocytes, its adhesion to VSMCs is another important factor in lesion development, which is primarily mediated by cell adhesion molecules such as VCAM-1 and ICAM-1 [32]. As shown in Fig. 4B and C, ICAM-1 and VCAM-1 mRNA levels were induced by PDGF in cultured VSMCs and 8-OHdG pretreatment significantly reduced PDGF-induced but not basal expression. MCP-1 mRNA levels also tended to decrease with 8-OHdG treatment but did not reach statistical significance (Fig. 4D).
Fig. 4.
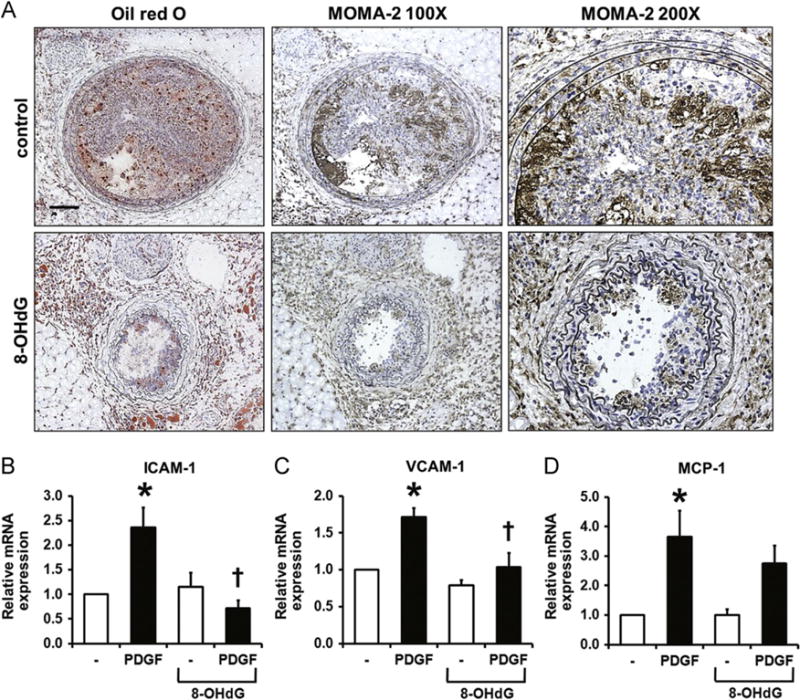
8-OHdG inhibits monocyte/macrophage infiltration in ApoE KO mice and adhesion molecule expression in cultured VSMCs. (A) Histological analysis of monocyte/macrophage infiltration was obtained by MOMA-2 staining in LCAs. Consecutive sections are shown to compare Oil Red O stained areas with MOMA-2 stained areas. Scale bar; 100 μm. Also, enlarged images of MOMA-2 staining are shown. (B) Real-time PCR results of ICAM-1, VCAM-1, and MCP-1 in VSMCs after 24 h PDGF-BB (10 ng/mL) treatment. 8-OHdG (100 μg/mL) was pretreated for 1 h before PDGF-BB treatment. Values are means±SE of 4 experiments. *P<0.05 vs. control, †P<0.05 vs. PDGF-BB treated cells. ICAM-1; Inter-cellular adhesion molecule 1, VCAM-1; vascular cell adhesion molecule, MCP-1; monocyte chemotactic protein-1.
8-OHdG attenuates ECM accumulation in ApoE KO mice and cultured VSMCs
ROS in VSMCs regulate not only cellular migration and proliferation but also accumulation of matrix proteins such as collagen [33,34]. As shown by Masson’s trichrome staining in Fig. 5A, partial ligation resulted in increased ECM accumulation, and 8-OHdG treatment significantly inhibited such accumulation. In cultured VSMCs, Ang II treatment significantly increased fibronectin and collagen type I secretion after 24 h of incubation, and pretreatment with 8-OHdG significantly diminished Ang II-induced ECM production (Fig. 5B). PDGF-induced mRNA expression of fibronectin and collagen type I was also significantly decreased by 8-OHdG treatment (Fig. 5C).
Fig. 5.
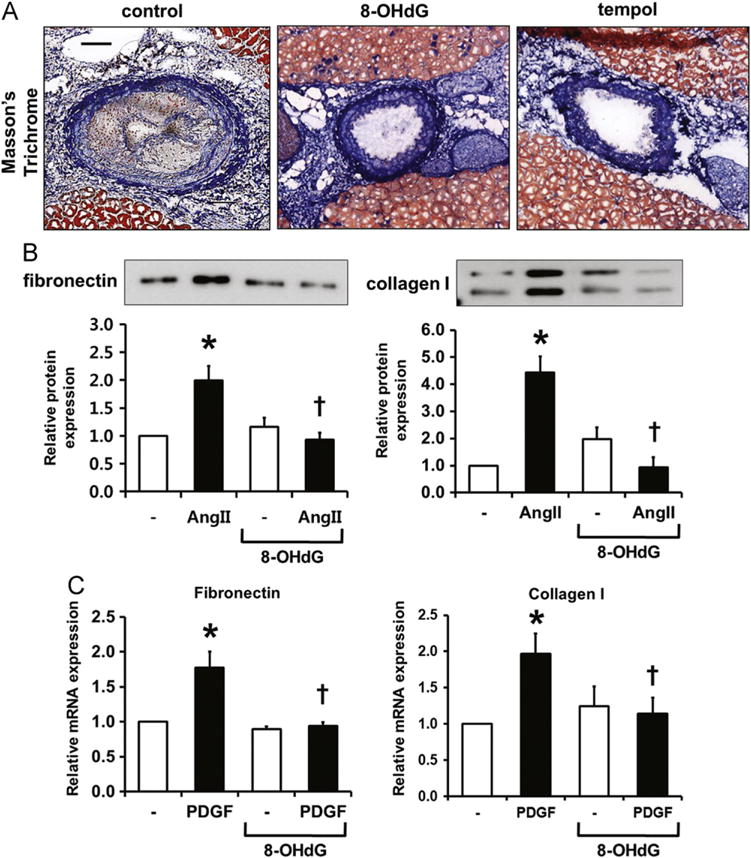
8-OHdG attenuates ECM accumulation in ApoE KO mice and cultured VSMCs. (A) Representative image of Masson’s trichrome staining in LCAs. Scale bar; 100 μm. (B) Measurement of fibronectin and collagen type I in culture media by Western blotting. Media was collected after 24 h treatment of Ang II (100 nM). (C) Real-time PCR results of fibronectin and collagen type I after 24 h PDGF-BB (10 ng/mL) treatment. 8-OHdG (100 μg/mL) was pretreated for 1 h before PDGF-BB treatment. Values are means±SE of 4 experiments. *P<0.05 vs. control, †P<0.05 vs. Ang II treated cells.
8-OHdG reduces Ang II-induced Rac1 activation and its downstream signaling in VSMCs
The inhibitory effects of 8-OHdG on vascular ROS, proliferation, migration, and ECM secretion shown so far have led us to examine the upstream mediator Rac1 for the effect of 8-OHdG on VSMCs. In line with our previous report that 8-OHdG suppressed Rac1 activation in leukocytes and macrophages [13–15], Ang II-induced Rac1–GTP levels were significantly inhibited by 8-OHdG (Fig. 6A and B). Activation of Rac1 by Ang II is known to activate the PAK/JNK pathway, which may also be involved in VSMC hypertrophy and migration [35]. To examine whether 8-OHdG blocks the downstream signaling of Rac1, Ang II-stimulated phosphorylation of JNK was observed in VSMCs. As a result, 8-OHdG pretreatment prevented Ang II-induced phosphorylation of JNK (Fig. 6A).
Fig. 6.
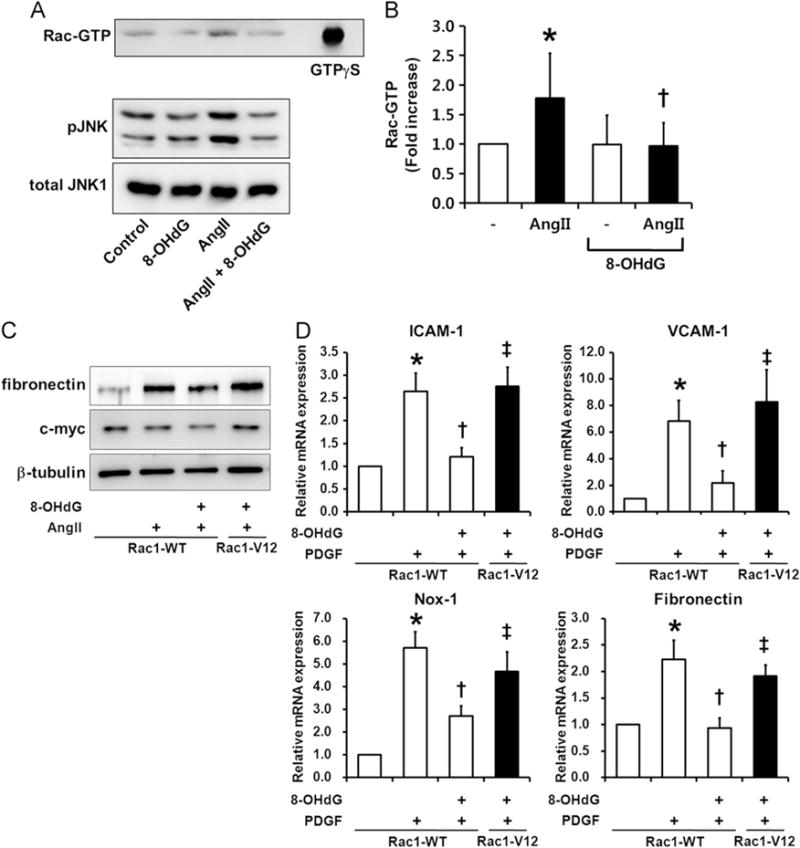
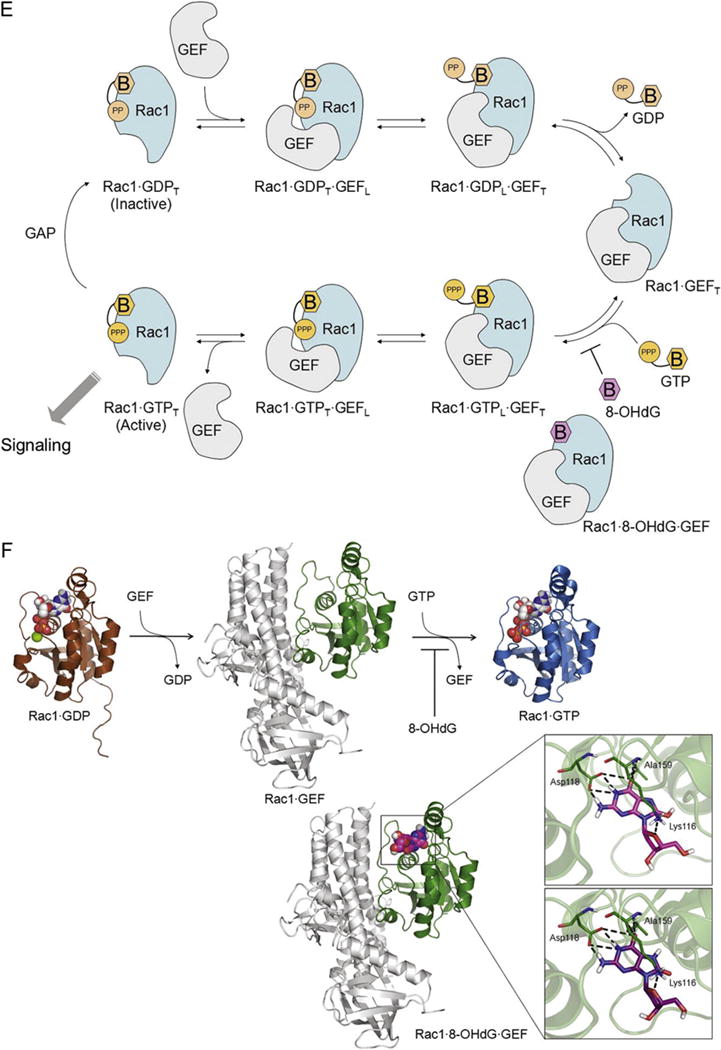
8-OHdG inhibits Rac1 activity. (A, B) Western blot analysis of Rac1-GTP and its downstream JNK phosphorylation. Values are means±SE of 3–4 experiments. *P<0.05 vs. control, †P<0.05 vs. Ang II (1 μm) treated cells. (C) Western blot analysis of VSMCs transfected with myc-tagged wild type Rac1 (Rac1-WT) or constitutively active Rac1 (Rac1-V12). Transfected cells were pretreated with 8-OHdG (100 μg/mL) for 1 h and then stimulated with Ang II (100 nM) for 24 h. Fibronectin was measured in the media collected. Anti-myc was used to monitor the transfection efficiency. (D) Real-time PCR results of ICAM-1, VCAM-1, Nox-1, and fibronectin in transfected cells. Transfected cells were pretreated with 8-OHdG (100 μg/mL) for 1 h and then stimulated with PDGF-BB (10 ng/mL) for 24 h. *P<0.05 vs. Rac1-WT, †P<0.05 vs. PDGF-BB treated Rac1-WT, ‡P<0.05 vs. PDGF-BB and 8-OHdG treated Rac1-WT. (E) Scheme for Rac1 regulation. Rac1 interacts with GDP and GTP (in orange) via their base (B) and phosphate moieties (PP for GDP and PPP for GTP). 8-OHdG (in magenta) could bind to the Rac1 GEF complex, stabilize the ternary complex of Rac1 8-OHdG GEF, and stop the activation of Rac1. The loose and tight interactions of Rac1 with the nucleotides and GEF are marked in subscript L and T, respectively. (F) Docking study showing 8-OHdG binding well at the base-binding site of Rac1 GEF, and the enlarged view of the detailed interactions. 8-OHdG and 8-oxodG are depicted in capped-stick with their carbon color in magenta and purple, respectively, and the interacting residues are in thin-stick with their carbon color in green. The hydrogen bonds are marked in black dashed lines. Rac1 structures bound with GDP(Mg), GTP(Mg), and GEF are represented in their secondary structures and colored in brown, sky-blue, and green, respectively. GEF is displayed in gray ribbons. The ligands are depicted in spacefill with their carbon color in white for GDP and GTP, and magenta for 8-OHdG. The non-polar hydrogens are undisplayed for clarity. GEF; guanosine exchange factor, GAP; GTPase-activating protein.
Constitutively active Rac1 reverses the protective effect of 8-OHdG in VSMCs
To confirm whether Rac1 is the key molecule responsible for the inhibitory effect of 8-OHdG on VSMC activation, cells were transiently transfected with vectors encoding myc-tagged constitutively active Rac1 (Rac1-V12) or its control (Rac1-WT). Electroporation resulted in efficient transfection of vectors determined by expression of myc at the size of Rac1 (approximately 20 kDa) by Western blotting (Fig. 6C). Expression of Rac1-V12, in which amino acid 12 was substituted with Val, completely reversed the effect of 8-OHdG to downregulate fibronectin, VCAM-1, ICAM-1, and Nox1 mRNA levels and fibronectin protein levels as shown in Ang II- or PDGF-stimulated Rac1-WT transfected cells (Fig. 6C and D).
Molecular docking study shows that 8-OHdG stabilizes Rac1–GEF complex
One question that remains to be answered is, “How can 8-OHdG inactivate Rac1?” To investigate the direct interaction of 8-OHdG with Rac1, a molecular docking study was carried out. Considering the biological data, the X-ray crystal structure of Rac1 complexed with GEF was selected. The docking result showed that 8-OHdG nicely binds to Rac1, by occupying the base-binding site and making good interactions with the surrounding residues including hydrogen bonds with Lys116, Asp118, and Ala159 (Fig. 6F). This docking result fits to the biological data and explains how 8-OHdG blocks Rac1–GTP binding (Fig. 6E). In the case of GTP, its phosphate group perturbs the binding interface of Rac1–GEF complex and drives GEF to be released. Without having the phosphate group, 8-OHdG fits well to the base-binding site of Rac1–GEF and stabilizes the ternary complex (i.e., Rac1–8-OHdG–GEF; Fig. 6). Consequently, the dissociation of GEF is interrupted and 8-OHdG would stop the activation of Rac1.
Discussion
In this study, we examined the anti-atherogenic effects of 8-OHdG using a partially ligated ApoE KO mouse model. We demonstrated for the first time that orally administered 8-OHdG prevents plaque formation with reduced superoxide production, ECM accumulation, and monocyte/macrophage infiltration in vivo and inhibits ROS generation, proliferation, migration, and ECM secretion of VSMCs in vitro. We also found that Rac1 is the key molecule responsible for 8-OHdG-mediated antiatherosclerotic effects in VSMCs.
When the guanine base in DNA is attacked by ROS, 8-oxo-Gua is formed. 8-Oxo-Gua in DNA can be detrimental because of its ability to induce transversion mutations [36]. To counteract the hazardous effects of 8-oxo-Gua, 8-OHdG is generated through multiple repair systems [37] and is not incorporated back into DNA. Although the role of endogenously formed 8-OHdG is largely unknown, we have recently rediscovered that exogenous 8-OHdG has anti-inflammatory and antioxidative effects in brain microglial cells and peritoneal macrophages by blocking the small G protein Rac1 [13–15].
In addition to the importance of Rac1 in phagocytic cells in the atherosclerotic process, a recent observation by Li et al. [38] indicated that VSMC-specific Rac1 KO mice are protected from neointimal formation after vascular injury, emphasizing the role of Rac1 in VSMC activation and development of atherosclerosis. Major elements of the signaling repertoire in VSMCs are known to be ROS dependent [5] and Rac1 is a critical component for Ang II-or PDGF-induced NADPH oxidase activation in VSMCs [17]. We have shown by Rac1 binding assay that 8-OHdG suppresses Ang II-induced activation of Rac1 (Fig. 6A) and subsequently that Ang II- and PDGF-induced ROS levels are downregulated by 8-OHdG (Fig. 2B, C, and D). Notably, there are challenges and limitations in using fluorescent probes developed so far for detecting and measuring ROS [39]. Dihydroethidium is a standard fluorophore for measuring ROS in damaged aorta [40–42]. To compare the results of 8-OHdG, tempol was used throughout the in vivo study. To verify our results in vitro, H2O2 and N-acetylcysteine were used as positive controls (Fig. 2B and C). Also, to overcome the limitation of DCF-DA staining, Amplex red was used to examine the effect of 8-OHdG on the amelioration of secreted ROS and showed that 8-OHdG significantly reduced H2O2 secretion in VSMCs (Fig. 2D). These results support the previous studies in vitro and in vivo, which include quenching of hydroxyl radical signal in ESR spectroscopy [43], strong inhibition of ROS-induced cleavage of DNA [44], and inhibition of nitrotyrosine formation in inflammatory lung tissue in ovalbumin-sensitized mice [21] by 8-OHdG. Together, they indicate that 8-OHdG suppresses oxidative stress in atherosclerotic lesion and VSMCs through antioxidative actions by ROS scavenging and/or inhibition of ROS formation. Nox1 mRNA expression in VSMCs induced by Ang II and PDGF is also responsible for stimulation of superoxide production [31,45] and it has been reported that Nox1 expression itself is regulated in a redox-sensitive manner, evidenced by treatment of antioxidants [46]. Moreover, deficiency of Rac1 not only blocks NADPH oxidase activation but also inhibits gp91phox expression levels in diabetic hearts [47], suggesting the importance of Rac1 in regulating NADPH oxidase subunits. As shown in Fig. 2E, PDGF-induced Nox1 mRNA expression in VSMCs was significantly inhibited by 8-OHdG, partly supporting the role of Rac1 in Nox1 regulation.
Rac1-mediated ROS generation results in initiation of various signaling pathways such as JNK (Fig. 6A), ERK, and p38 MAPK [7] to activate VSMCs. Our results have shown that antioxidative effects of 8-OHdG led to the attenuation of the deleterious effects of Ang II and PDGF on VSMCs including adhesion molecule expression (Fig. 4B and C), proliferation (Fig. 3), and ECM accumulation (Fig. 5B). Consistently, oral administration of 8-OHdG inhibited atherosclerotic development, including monocyte/macrophage infiltration (Fig. 4A) and ECM accumulation (Fig. 5A), in mouse LCA.
The Rac1–ROS signaling pathway is a crucial mediator of cardiovascular disease and therefore, 8-OHdG has the potential to be an important therapeutic molecule. However, because Rac1 plays an important role not only in the vasculature but in the whole body, the toxicity of exogenous 8-OHdG should be considered. 8-OHdG treatment by gavage had no effect on body weight or AST levels after 16 day of treatment (Supplementary Table 1). In addition, MTT assay using VSMCs treated with 8-OHdG alone revealed that 8-OHdG did not affect cell viability up to 500 μg/ml treatment (Supplementary Fig. 2). In the previous studies we did not observe any histological changes in lung, liver, and intestines of mice with even much higher doses than 30 mg/kg [21,48,49]. Although 30 mg/kg is a fairly large dose in clinical terms, the dose of numerous pharmaceutical modifiers such as statins and fibrates tested in mouse models of atherosclerosis so far has ranged generally between 10 and 100 mg/kg/day [50]. Considering the utilization of 8-OHdG to treat atherosclerosis in humans, lower doses should be tested possibly in preclinical processes. To eliminate the possible off-target effects of 8-OHdG at the cellular level, constitutively active Rac1 (Rac1-V12) was transfected into VSMCs. mRNA and protein expression showed that mutant Rac1 significantly reversed the protective effects of 8-OHdG (Fig. 6C and D). Another concern is whether 8-OHdG is salvaged for DNA synthesis because 8-oxo-Gua in DNA mismatches with adenine, whereas guanine matches with cytosine, thus causing conversion of GC to AT [36,51]. However, 8-OHdG cannot become 8-OH-dGTP, a DNA polymerase substrate, because 8-OH-dGMP is not a substrate for guanylate kinase [52], which converts 8-OH-dGMP into 8-OH-dGDP. In the experiment using the radiolabeled 8-OHdG [53], we confirmed that 8-OHdG was not incorporated into DNA. Consistent with these findings, a series of genetic toxicity studies showed negative results (unpublished data).
Molecular modeling plays an important role in drug discovery and is widely utilized to predict protein–ligand interactions at the molecular and atomic levels. Taken together with the experimental biological data, a compound’s mode of action could be uncovered [54]. Based on our biological data, we deduced that 8-OHdG would bind to the Rac1–GEF complex and stabilize the ternary complex of Rac1–GEF–8-OHdG as shown in Fig. 6E. Our results confirmed that 8-OHdG could directly bind to the Rac1–GEF complex by occupying the base-binding site and making interactions with the surrounding residues (Fig. 6F). This is in line with the previous result that showed 8-OHdG inhibits Rac1 activation without influencing Rac–GEF activity in LPS-stimulated RAW 264.7 cells [43]. Recognition of the 8-OHdG binding site would enable the development of a more potent and safer drug for clinical use.
To date, several experimental molecules have been developed as inhibitors of oxidative stress, many of which focused on blockade of NADPH oxidases [7]. However, the majority of the compounds showed limited potential for clinical therapeutics either because they cannot be administered orally or because of a lack of specificity for NADPH oxidases. Also, many other studies regarding antioxidant vitamin therapy have also failed to show clinical benefit [55]. Therefore, development of 8-OHdG as a novel inhibitor of Rac1 would certainly be useful for treating atherosclerosis and a broad range of other diseases associated with NADPH oxidase activation.
Other studies regarding the exogenous treatment of 8-OHdG have been recently reported in several animal models by our group and others. Kim et al. [21] have demonstrated that 8-OHdG can inhibit allergy-induced inflammation and remodeling in airway and lung tissues through Rac inactivation. Also, the in vivo pretreatment efficacy of exogenous 8-OHdG in a rat model of water immersion restraint stress-induced gastritis has been documented [43]. So far, the effect of 8-OHdG has been mostly focused on inhibition of inflammation-provoking activities of Rac1 in phagocytes. This study has expanded the concept to nonimmune cells, which raises the possibility that 8-OHdG could be useful in both ways.
The pathogenesis of atherosclerosis is a complex process due to the diverse cellular compositions of blood vessels. Apart from the effects of 8-OHdG on VSMCs, it would be interesting to observe the effects of 8-OHdG on endothelial cells, because endothelial dysfunction precedes the formation of lesions in early atherosclerosis [56].
In summary, we have provided the first evidence that 8-OHdG prevents the development of advanced lesions in partially ligated ApoE KO mice through inhibition of Rac1 and related functions with beneficial effects in ameliorating VSMC activation and ROS generation. This points to a novel mechanism of action of synthetic 8-OHdG to reduce oxidative stress-associated pathogenic process and emphasizes a new therapeutic avenue to benefit atherosclerosis.
Supplementary Material
Acknowledgments
This work was supported, in part, by the Ministry of Education, Science, and Technology (MEST, 314-2008-1-E00056 and 2011-0028885) and the WCU project (R31-2008-000-10010-0). Joo Young Huh, Yoonji Lee, Junghyun Lee, and Boyeon Kim were supported by the Brain Korea 21 Project.
Abbreviations
- 8-OHdG
8-hydroxy-2-deoxyguanosine
- 8-oxodG
8-oxo-2-deoxyguanosine
- 8-oxo-Gua
8-hydroxyguanine
- Ang II
angiotensin II
- AST
aspartate aminotransferase
- DHE
dihydroethidium
- ECM
extracellular matrix
- GEF
guanosine exchange factor
- ICAM-1
intracellular adhesion molecule-1
- KO
knockout
- LCA
left common carotid artery
- MCP-1
monocyte chemotactic protein-1
- PDGF
platelet-derived growth factor
- RCA
right common carotid artery
- ROS
reactive oxygen species
- VCAM-1
vascular cell adhesion molecule-1
- VSMC
vascular smooth muscle cell
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freerad biomed.2012.03.023.
References
- 1.Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 3.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 5.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 7.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 8.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol Chem. 2006;387:373–379. doi: 10.1515/BC.2006.050. [DOI] [PubMed] [Google Scholar]
- 9.Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 11.Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, Nicotera T. Oxidative damage to DNA in diabetes mellitus. Lancet. 1996;347:444–445. doi: 10.1016/s0140-6736(96)90013-6. [DOI] [PubMed] [Google Scholar]
- 12.Broedbaek K, Weimann A, Stovgaard ES, Poulsen HE. Urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine as a biomarker in type 2 diabetes. Free Radic Biol Med. 2011;51:1473–1479. doi: 10.1016/j.freeradbiomed.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Taek Han S, Choi SW, Sung SY, You HJ, Ye SK, Chung MH. Inhibition of rac and rac-linked functions by 8-oxo-2′-deoxyguanosine in murine macrophages. Free Radic Res. 2009;43:78–84. doi: 10.1080/10715760802609432. [DOI] [PubMed] [Google Scholar]
- 14.Kim HS, Ye SK, Cho IH, Jung JE, Kim DH, Choi S, Kim YS, Park CG, Kim TY, Lee JW, Chung MH. 8-Hydroxydeoxyguanosine suppresses NO production and COX-2 activity via Rac1/STATs signaling in LPS-induced brain microglia. Free Radic Biol Med. 2006;41:1392–1403. doi: 10.1016/j.freeradbiomed.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 15.Kim DH, Cho IH, Kim HS, Jung JE, Kim JE, Lee KH, Park T, Yang YM, Seong SY, Ye SK, Chung MH. Anti-inflammatory effects of 8-hydroxydeoxyguanosine in LPS-induced microglia activation: suppression of STAT3-mediated intercellular adhesion molecule-1 expression. Exp Mol Med. 2006;38:417–427. doi: 10.1038/emm.2006.49. [DOI] [PubMed] [Google Scholar]
- 16.Hordijk PL. Regulation of NADPH oxidases: the role of rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 17.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 18.Kong G, Lee S, Kim KS. Inhibition of rac1 reduces PDGF-induced reactive oxygen species and proliferation in vascular smooth muscle cells. J Korean Med Sci. 2001;16:712–718. doi: 10.3346/jkms.2001.16.6.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–1543. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 21.Kim JS, Kim DY, Lee JK, Ro JY, Chung MH. 8-Oxo-2′-deoxyguanosine suppresses allergy-induced lung tissue remodeling in mice. Eur J Pharmacol. 2011;651:218–226. doi: 10.1016/j.ejphar.2010.10.087. [DOI] [PubMed] [Google Scholar]
- 22.San Martin A, Du P, Dikalova A, Lassegue B, Aleman M, Gongora MC, Brown K, Joseph G, Harrison DG, Taylor WR, Jo H, Griendling KK. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2007;292:H2073–2082. doi: 10.1152/ajpheart.00943.2006. [DOI] [PubMed] [Google Scholar]
- 23.Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am J Pathol. 2002;160:2145–2155. doi: 10.1016/S0002-9440(10)61163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 25.Ohmi K, Masuda T, Yamaguchi H, Sakurai T, Kudo Y, Katsuki M, Nonomura Y. A novel aortic smooth muscle cell line obtained from p53 knock out mice expresses several differentiation characteristics. Biochem Biophys Res Commun. 1997;238:154–158. doi: 10.1006/bbrc.1997.7218. [DOI] [PubMed] [Google Scholar]
- 26.Nicosia RF, Ottinetti A. Growth of microvessels in serum-free matrix culture of rat aorta: a quantitative assay of angiogenesis in vitro. Lab Invest. 1990;63:115–122. [PubMed] [Google Scholar]
- 27.Rapley J, Tybulewicz VL, Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008;9:655–661. doi: 10.1038/embor.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price MO, Atkinson SJ, Knaus UG, Dinauer MC. Rac activation induces NADPH oxidase activity in transgenic COSphox cells, and the level of superoxide production is exchange factor-dependent. J Biol Chem. 2002;277:19220–19228. doi: 10.1074/jbc.M200061200. [DOI] [PubMed] [Google Scholar]
- 29.Grizot S, Faure J, Fieschi F, Vignais PV, Dagher MC, Pebay-Peyroula E. Crystal structure of the Rac1–RhoGDI complex involved in NADPH oxidase activation. Biochemistry. 2001;40:10007–10013. doi: 10.1021/bi010288k. [DOI] [PubMed] [Google Scholar]
- 30.Lapouge K, Smith SJ, Walker PA, Gamblin SJ, Smerdon SJ, Rittinger K. Structure of the TPR domain of p67 phox in complex with rac.GTP. Mol Cell. 2000;6:899–907. doi: 10.1016/s1097-2765(05)00091-2. [DOI] [PubMed] [Google Scholar]
- 31.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 32.Braun M, Pietsch P, Schror K, Baumann G, Felix SB. Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res. 1999;41:395–401. doi: 10.1016/s0008-6363(98)00302-2. [DOI] [PubMed] [Google Scholar]
- 33.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 34.Park J, Chang HK, Ha H, Kim MS, Ahn HJ, Kim YS. Mycophenolic acid inhibits cell proliferation and extracellular matrix synthesis in rat vascular smooth muscle cells through direct and indirect inhibition of cellular reactive oxygen species. J Surg Res. 2008;150:17–23. doi: 10.1016/j.jss.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 35.Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, Eguchi S. Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension. 2006;48:534–540. doi: 10.1161/01.HYP.0000237975.90870.eb. [DOI] [PubMed] [Google Scholar]
- 36.Moriya M. Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C-T.A transversions in simian kidney cells. Proc Natl Acad Sci USA. 1993;90:1122–1126. doi: 10.1073/pnas.90.3.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooke MS, Evans MD, Dove R, Rozalski R, Gackowski D, Siomek A, Lunec J, Olinski R. DNA repair is responsible for the presence of oxidatively damaged DNA lesions in urine. Mutat Res. 2005;574:58–66. doi: 10.1016/j.mrfmmm.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Hiroi Y, Liao JK. Critical role of smooth muscle Rac1 in neointimal formation after vascular injury. Circulation. 2010;122:10021. [Abstract] [Google Scholar]
- 39.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuhmacher S, Foretz M, Knorr M, Jansen T, Hortmann M, Wenzel P, Oelze M, Kleschyov AL, Daiber A, Keaney JF, Jr, Wegener G, Lackner K, Munzel T, Viollet B, Schulz E. a1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arterioscler Thromb Vasc Biol. 2011;31:560–566. doi: 10.1161/ATVBAHA.110.219543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ., Jr Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. 2011;216:321–326. doi: 10.1016/j.atherosclerosis.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ock CY, Hong KS, Choi KS, Chung MH, Kim YS, Kim JH, Hahm KB. A novel approach for stress-induced gastritis based on paradoxical antioxidative and anti-inflammatory action of exogenous 8-hydroxydeoxyguanosine. Biochem Pharmacol. 2010;81:111–122. doi: 10.1016/j.bcp.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 44.Kim JE, Choi S, Yoo JA, Chung MH. 8-Oxoguanine induces intramolecular DNA damage but free 8-oxoguanine protects intermolecular DNA from oxidative stress. FEBS Lett. 2004;556:104–110. doi: 10.1016/s0014-5793(03)01385-1. [DOI] [PubMed] [Google Scholar]
- 45.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimoto T, Fukai N, Sato R, Sugiyama T, Ozawa N, Shichiri M, Hirata Y. Antioxidant effect of adrenomedullin on angiotensin II-induced reactive oxygen species generation in vascular smooth muscle cells. Endocrinology. 2004;145:3331–3337. doi: 10.1210/en.2003-1583. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. 2010;59:2033–2042. doi: 10.2337/db09-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ock CY, Kim EH, Hong H, Hong KS, Han YM, Choi KS, Hahm KB, Chung MH. Prevention of colitis-associated colorectal cancer with 8-hydroxydeoxyguanosine. Cancer Prev Res (Philadelphia) 2011;4:1507–1521. doi: 10.1158/1940-6207.CAPR-11-0161. [DOI] [PubMed] [Google Scholar]
- 49.Ro JY, Kim DY, Lee SH, Park JW, Chung MH. Effects of 7,8-dihydro-8-oxo-deoxyguanosine on antigen challenge in ovalbumin-sensitized mice may be mediated by suppression of rac. Br J Pharmacol. 2009;158:1743–1752. doi: 10.1111/j.1476-5381.2009.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 51.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 52.Hayakawa H, Taketomi A, Sakumi K, Kuwano M, Sekiguchi M. Generation and elimination of 8-oxo-7,8-dihydro-2′-deoxyguanosine 5′-triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry. 1995;34:89–95. doi: 10.1021/bi00001a011. [DOI] [PubMed] [Google Scholar]
- 53.Kim JE, Chung MH. 8-Oxo-7,8-dihydro-2′-deoxyguanosine is not salvaged for DNA synthesis in human leukemic U937 cells. Free Radic Res. 2006;40:461–466. doi: 10.1080/10715760600570539. [DOI] [PubMed] [Google Scholar]
- 54.Pfisterer PH, Wolber G, Efferth T, Rollinger JM, Stuppner H. Natural products in structure-assisted design of molecular cancer therapeutics. Curr Pharm Des. 2010;16:1718–1741. doi: 10.2174/138161210791164027. [DOI] [PubMed] [Google Scholar]
- 55.Yokoyama M. Oxidant stress and atherosclerosis. Curr Opin Pharmacol. 2004;4:110–115. doi: 10.1016/j.coph.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.