

Abstract
Knowledge on cellular differentiation pathways is critical to understanding organ development, homeostasis, and disease. Studying cell differentiation and cell fate restrictions in these contexts can be done through lineage tracing experiments, which entail permanent labeling of a cell and all its progeny. Recent lineage experiments within the cardiovascular system have uncovered unexpected findings on cellular origins during organogenesis and cell plasticity during disease. For example, there is increasing evidence that multiple progenitor sources exist for a single cell type, and that cells have remarkable expansive capacities under disease settings. Here, we summarize some recent findings in the cardiovascular system and highlight where there is evidence that the underlying concepts are a widespread phenomenon used by other organ systems.
Keywords: genetic lineage tracing, clonal analysis, cardiovascular system, progenitor cells
Genetic Lineage Tracing
Today, the most common fate mapping method in vertebrate species is genetic lineage tracing using the Cre/loxP system (Kretzschmar and Watt, 2012). Cre recombinase, an enzyme that catalyzes recombination between two specific DNA loxP sequences, can be expressed in specific cell types through inserting the Cre gene into the genome under the control of a tissue specific enhancer/promoter. The Cre allele is then coupled with a reporter allele that contains a transcriptional stop sequence flanked by loxP sites 5′ to a marker gene. In cells expressing Cre, there is recombination of the loxP sites excising the stop sequence, resulting in the permanent expression of the marker gene in Cre-expressing cell and all of its progeny. The most common markers are β-galactosidase and fluorescent proteins, which can be followed through histological examination. In addition to alleles that express a single lineage marker upon recombination, reporters that randomly express a variety of different fluorescent colors also exist, such as Brainbow (Livet et al., 2007), Rainbow (Rinkevich et al., 2011), Confetti (Snippert et al., 2010), etc. Multicolor recombination strategies mark single progenitor cells with distinct colors so that: (1) clonal level analysis can be performed and (2) multiple clonal populations can be followed in a single animal. The cell type-specific expression of Cre generates spatial regulation, but temporal control can also be introduced by fusing Cre to the estrogen receptor (ER). A CreER is inactive in the absence of its ligand so that labeling can be restricted to a time period in which a ligand has been applied. Cre recombinase technology has become a ubiquitous and essential tool for studying cell biology during development and in the adult in multiple organisms.
Despite the power and sophistication of available lineage tracing tools, there are important caveats to keep in mind. The interpretation of fate mapping results relies on the specificity of Creinduced labeling. Expression in other cell types and leaky expression from the genetic construct give rise to misleading conclusions. In addition, it is rare for any enhancer/promoter to be specifically expressed in only one cell type throughout development or in the adult animal. Thus, there must be a careful evaluation of the extent of Cre expression in each transgenic line before it is used for mapping cell fates. Methods to increase confidence in Cre specificity include the above-described CreER to temporally restrain labeling. Confidence in progenitor cell identity can also be increased by clonal analysis where single cells are initially labeled and progeny are clonally related. Although caveats exist with genetic lineage tracing, they are often surmountable with careful experimentation and produce important information on the cell differentiation pathways in development and disease. In addition to contemporary Cre-mediated lineage tracking methods, genome editing followed by next generation deep sequencing has allowed scientists to mark individual cells and construct lineage trees based on shared mutations in their DNA barcodes (GESTALT, genome editing of synthetic target arrays for lineage tracing) (McKenna et al., 2016).
Since the advent of Cre technology, the number of cell type specific Cre lines has exploded, mostly due to their use in creating tissue-specific gene knockouts. A common side effect has been the unexpected discovery of novel lineage relationships. Upon developing a Cre mouse line, one must first confirm the pattern of recombination by crossing it to a Cre reporter, an experiment that often leads to surprising findings on cell differentiation. One theme that is emerging from these observations is that multiple progenitor cells exist for a single tissue type. Another is some unpredicted cellular expansion and plasticity during disease models. Below, we outline such recent examples in the cardiovascular system.
Multiple Progenitors Exist for Organ-Specific Cardiovascular Cell Types
Coronary Vessels
Coronary arteries provide blood to ventricular heart muscle, and their health is critical to cardiomyocyte function. Thus, there is much interest in understanding how these vessels form, particularly for regenerative medicine purposes. Coronary arteries are composed of three major cell types: endothelial cells lining the vessel lumen, smooth muscle cells covering the vessel tube, and adventitial fibroblasts forming the outermost layer. An effective regeneration response must account for all of these cell types, and the production of new Cre lines has recently refined our understanding of how coronary cells emerge from multiple progenitor sources.
Coronary Endothelial Cells
To date, three different sources for the endothelial cells that line coronary vessels in mice have been identified, and their contribution has recently been a subject of much debate. Early lineage analyses performed in the 1990s using cross species tissue transplantations in avian embryos indicated that coronary vessels arise from an extracardiac progenitor source called the proepicardium (Poelmann et al., 1993; Männer, 1999). This structure is a group of transient embryonic cells attached to the venous pole of heart that migrate onto the heart and give rise to the epicardium (the outer layer of the heart) and other stromal cells within ventricle muscle (Mikawa and Gourdie, 1996). Following these experiments, the progenitor capacity of the proepicardium was assumed to occur in other species, including humans, and it was concluded that coronary vessels derive from the proepicardium.
However, a surprising finding occurred with the production of Cre mouse lines that were expressed in the proepicardium (Wt1-Cre, Tbx18-Cre, and others). Crossing proepicardial Cre lines to a reporter did not lead to lineage labeling of the majority of coronary endothelial cells, but instead primarily traced coronary smooth muscle and cardiac fibroblasts (Wilm et al., 2005; Cai et al., 2008; Zhou et al., 2008). Subsequent experiments in mice used histological observations and clonal labeling of endothelial cells (VE-Cadherin-CreER) to conclude that the coronary endothelial layer develops primarily through angiogenic sprouting of the venous inflow vessel of the heart called the sinus venosus with contributions from the endocardium that buds out from the heart lumen (Red-Horse et al., 2010). In parallel studies, fortuitous observations using an Nfatc1-Cre line, which was originally produced to study the endocardium, showed that a large portion of endocardial cells in mice contribute to the coronary endothelial cell layer (Wu et al., 2012). The sinus venosus was later found to also contribute to a significant portion of the coronary endothelium through bulk lineage tracing of the sinus venosus using Apj-CreER mice (Chen et al., 2014).
These two endothelial sources migrate onto the heart during embryonic development, and, through branching morphogenesis, create a vascular plexus that ultimately remodels into mature coronary arteries, capillaries, and veins. Although initial proepicardial Cre lines did not detect robust coronary endothelial lineage labeling, two other Cre lines were produced for studying other aspects of murine heart and organ development- Scx-Cre and Sema3D-Cre. These mouse lines lineage labeled only a subset of the proepicardium demonstrating a previously unappreciated heterogeneity to this tissue. They also labeled a minor percentage of coronary endothelial cells, although the details of when and how proepicardial cells converted was not described (Katz et al., 2012). In total, these lineage data provided evidence that the sinus venosus, endocardium, and proepicardium are progenitors for the coronary endothelial cell layer.
Following the publication of these seemingly disparate reports, controversy surrounded the source of coronary endothelial cells because it was not obvious why more than one progenitor would be used. Confidence in the existence of multiple sources was gained when a single study analyzed vessels arising from all three sources side-by-side in the whole embryonic mouse heart (Chen et al., 2014). The results showed that the sinus venosus (ApjCreER-traced) and endocardium (Nfatc1-creER-traced) contribute to the majority of coronary endothelial cells in complementary regions of the heart, while the proepicardium produced a smaller portion more equally throughout. Interestingly, the compartmentalization between sinus venosus and endocardium was not segregated by vessel subtype, but rather by heart region. The sinus venosus gives rise to all coronary vessel types (artery/capillary/vein) in a dorsal-to-ventral gradient where they infiltrate the heart muscle in an outside-in direction.
In contrast, the endocardium has a ventral-to-dorsal bias infiltrating the ventricles from the inside out (Chen et al., 2014). The compartmentalized contribution of sinus venosus- and endocardial-derived coronary vessels continues during early postnatal growth when the vasculature of the inner myocardium that is added after birth arises largely from the endocardium (Tian et al., 2014). There have been some disputes over the extent of sinus venosus versus endocardial contribution during embryonic mouse development because the original Nfatc1-Cre exhibits some sinus venosus labeling. A more specific endocardial Cre line, Npr3CreER, was reported not to label coronary vessels in the ventricular “free walls,” but still heavily traced mouse embryonic coronary vessels in the ventricular septum and ventral aspect, regions of the myocardium that are also importance to heart function (Zhang et al., 2016). Therefore, it is becoming established that the endothelial cell layer of the coronary vasculature in mice arises from multiple progenitors that migrate onto the heart from opposite sides, merge, and remodel to form its mature circulatory network.
Coronary Mural Cells (Pericytes and Smooth Muscle)
This unexpected assembling of multiple progenitors is not restricted to the coronary endothelial layer, but is also the case for coronary vessel mural cells. In contrast to the lack of prominent endothelial lineage labeling in many proepicardially expressed murine Cre lines (Tbx18-Cre, Wt1-Cre, Gata5-Cre, Tcf21-CreER), they heavily label coronary pericytes and smooth muscle indicating that a large portion of mural cells arise from the proepicardium/epicardium (Merki et al., 2005; Zhou et al., 2008; Cai et al., 2008; Acharya et al., 2012; Katz et al., 2012). Some smooth muscle cells proximal to the outflow tract and within the septum in mice can also be traced from Wnt1+ neural crest cells (Jiang et al., 2000), which in chicks arises from preotic neural crest (Arima et al., 2012). These neural crest cells are converted to coronary artery smooth muscle in response to endothelin signaling (Edn1/Ednra), and the absence of Ednra results in coronary artery dilations specifically within the septum (Arima et al., 2012).
Paralleling observations on the endothelial layer, there is also a third source for coronary artery mural cells in mice: the endocardium (Chen et al., 2016). This was discovered during characterization of a PDGFRβ-CreER line generated to study vascular mural cells. Induction of Cre activity before mural cells are present in the heart lineage labeled some cardiac pericytes and smooth muscle. This occurred because PDGFRβ is expressed in the cushion mesenchyme in the atrioventricular canal and outflow tract that is the precursor to cardiac valves. The cushion mesenchyme arises from endocardial cells that undergo an endothelial-to-mesenchymal transition (EndoMT) early in heart development. These endocardial-derived mesenchymal cells migrate into the myocardium and differentiate into coronary pericytes and smooth muscle in response to Wnt-Fzd4-β-catenin signaling. Also in analogy with the endothelial layer, smooth muscle cells display region-specific contributions with those derived from the endocardium being much more prominent in the ventricular septum than the lateral sides of the embryonic heart. It has been appreciated for over a decade that the smooth muscle of vessels within the different segments of the embryo have different origins (Majesky, 2007), and we can now extend this concept to vessels within a single organ. Thus, there are at least two cell types of the mature coronary vasculature in mice that develop through combining different progenitor sources that arise through distinct morphogenic events and cellular conversions.
Why are there multiple sources for cells of the coronary vasculature? One possibility is that it might be more efficient to vascularize the rapidly growing ventricles when progenitors are simultaneously infiltrating the heart from different locations. It could also be that alternative progenitors provide a source of compensatory cells that are mobilized in the presence of mutations that affect normal coronary growth. Answers to these questions may arise from investigations into the molecular control of coronary development. Data for coronary endothelial cells show that at least some of the signals are not shared between the sinus venosus and endocardium pathways. Sinus venosus-derived vessel sprouting requires vascular endothelial growth factor C (VEGF-C), which is expressed in the epicardium, while VEGF-C-deficient hearts have normal vascular growth in endocardial-derived areas (Chen et al., 2014).
Conversely, VEGF-A is critical for the endocardial-to-coronary pathway, but may not be required for sinus venosus-derived veins (Wu et al., 2012). Distinct molecular requirements bring up the possibility that one source could compensate for the other, providing robustness to organ development. Indeed, an alternative smooth muscle cell source in mice can repopulate coronary arteries in hearts where PDGRβ or Rbpj has been deleted from the epicardium, although at severely delayed kinetics (Mellgren et al., 2008; Wei et al., 2015). Of interest, upon epicardial Rbpj deficiency, the presence of compensatory smooth muscle cells rendered coronary arteries in mice more susceptible to disease during adult stages (Wei et al., 2015). The capacity for cellular compensation within the vasculature may be a general feature of the vasculature. Ablation of the Tcf21+Nkx2.5+ pharyngeal arch artery progenitors in zebrafish does not inhibit the formation of these arteries because compensatory endothelial cells from the head vasculature migrate in and make up for the losses in the normal pathway (Nagelberg et al., 2015). Future experiments should begin to shed light on the biological consequences of multiple coronary progenitors, their dynamics in the face of genetic mutations, and their respective activities following an injury response.
Cardiac Fibroblasts
In addition to coronary vessels, the mouse heart contains a large number of resident fibroblasts, and there is great interest in understanding their developmental origins due to their role in cardiac disease. Following myocardial infarction or chronic pressure loading, fibroblasts are activated and secrete excessive extracellular matrix, leading to fibrosis, pathological myocardial remodeling, and heart failure (Banerjee et al., 2006). Two studies performed extensive lineage analysis of cardiac fibroblasts within the ventricles using an array of available Cre lines (Ali et al., 2014; Moore-Morris et al., 2014). They both found that in mice the majority (∼80%) of cardiac fibroblasts were fate mapped from the epicardium, which was consistent with previous reports (Mikawa and Gourdie, 1996; Wilm et al., 2005; Cai et al., 2008; Zhou et al., 2008).
However, the remaining fifth arose from endothelial cells (Tie2-Cre-traced), and it was concluded that these arose from EndoMT (Ali et al., 2014; Moore-Morris et al., 2014). Trans-differentiation of endothelial cells into fibroblasts was first described as occurring during the fibrotic response using a Tie1-Cre line in mice (Zeisberg et al., 2007). However, Moore-Morris et al. found that the Tie2Cre-traced fibroblasts in the resting mouse heart developed in a pathway similar to coronary mural cells, through first differentiating into cushion mesenchyme, and that they were also most numerous in the ventricular septum. The large contribution of cushion mesenchyme to fibroblasts and mural cells in the septum could reflect the fact that this region is inside the heart, more distant from the epicardium that serves as progenitors for most of these cells within the ventricular free walls.
These studies also addressed the question of whether cardiac fibroblasts from distinct sources behave differently in response to disease. They found that epicardial- and endothelial-derived fibroblasts had similar proliferative and transcriptional changes in response to trans aortic constriction, a mouse model of pathological pressure overloading (Ali et al., 2014; Moore-Morris et al., 2014). In contrast, another study performed lineage analysis of Periostin + myofibroblasts that differentiate from resting fibroblasts following experimental myocardial infarction in mice (Kanisicak et al., 2016). The data indicated that myofibroblasts arise only from Tcf21 + epicardial-derived fibroblasts, but not those from endothelial cells through EndoMT. It will be interesting to identify the signals that drive the development of fibroblasts from their two very different progenitors, and how these pathways predispose them to changes that contribute to disease.
Not only can endothelial cells become fibroblasts, but the reverse may also be true. Col1α2-CreERT was used to lineage label fibroblasts, and when mouse hearts were subjected to ischemia reperfusion up to 40% of the endothelial cells near the injury were lineage labeled and incorporated in perfused vessels (Ubil et al., 2014). This fibroblast-to-endothelial transition (MEndoT) was mediated by stress-induced p53, which activated the expression of endothelial differentiation genes such as Hoxa9 and Hoxd3 in an in vitro model. Inhibition or activation of MEndoT following myocardial infarction through modulating p53 decreased or increased, respectively, the recovery of heart function. These studies suggest that injury could incite unique cellular plasticity not observed during normal organogenesis.
Lymphatic Vessels
The lymphatic vasculature is a second circulatory system that complements the blood vasculature by returning extracellular fluid to the blood and organizing the immune response. Lymphatic vessels consist of lymphatic endothelial cell (LEC)-lined capillaries that feed into larger lymphatic collecting ducts that associate with smooth muscle. Data compiled over the past 2 decades have established a model on how the lymphatic vasculature develops. This process begins at embryonic day 9.5 in the mouse when a small proportion of cells within the endothelial layer of the cardinal vein begin to express the transcription factor Prox1 and migrate outward in response to VEGF-C (Yang and Oliver, 2014). These LEC progenitors assemble into lymph sacs that subsequently migrate throughout the body, giving rise to lymphatic vessels that are spread throughout the entire organism. This work led to the widespread acceptance that trans-differentiation of venous EC into LECs was the sole source of the entire lymphatic vasculature in mammals (Yang and Oliver, 2014). Recently, lineage tracing experiments have shown that the venous pathway is not the only source, but is complemented by LECs differentiating from other progenitor cell types.
The first questioning of venous cells as the sole source of LECs occurred when studies deleting Prox1 using venous expressed Cre lines resulted in residual lymphatic vessels in certain regions. This strategy depleted all lymphatic vessels in the cervical and thoracic skin, but not those in the lumbar areas of the mouse (Martinez-Corral et al., 2015). A histological analysis of lymphatic vessels in the lumbar area observed the presence of isolated cells expressing LEC markers that rapidly coalesce, instead of a pattern suggesting progressive sprouting from preformed vessels, which was observed in the cervical and thoracic regions. The isolated LEC clusters were not lineage labeled with a Cre expressed in veins (Tie2-Cre), establishing that they derive from an alternative, yet unidentified, source.
Nonvenous derived lymphatics were also observed in the murine mesentery, a membrane of the peritoneum that encloses the intestines (Stanczuk et al., 2015). The mesenteric lymphatics also initially appeared as isolated cell clusters that subsequently coalesced into vessels. These vessels were traced by Pdgfb-CreER, which does not label venous-derived LECs. Instead, the labeling regimen used (embryonic tamoxifen dosing) specifically induced recombination in hemogenic arteries containing endothelial cells that bud off hematopoietic stem cells during development. Further evidence for hemogenic endothelium being the source of nonvenous LECs was provided when cKit-CreER, but not Vav1-Cre (hematopoietic specific expression), was able to fate map these cells. As in the coronary vasculature, there are distinct mechanisms that regulate the formation and maturation of LEC progenitors from different sources. Pitx2 is specifically upstream of the lymphatic vessels located where the non–venous-derived LECs are found in mice (Mahadevan et al., 2014). However, further research is required to fully understand the similarities and differences between the various merging LEC populations.
Other studies analyzing organ-specific lymphatic vessels have found a similar heterogeneity in LECs of the mouse heart (Klotz et al., 2015). The majority of cardiac lymphatic vessels arrive through sprouting from the venous-derived lymphatic sac as indicated by lineage labeling with Tie2Cre. However, approximately 1/7th of LECs in this organ were found to be lineage labeled with the hematopoietic Cre line Vav1-Cre, but not other Cre lines that are expressed in a collection of other cardiac progenitor cell types. Prox1 was critical for each source to contribute to cardiac lymphatics, although deletion of Prox1 in venousderived lymphatic vessels was the only manipulation that resulted in significant defects in cardiac LEC numbers. The authors used a yolk-sac culture model and precise timing of Csf1-CreER induction in vivo to arrive at the conclusion that the non-venous LECs may emerge from the hemogenic endothelium of the yolk sac. Interestingly, lymphatic vessels increased in a mouse model of myocardial infarction, and treatment with VEGF-C led to a better cardiac function following injury (Klotz et al., 2015). Given these finding, it is likely that more unexpected sources will be identified as the nature of other organ-specific lymphatic beds are investigated.
Surprising findings on the origins of LECs have also been observed in zebrafish. Using an elegant genetic/spatiotemporal lineage labeling technique and time-lapse observations in zebrafish embryos, it was shown that venous-derived lymphatic vessels differentiate from a specialized cell that resides in the wall of the posterior cardinal vein (PCV) (Nicenboim et al., 2015). Although located in the vein, these progenitors were found not to be venous endothelial cells, but rather dedicated angioblasts expressing an arterial marker that give rise to lymphatic and intestinal vessels (Hen et al., 2015). The authors could discover this by following the reverse migration paths of zebrafish lymphatics in time-lapse videos, which revealed that LECs arise from cells located within the ventral region of the PCV.
The authors then specifically fate mapped cells within either the ventral or dorsal portion of the cardinal vein using laser photo-conversion of a genetically encoded Kaede protein (which irreversibly turns from red to green) expressed in the endothelium. This resulted in lineage labeling the lymphatics and intestinal vessels. Localizing the angioblasts lead to the search for factors expressed in adjacent tissues and the identification of Wnt5b emanating from the endoderm underlying the cardinal vein as activating lymphatic development. This ability to combine real time visualization with the lineage tracing of cells expressing specific enhancers/promoters and in specific locations makes zebrafish a powerful experimental model for developmental and stem cell biology. It will be interesting to investigate whether similar angioblast niches exist within vessels in the embryo and adult of other species and whether they are expanded during regenerative responses.
Is the Existence of Multiple Progenitors for Single Cell Types a General Phenomenon?
The identification of alternative progenitors for cells of the cardiovascular system was stumbled upon through analysis of different lineage experiments and conundrums over the penetrance of knockout phenotypes (Fig. 1; Table 1). Will similar experiments in other systems reveal this as a widespread phenomenon? It is known that both the pancreas and liver develop from two spatially distinct regions of the endoderm that later merge to produce the mature organ in mice (Zaret, 2008). Of interest, the different liver and pancreas buds respond to different signals such that mutations or molecular manipulations frequently specifically affect one part of the developing organ (Harrison et al., 1999; Li et al., 1999; Haumaitre et al., 2005; Wang et al., 2015). Further experiments should investigate whether the organ systems possessing multiple progenitors can compensate for each other, which, in addition to genetic compensation mechanisms (Rossi et al., 2015), could perhaps explain the incomplete penetrance and lack of phenotypes in different knockout animals.
Fig. 1.
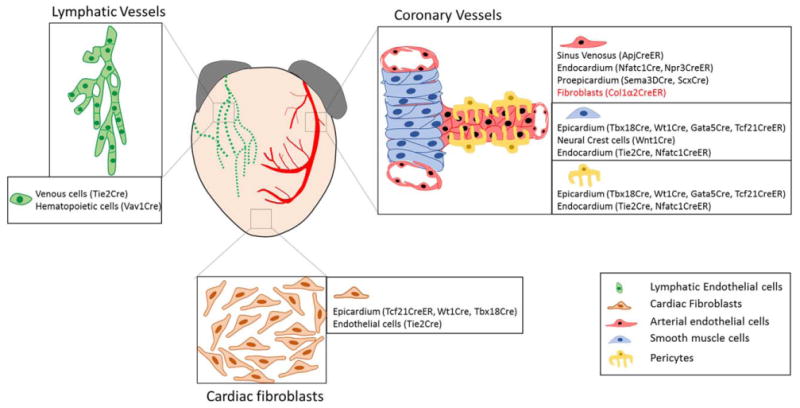
Multiple sources for cardiac cells. Genetic lineage tracing reveals that multiple cellular sources contribute to the development of different cardiac cell types. (1) Lymphatic endothelial cells arise from both venous and hematopoietic progenitors. (2) Cardiac fibroblasts originate from epicardial and endocardial cells. (3) Coronary endothelial cells originate from sinus venosus, endocardium, and proepicardium. (4) Coronary mural cells (pericytes and smooth muscle cells) arise from the epicardium, endocardium and neural crest cells. Under pathological conditions (in red), resident fibroblasts proliferate to give rise to activated cardiac fibroblasts, and coronary endothelial cells and may undergo a mesenchymal-to-endothelial transition (MEndT) to produce coronary vessels. The Cre transgenes used in labeling corresponding lineages are mentioned in brackets next to the cellular sources.
TABLE 1.
Summary of Cre Driver Lines and Associated Studies Suggesting Multiple Progenitor Cell Types in Development of Cardiac Lymphatic Vessels, Coronary Vessels, and Cardiac Fibroblasts in Micea
| Progeny cells | Progenitor cells | Cre line | References |
|---|---|---|---|
| Lymphatic endothelial cells | Venous cells | Tie2 Cre | Klotz et al., 2015 |
| Hematopoietic cells | Vav1 Cre | Klotz et al., 2015 | |
| Coronary endothelial cells | Sinus venosus | ApjCreER | Chen et al., 2014 |
| Endocardium | Nfatc1Cre | Chen et al., 2014 | |
| Npr3CreER | Zhang et al., 2016 | ||
| Proepicardium | Sema3DCre | Katz et al., 2012 | |
| ScxCre | Katz et al., 2012 | ||
| Fibroblasts | Col1a2CreER | Ubil et al., 2014 | |
| Coronary SMCs | Epicardium | Tbx18Cre | Cai et al., 2008 |
| Wt1Cre | Zhou et al., 2008 | ||
| Gata5Cre | Merki et al., 2005 | ||
| Tcf21CreER | Acharya et al., 2012 | ||
| Neural crest cells | Wnt1Cre | Jiang et al., 2000 | |
| Endocardium | Tie2Cre | Chen et al., 2016 | |
| Nfatc1CreER | Chen et al., 2016 | ||
| Coronary pericytes | Epicardium | Tbx18Cre | Cai et al., 2008 |
| Wt1Cre | Zhou et al., 2008 | ||
| Gata5Cre | Merki et al., 2005 | ||
| Tcf21CreER | Acharya et al., 2012 | ||
| Endocardium | Tie2Cre | Chen et al., 2016 | |
| Nfatc1CreER | Chen et al., 2016 | ||
| Cardiac fibroblasts | Epicardium | Tcf21CreER | Acharya et al., 2012 |
| Wt1Cre | Zhou et al., 2008 Moore-Morris et al., 2014 | ||
| Tbx18Cre | Cai et al., 2008 Ali et al., 2014 Moore-Morris et al., 2014 | ||
| Endothelial cells | Tie2Cre | Ali et al., 2014 Moore-Morris et al., 2014 | |
| Resident fibroblasts | PostnCreER | Kanisicak et al., 2016 |
References in italic type show studies performed in a pathological setting.
Clonal Analysis Reveals the Progenitor Capacity of Cardiovascular Cells
Obtaining high-resolution lineage information retrospectively from genetic tracing experiments can be difficult in complex tissue environments. One technique to better understand how single cells behave is to perform lineage tracing on the clonal level. These experiments have unveiled some unpredicted progenitor cell biology, particularly within the cardiovascular system.
Compartment Boundaries Vs. Retained Multipotency
To study the mechanisms guiding cell differentiation and fate restriction, it is critical to understand when and where important specification events occur. A predominant model in the cardiac field was that a multipotent progenitor for the cell types in the heart exists, but during which window in development was not clear (Laugwitz et al., 2008). Recent clonal level lineage analyses have uncovered that murine cardiac progenitors are allocated to the different chambers of the heart (e.g., separated into the first and second heart field) much earlier than anticipated, during gastrulation. Using a transiently expressed Cre line (Mesp1-Cre) and multicolor labeling strategies, the fate and organization of clusters of cells arising from single mesodermal cardiac precursors present at gastrulation could be followed in mice (Devine et al., 2014; Lescroart et al., 2014). Clonally labeled cells arising from these progenitors did not span the left and right ventricles of the heart, but were restricted to one or the other chamber, indicating the establishment of a previously unrecognized compartment boundary very early in development.
In addition, statistical modeling and quantitative analysis of the clones calculated the existent of ∼250 Mesp1+ original cardiac progenitors in mice (Chabab et al., 2016). Appreciation of the early specification of these cells allowed the further characterization of a chromatin remodeling factor, Smarc3, in the cardiac differentiation pathway. Expression of Smarc3 defined a new progenitor stage between the Mesp1+ step and the expression of Nkx2.5 and Tbx5 in mice (Devine et al., 2014). Given the prominent role compartment boundaries can have on organizing development (Dahmann et al., 2011), it will be interesting to discover just how many vertebrate organs are patterned by early cell fate allocations. Indeed, a highly scaled version of clonal labeling where genome editing was used to individually barcode thousands of cells in a way that preserves lineage information (GESTALT) has described that the majority of cells in individual Zebrafish organs arise from a small number of early progenitors (McKenna et al., 2016).
The opposite of very early fate restrictions is the persistence of mutipotent progenitor cells that can give rise to various cell types. These cells can have transient activity during development or retain their multipotency to support tissue homeostasis in the adult. Clonal analysis is also critical in unraveling the lineage capabilities and progression of these cell types (Blanpain and Simons, 2013). One recently described example in the mammalian cardiovascular system is the existence of a multipotent cardiopulmonary progenitor whose differentiation capacity is retained until mid-gestation when it develops into the cell types that form the vascular connections between the heart and lungs (Peng et al., 2013). All cell layers of pulmonary arteries and veins proximal to the heart develop independent of lung formation from an individual progenitor located near the anterior foregut that expresses Wnt2, Gli1, and Isl1. Differentiation into the cardiopulmonary vessels requires Shh secreted by adjacent foregut endoderm. Understanding the developmental road map of these types of progenitors at the single cell level, and how their pathways can be recapitulated in culture, could have great impacts for regenerative medicine (Loh et al., 2016).
Clonal Expansion in Pathological Vascular Remodeling
Clonal level lineage labeling in different disease models has revealed the surprising finding that a few or even single cells are selected to massively expand in a way that contributes to vascular disease. Pulmonary hypertension (PH) is a disease characterized by increased blood pressure in the vasculature of the lung caused by extensive thickening of the pulmonary artery wall. Progression of the disease can interfere with lung function and lead to fatal heart disease. Hypoxia-induced PH involves excessive muscularization of distal arterioles that are normally not covered by smooth muscle cells. Based on known smooth muscle biology, it was predicted that the neomuscularization arises through dedifferentiation and expansion of a population of preexisting smooth muscle cells in mice (Sheikh et al., 2014).
However, clonal analysis of existing smooth muscle using a multicolor Rainbow Cre reporter established that the new muscularization of distal arteries arises from the expansion of just a single smooth muscle cell located at the muscular/nonmuscular border of the artery in mice (Sheikh et al., 2015). It was discovered that these single cells express markers of an immature smooth muscle phenotype, specifically, KLF4+, SMα+, PDGFRβ+, and they require KLF4 and PDGFB signaling for their expansion onto distal arterioles. Thus, the interrogation of cell expansion at the single cell level was critical to the identification of a “primed” progenitor cell responsible for a disease phenotype.
Endothelial cells that line blood vessels have also been observed to clonally expand under pathological conditions (Corey et al., 2016). Multicolor labeling of endothelial cells in the adult mice using the same Rainbow Cre reporter detected only single to very small clusters of cells expressing the same color, reflecting the general quiescence of the vasculature under normal conditions. The authors then investigated endothelial expansion during tumor angiogenesis. Upon implantation of B16 melanoma cells, growing tumors were colonized by endothelial cells derived from only one to a few clones. These clonally expanded endothelial cells were highly similar to their parent endothelial cells at the transcriptional level except for a select subset of genes, particularly leukocyte adhesion molecules and chemokines. It remains to be determined if the clonal nature of the tumor vasculature reflects the initial growth of only a few cells during the angiogenesis phase or the selection over time of subtypes that are immunologically advantageous to the tumor. The mechanisms underlying either possibility will reveal fascinating and important biology.
References
- Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AM, Volz KS, Tang Z, Red-Horse K, Ardehali R. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115:625–635. doi: 10.1161/CIRCRESAHA.115.303794. [DOI] [PubMed] [Google Scholar]
- Arima Y, Miyagawa-Tomita S, Maeda K, Asai R, Seya D, Minoux M, Rijli FM, Nishiyama K, Kim KS, Uchijima Y, Ogawa H, Kurihara Y, Kurihara H. Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat Commun. 2012;3:1267. doi: 10.1038/ncomms2258. [DOI] [PubMed] [Google Scholar]
- Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Anna N Y Acad Sci. 2006;1080:76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Simons BD. Unravelling stem cell dynamics by lineage tracing. Nat Rev Mol Cell Biol. 2013;14:489–502. doi: 10.1038/nrm3625. [DOI] [PubMed] [Google Scholar]
- Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104–108. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabab S, Lescroart F, Rulands S, Mathiah N, Simons BD, Blanpain C. Uncovering the number and clonal dynamics of MESP1 progenitors during heart morphogenesis. Cel Rep. 2016;14:1–10. doi: 10.1016/j.celrep.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivelä R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K, Red-Horse K. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development. 2014;141:4500–4512. doi: 10.1242/dev.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang H, Liu Y, Adams S, Eilken H, Stehling M, Corada M, Dejana E, Zhou B, Adams RH. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat Commun. 2016;7:12422. doi: 10.1038/ncomms12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey DM, Rinkevich Y, Weissman IL. Dynamic patterns of clonal evolution in tumor vasculature underlie alterations in lymphocyte-endothelial recognition to foster tumor immune escape. Cancer Res. 2016;76:1348–53. doi: 10.1158/0008-5472.CAN-15-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmann C, Oates AC, Brand M. Boundary formation and maintenance in tissue development. Nat Rev Genet. 2011;12:43–55. doi: 10.1038/nrg2902. [DOI] [PubMed] [Google Scholar]
- Devine WP, Wythe JD, George M, Koshiba-Takeuchi K, Bruneau BG. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. Elife. 2014;3:e03848. doi: 10.7554/eLife.03848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23:71–75. doi: 10.1038/12674. [DOI] [PubMed] [Google Scholar]
- Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A. 2005;102:1490–1495. doi: 10.1073/pnas.0405776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hen G, Nicenboim J, Mayseless O, Asaf L, Shin M, Busolin G, Hofi R, Almog G, Tiso N, Lawson ND, Yaniv K. Venous-derived angioblasts generate organ-specific vessels during zebrafish embryonic development. Development. 2015;142:4266–4278. doi: 10.1242/dev.129247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz TC, Singh MK, Degenhardt K, Rivera-Feliciano J, Johnson RL, Epstein JA, Tabin CJ. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev Cell. 2012;22:639–650. doi: 10.1016/j.devcel.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62–67. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Laugwitz KL, Moretti A, Caron L, Nakano A, Chien KR. Islet1 cardiovascular progenitors: a single source for heart lineages? Development. 2008;135:193–205. doi: 10.1242/dev.001883. [DOI] [PubMed] [Google Scholar]
- Lescroart F, Chabab S, Lin X, Rulands S, Paulissen C, Rodolosse A, Auer H, Achouri Y, Dubois C, Bondue A, Simons BD, Blanpain C. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat Cell Biol. 2014;16:829–840. doi: 10.1038/ncb3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23:67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, Shyh-Chang N, Fernhoff NB, George BM, Wernig G, Salomon RE, Chen Z, Vogel H, Epstein JA, Kundaje A, Talbot WS, Beachy PA, Ang LT, Weissman IL. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell. 2016;166:451–467. doi: 10.1016/j.cell.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan A, Welsh IC, Sivakumar A, Gludish DW, Shilvock AR, Noden DM, Huss D, Lansford R, Kurpios NA. The left-right Pitx2 pathway drives organ-specific arterial and lymphatic development in the intestine. Dev Cell. 2014;31:690–706. doi: 10.1016/j.devcel.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- Martinez-Corral I, Ulvmar MH, Stanczuk L, Tatin F, Kizhatil K, John SWM, Alitalo K, Ortega S, Makinen T. Nonvenous origin of dermal lymphatic vasculature. Circ Res. 2015;116:1649–1654. doi: 10.1161/CIRCRESAHA.116.306170. [DOI] [PubMed] [Google Scholar]
- Männer J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anat Rec. 1999;255:212–226. doi: 10.1002/(sici)1097-0185(19990601)255:2<212::aid-ar11>3.3.co;2-o. [DOI] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353:aaf7907. doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, Kazi MN, Ruiz FR, Pu WT, Tallquist MD. Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res. 2008;103:1393–1401. doi: 10.1161/CIRCRESAHA.108.176768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol. 1996;174:221–232. doi: 10.1006/dbio.1996.0068. [DOI] [PubMed] [Google Scholar]
- Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J Clin Invest. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelberg D, Wang J, Su R, Torres-Vázquez J, Targoff KL, Poss KD, Knaut H. Origin, specification, and plasticity of the great vessels of the heart. Curr Biol. 2015;25:2099–2110. doi: 10.1016/j.cub.2015.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicenboim J, Malkinson G, Lupo T, Asaf L, Sela Y, Mayseless O, Gibbs-Bar L, Senderovich N, Hashimshony T, Shin M, Jerafi-Vider A, Avraham-Davidi I, Krupalnik V, Hofi R, Almog G, Astin JW, Golani O, Ben-Dor S, Crosier PS, Herzog W, Lawson ND, Hanna JH, Yanai I, Yaniv K. Lymphatic vessels arise from specialized angioblasts within a venous niche. Nature. 2015;522:56–61. doi: 10.1038/nature14425. [DOI] [PubMed] [Google Scholar]
- Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, Evans SM, Morrisey EE. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature. 2013;500:589–592. doi: 10.1038/nature12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelmann RE, Gittenberger-De Groot AC, Mentink MM, Bökenkamp R, Hogers B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ Res. 1993;73:559–568. doi: 10.1161/01.res.73.3.559. [DOI] [PubMed] [Google Scholar]
- Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y, Lindau P, Ueno H, Longaker MT, Weissman IL. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature. 2011;476:409–413. doi: 10.1038/nature10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DY. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- Sheikh AQ, Lighthouse JK, Greif DM. Recapitulation of developing artery muscularization in pulmonary hypertension. Cel Rep. 2014;6:809–817. doi: 10.1016/j.celrep.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med. 2015;7:308ra159. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Stanczuk L, Martinez-Corral I, Ulvmar MH, Zhang Y, Laviña B, Fruttiger M, Adams RH, Saur D, Betsholtz C, Ortega S, Alitalo K, Graupera M, Mäkinen T. cKit lineage hemogenic endothelium-derived cells contribute to mesenteric lymphatic vessels. Cel Rep. 2015;10:1708–1721. doi: 10.1016/j.celrep.2015.02.026. [DOI] [PubMed] [Google Scholar]
- Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, He L, Yang Z, Yan Y, Yang X, Zhong TP, Pu WT, Zhou B. Vessel formation. De novo formation of a distinct coronary vascular population in neonatal heart. Science. 2014;45:90–94. doi: 10.1126/science.1251487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubil E, Duan J, Pillai ICL, Rosa-Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M, Vondriska TM, Stefani E, Deb A. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–590. doi: 10.1038/nature13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Rhee S, Palaria A, Tremblay KD. FGF signaling is required for anterior but not posterior specification of the murine liver bud. Dev Dyn. 2015;244:431–443. doi: 10.1002/dvdy.24215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K, Díaz-Trelles R, Liu Q, Diez-Cuñado M, Scimia MC, Cai W, Sawada J, Komatsu M, Boyle JJ, Zhou B, Ruiz-Lozano P, Mercola M. Developmental origin of age-related coronary artery disease. Cardiovasc Res. 2015;107:287–294. doi: 10.1093/cvr/cvv167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm B, Ipenberg A, Hastie ND, Burch JBE, Bader DM. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development. 2005;132:5317–5328. doi: 10.1242/dev.02141. [DOI] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O'Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang CP, Zhou B. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Oliver G. Development of the mammalian lymphatic vasculature. J Clin Invest. 2014;124:888–897. doi: 10.1172/JCI71609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nat Rev Genet. 2008;9:329–340. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zhang H, Pu W, Li G, Huang X, He L, Tian X, Liu Q, Zhang L, Wu SM, Sucov HM, Zhou B. Endocardium minimally contributes to coronary endothelium in the embryonic ventricular free walls. Circ Res. 2016;116:1880–1893. doi: 10.1161/CIRCRESAHA.116.308749. [DOI] [PubMed] [Google Scholar]
- Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]