

Abstract
Esterified cholesterol (EC) and triglycerides, contained within lipoproteins taken up by cells, are hydrolysed by lysosomal acid lipase (LAL) in the late endosomal/lysosomal (E/L) compartment. The resulting unesterified cholesterol (UC) is transported via Niemann-Pick type C2 and C1 into the cytosolic compartment where it enters a putative pool of metabolically active cholesterol that is utilized in accordance with cellular needs. Loss-of-function mutations in LIPA, the gene encoding LAL, result in dramatic increases in tissue concentrations of EC, a hallmark feature of Wolman Disease and Cholesteryl Ester Storage Disease (CESD). The lysosomal sequestration of EC causes cells to respond to a perceived deficit of sterol by increasing their rate of cholesterol synthesis, particularly in the liver. A similar compensatory response occurs with treatments that disrupt the enterohepatic movement of cholesterol or bile acids. Here we measured rates of cholesterol synthesis in vivo in the liver and small intestine of a mouse model for CESD given the cholesterol absorption inhibitor ezetimibe from weaning until early adulthood. Consistent with previous findings, this treatment significantly reduced the amount of EC sequestered in the liver (from 132.43 ± 7.35 to 70.07 ± 6.04 mg/organ) and small intestine (from 2.78 ± 0.21 to 1.34 ± 0.09 mg/organ) in the LAL-deficient mice even though their rates of hepatic and intestinal cholesterol synthesis were either comparable to, or exceeded those in matching untreated Lal−/− mice. These data reveal the role of intestinal cholesterol absorption in driving the expansion of tissue EC content and disease progression in LAL deficiency.
Keywords: esterified cholesterol, liver, enterocyte, cholesterol absorption, cholesterol sequestration
Graphical abstract
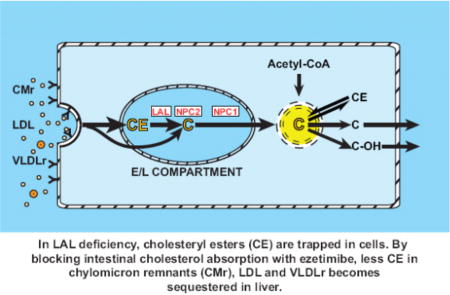
1. Introduction
There is a vast literature defining the role that the liver and small intestine play in the regulation of whole-body cholesterol metabolism and the plasma lipoprotein composition [1–6]. Like most other organs, the liver and small intestine actively synthesize cholesterol although at rates that vary with multiple factors. This is especially true in the liver where the synthesis rate can fluctuate over an extraordinary range in response to shifts in dietary cholesterol intake or treatment with different classes of cholesterol-lowering agents, particularly statins, bile acid sequestrants and cholesterol absorption inhibitors [7–12]. Changes in cholesterol synthesis rates can also occur because of mutations in genes that encode enzymes at specific steps in the biosynthetic pathway [13], or that regulate the absorption, transport, degradation, storage and excretion of cholesterol [14–18]. Mutations in three particular genes, LIPA, NPC1 and NPC2, which together regulate cholesterol trafficking through the late endosomal/lysosomal compartment of every cell, have far reaching consequences in most organ systems. In the case of NPC2, and especially NPC1, mutations lead to multisystem disease including neurodegeneration, liver failure and pulmonary dysfunction [19]. Mutations in LIPA, the gene that encodes LAL, result in either Wolman disease (WD), or cholesteryl ester storage disease (CESD). Whereas WD is a severe, early onset illness caused by complete loss of LAL activity, CESD is a milder, later-onset disease resulting from partial LAL deficiency [20–22].
In NPC1 or NPC2 deficiency, unesterified cholesterol (UC) becomes sequestered in the late endosomal/lysosomal compartment thus preventing its movement elsewhere in the cell for further utilization. Such entrapment of UC causes a perceived shortage of cholesterol in cells leading to a compensatory increase in cholesterol synthesis [14]. The same scenario is seen in LAL deficiency except that in this disorder it is esterified cholesterol (EC) that is sequestered in the E/L compartment [20–23]. Our studies in a mouse model for CESD showed a profound compensatory increase in hepatic cholesterol synthesis [23]. The greatly elevated rate per gram of liver, combined with the pronounced increase in liver mass, were largely responsible for driving the elevation in whole-body cholesterol synthesis in this model even though increased synthesis in other organs, including the small intestine, contributed to this elevation.
Although an enzyme replacement therapy (ERT), Sebelipase alfa, is now available for treating LAL deficiency [24, 25], various classes of cholesterol-lowering agents have proven useful for managing the dyslipidemia often seen in CESD patients [26–28]. One of these is ezetimibe, a potent and selective inhibitor of intestinal sterol absorption that is widely used in combination with statins, or as a monotherapy, for dyslipidemia management in the general population [29, 30]. A more recent study demonstrated efficacy of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia [31]. Ezetimibe is now an established therapy for sitosterolemia, a rare sterol storage disease [32]. It has also been shown to reduce biliary cholesterol saturation in humans and animal models [7, 8, 33]. Studies from multiple labs using various types of animal models for non-alcoholic fatty liver disease (NAFLD) implied a therapeutic benefit of ezetimibe in this disorder [34–36]. However, a recent clinical trial in patients with non-alcoholic steatohepatitis (NASH) given ezetimibe did not find a reduction in liver fat content [37]. Although the mouse model for CESD that we acquired does not manifest elevated plasma total cholesterol levels, we used it for an exploratory study to determine whether the imposition of a chronic block of intestinal cholesterol absorption by ezetimibe from the time of weaning had any impact on liver mass and EC content by the time the mutants reached early adulthood [38]. The treated mutants showed a reduction in both their degree of hepatomegaly and the level of EC sequestration in the liver, along with a clear fall in plasma ALT activity. These decisive effects raised several questions, particularly as blocking cholesterol absorption with ezetimibe is known to lead to a dramatic compensatory increase in hepatic, intestinal and whole-body cholesterol synthesis in other types of animals models [7, 8] and humans [39].
The principal objective of these studies then was to use an established in vivo technique to determine what happens to the already elevated rates of cholesterol synthesis in the liver and small intestine in LAL-deficient mice when they have a sustained pharmacological block of their cholesterol absorption pathway. This was part of a broader goal of better understanding the interrelationship between intestinal and hepatic cholesterol metabolism in CESD, and of further exploring the potential use of ezetimibe as an adjunctive therapy for this rare disorder.
2. Materials and Methods
2.1 Animals and diets
Lal+/+ and Lal−/− mice were generated from heterozygous breeding stock, all on an FVB/N strain background. The litters were weaned at 21 days and genotyped before that age using an ear notch. Except for an initial experiment using different doses of ezetimibe, all studies were carried out in females only. The ground form a cereal-based, low-cholesterol rodent chow diet (No. 7001, Envigo:Teklad, Madison, WI) was used in all experiments. This formulation had an inherent cholesterol content of 0.02% (w/w) and a crude fat content of not less than 4% [38]. In our previously published experiments with ezetimibe treated Lal-deficient mice, only males were studied, and the dose of ezetimibe was set at ~20 mg / day / kg bw. This dose was determined on the basis of a food intake of approximately 160 g / day / kg bw that was measured in another project using young adult Ldlr−/− mice and their wildtype controls given the chow diet alone [7]. For the current studies we first investigated the impact of varying the dose of ezetimibe on liver mass and cholesterol content in Lal-deficient mice. As shown in Table 1, a modest but significant reduction in the degree of hepatomegaly and hepatic total cholesterol concentration was seen at a dose of 5 mg / day / kg bw. A dose of 20 mg / day / kg bw was considerably more efficacious. Doubling this dose resulted in about the same degree of reduction in hepatic cholesterol content. Therefore, it was determined that 20 mg / day / kg bw would be used in the subsequent studies. In all experiments, the mice were housed in groups of 3 or 4 per cage, their food intake and stool output were monitored daily, and their body weights were measured at weekly intervals. The period of ezetimibe treatment was 28 to 30 days starting on the day of weaning. Hence, their age at the time of study was 49–51 days. The mice were provided their respective diets ad libitum and studied in the fed state towards the end of the dark phase of the lighting cycle. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Table 1.
Impact of different doses of ezetimibe on liver mass and cholesterol content in mice with LAL deficiency.
| Gender | Genotype | Approx. dose of ezetimibe (mg/ day/ kg body wt) | No. of mice | Body wt (g) | Liver wt (g) | Hepatic total cholesterol concentration (mg/ g) | Whole-liver cholesterol content (mg/ organ) |
|---|---|---|---|---|---|---|---|
| Male | Lal+/+ | 0 | 4 | 24.3 ± 1.3 | 1.33 ± 0.09 | 2.14 ± 0.06 | 2.83 ± 0.14 |
| Lal−/− | 0 | 4 | 24.3 ± 0.5 | 2.41 ± 0.08 | 57.4 ± 3.3 | 138.5 ± 9.1 | |
| Lal−/− | 5 | 4 | 24.5 ± 1.1 | 2.29 ± 0.17 | 41.3 ± 2.5* | 95.2 ± 10.3* | |
| Lal−/− | 20 | 4 | 23.5 ± 1.2 | 1.95 ± 0.17* | 37.3 ± 1.1* | 72.5 ± 4.9* | |
|
| |||||||
| Female | Lal+/+ | 0 | 6 | 20.9 ± 0.2 | 1.08 ± 0.03 | 2.45 ± 0.04 | 2.63 ± 0.04 |
| Lal−/− | 0 | 6 | 21.3 ± 0.4 | 2.39 ± 0.09 | 59.1 ± 2.4 | 141.0 ± 7.7 | |
| Lal−/− | 40 | 6 | 18.8 ± 0.6* | 1.70 ± 0.03* | 46.2 ± 1.9* | 78.4 ± 3.2* | |
All mice were weaned at 21 days and thereafter fed ad libitum a rodent cereal-based diet either alone or containing ezetimibe at different doses until 50 days of age.
Values are mean ± SEM for the number of mice indicated.
p<0.05 compared to the untreated Lal−/− mice of the same gender.
2.2 Measurement of rate of cholesterol and fatty acid synthesis in the liver and small intestine, and of cholesterol synthesis in the whole animal
These rates were measured in vivo using [3H]water as detailed elsewhere [15, 40]. Custom generated [3H]water at a concentration of 5 Ci/ml (PerkinElmer Life Sciences) was subsequently diluted in sterile sodium chloride solution (0.9% w/v) to ~200 mCi/ml. One h after the mice were administered ~40 mCi of [3H]water (~0.2ml) intraperitoneally, the liver and whole small intestine were removed, rinsed, blotted, and weighed. They were then saponified and the labeled sterols extracted and quantitated as described [40]. The rate of cholesterol synthesis in each organ was calculated as nmol of water incorporated into sterols / h / g wet weight of tissue (nmol/h/g). In one study, the residual carcass was digested and its labeled sterols isolated. The combined [3H]sterol contents of the liver, small intestine and carcass yielded a measure of whole-animal sterol synthesis (nmol/h). The rate of hepatic and intestinal fatty acid synthesis was determined with the same tissue extracts used for isolation of the tritiated sterols [15]. These rates were expressed in the same way as those for cholesterol synthesis in these organs (nmol/h/g).
2.3 Quantitation of total, unesterified and esterified cholesterol in tissue and plasma, and of plasma ALT activity
After exsanguination, the liver and entire small intestine were removed, rinsed, blotted and weighed. Depending on the planned measurements, aliquots of liver, and the whole small intestine were placed in chloroform:methanol (2:1 v/v) for determination of the tissue concentrations (mg/g) of esterified (EC) and unesterified cholesterol (UC) using a combination of column and gas chromatography as described [41]. When only a direct measure of the total cholesterol (TC) concentration was required, an aliquot of liver tissue or plasma was digested directly in alcoholic KOH. All cholesterol quantitation was done using gas chromatography with pure stigmastanol as the internal standard [40]. Plasma total cholesterol concentrations were expressed as mg/dl. For the liver and small intestine, the total cholesterol concentration was expressed as mg/g tissue. To obtain whole-organ cholesterol content (mg/organ), the total cholesterol concentration was multiplied by the respective whole organ weight. The measurement of whole-animal cholesterol content required digestion and extraction of the residual carcass, which included those regions of the gastrointestinal tract separate from the small intestine. Those regions were excised, rinsed, and then added back to the remaining residual carcass, which after weighing, was digested in alcoholic KOH on a steambath. The carcass extract was filtered through gauze sponges into a 100-ml volumetric flask. Aliquots of this extract were subsequently used for measurement of total cholesterol concentration (mg/g). Multiplication of this value by the mass of the carcass provided a measure of its entire cholesterol content. Summation of the cholesterol contents for the liver, small intestine and carcass yielded the whole-body cholesterol content (mg/animal). Hepatic and intestinal triglyceride concentrations were determined using a combination of column chromatography and an enzymatic-colorimetric assay [42]. Plasma alanine aminotransferase (ALT) activities (units/L) were measured by a commercial laboratory.
2.4 Relative mRNA expression analysis
Aliquots of liver and the mucosal scrapings from the entire length of the small intestine were quickly frozen in liquid nitrogen. mRNA levels were measured by real-time qPCR using an ABI Prism 7900 Sequence Detection System. The primer sequences used to measure the mRNA levels for genes of specific interest are given in earlier publications [23, 41, 43]. All analyses were determined by the comparative cycle number at threshold method with cyclophilin as the internal control [44]. The mRNA levels were normalized to cyclophilin and values for each Lal−/− mouse on no treatment, and for each Lal+/+ and Lal−/− mouse on ezetimibe, were then expressed relative to that obtained for untreated Lal+/+ mice, which in each case was arbitrarily set at 1.0.
2.5 Analysis of data
Values are mean ± SEM for the specified number of animals. GraphPad Prism 6.02 software (GraphPad, San Diego, CA) was used to perform all statistical analyses. Differences between mean values were tested for statistical significance (p < 0.05) by a two-way analysis of variance (ANOVA) with genotype and treatment as variables.
3. Results
3.1 Degree of hepatomegaly in Lal−/− mice was reduced by ezetimibe treatment
The data in Fig.1A show ezetimibe had no impact on the body weights of mice of either genotype. Although hepatomegaly persisted in the treated Lal−/− mice, it was nevertheless significantly reduced compared to that seen in matching untreated mutants (Fig.1B). In contrast to that of the liver, the mass of the small intestine increased only moderately in the Lal−/− mice (Fig.1C). There was a marginal contraction (p > 0.05) in intestinal weight in the treated mutants.
Fig. 1.
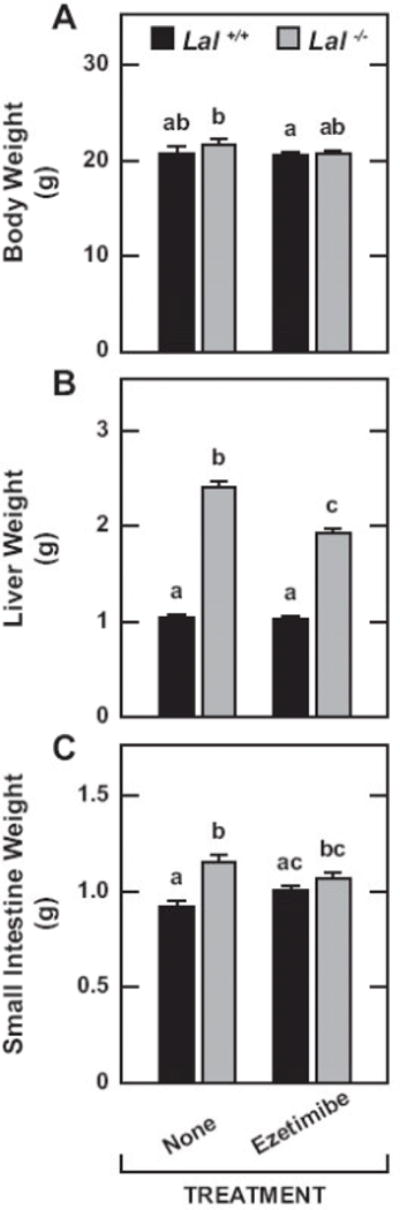
Body, liver and small intestine weights in Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days of age. All mice were females fed a basal chow diet either alone or containing ezetimibe at a level of 0.0125% w/w which corresponded to an approximate dose of 20 mg/ day/ kg bw. These mice were then used mainly for the measurement of either the rate of cholesterol synthesis in the liver, small intestine and whole body, or for other parameters including the concentration of esterified and unesterified cholesterol in the liver and small intestine. Values are the mean ± SEM of data from 10 untreated mice of each genotype, and 11 of each genotype given ezetimibe. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
3.2 Ezetimibe markedly increased hepatic and whole-animal cholesterol synthesis in the in Lal+/+ mice but not their Lal−/− counterparts
The cholesterol synthesis rates in Fig.2 are for the liver and small intestine, and are presented per gram of tissue (Fig.2A and 2D) as well as on a whole-organ basis (Fig.2B and 2E). This was done partly to better define the impact that changing rates of synthesis in the small intestine, and liver particularly, had on synthesis in the animal as a whole (Fig.3). In the mutants, the rate of synthesis in the liver was greatly elevated and remained so with ezetimibe treatment (Fig.2A and 2B). Although not statistically significant, the rate of synthesis per gram of liver in the treated mutants was marginally higher than in their untreated Lal−/− littermates (Fig.2A). This was partially offset by their reduced liver mass (Fig.1B). Hence, whole-liver synthesis rates in the untreated and treated Lal-deficient mice were virtually identical (Fig.2B). In contrast, in the treated Lal+/+ mice, the rate of hepatic cholesterol synthesis, expressed either per gram (Fig.2A) or per organ (Fig.2B), increased ~7-fold compared to that in Lal+/+ mice on the chow diet only. A different pattern of response was seen for the small intestine in both the wildtype and mutant mice. Here, the rate of cholesterol synthesis, expressed either per gram (Fig.2D) or per whole organ (Fig.2E), was significantly higher in both the Lal+/+ and Lal−/− mice given ezetimibe compared to their matching controls fed the basal diet alone.
Fig. 2.
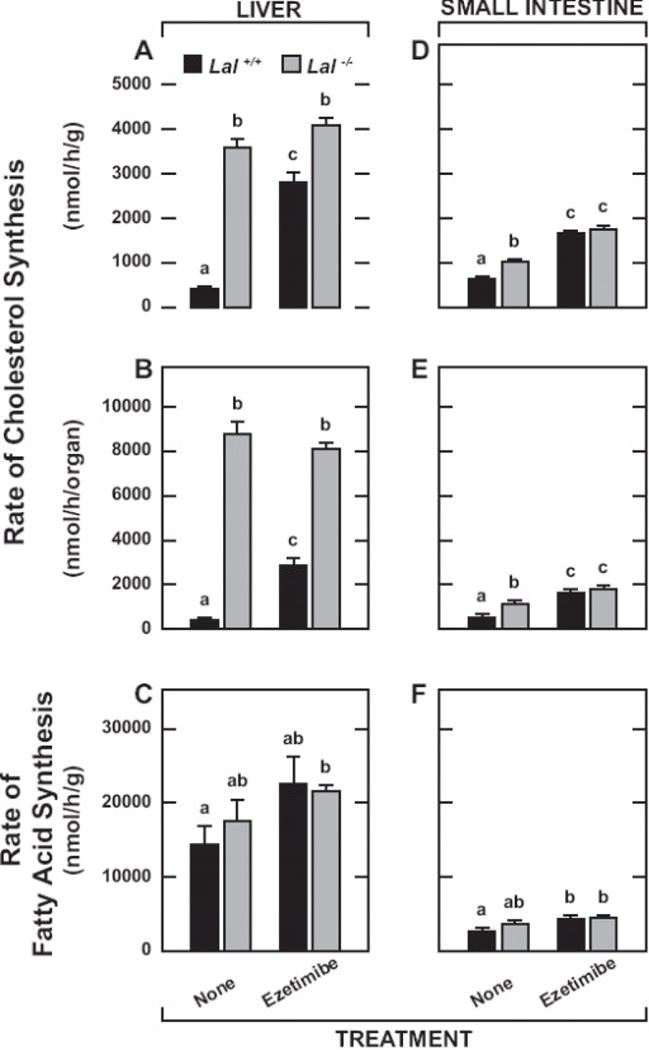
Rates of hepatic and intestinal cholesterol and fatty acid synthesis in Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days of age. As described in Materials and Methods, these mice received on i.p. injection of [3H]water and were anesthetized and exsanguinated 1 h later. The liver, small intestine and remaining carcass were taken for the measurement of [3H]sterol content. For the liver and small intestine, the content of [3H]fatty acids was also determined. The cholesterol synthesis rates represent the nmol of water incorporated into digitonin-precipitable sterols per gram wet weight of tissue (A and D), or per whole organ (B and E) per h. The rates of hepatic and intestinal fatty acid synthesis (C and F, respectively) are expressed per gram wet weight of tissue per h. Values are the mean ± SEM of data from 4 mice of each genotype fed the basal diet alone, and 5 Lal+/+ and 5 Lal−/− mice given ezetimibe. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
Fig. 3.
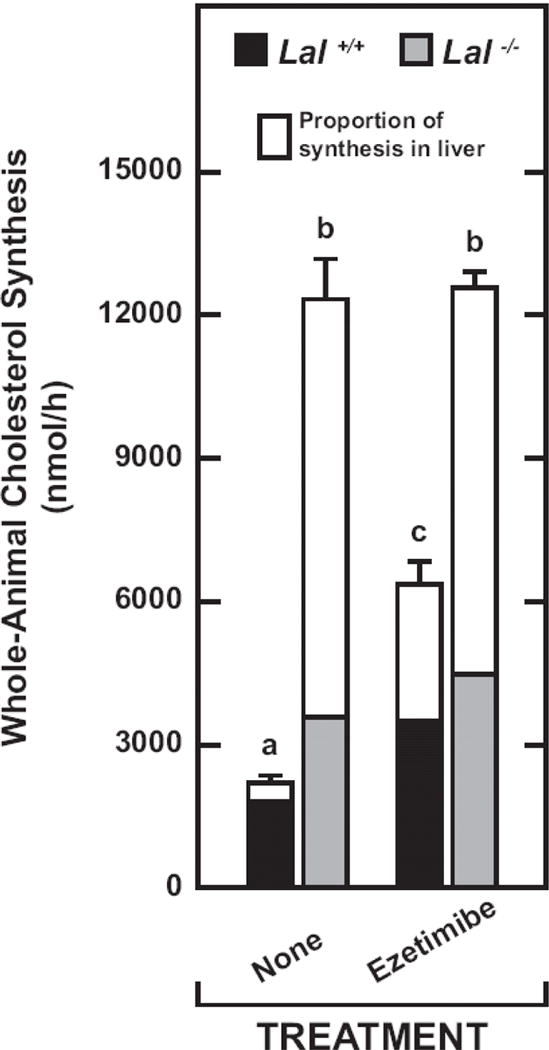
Contribution of the liver to whole-animal cholesterol synthesis in Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days. These data were derived from the mice used in the study described in Fig. 2. In addition to the liver and whole small intestine, the remaining carcass for each mouse was used also to determine the [3H]sterol content. These data were then used to calculate the rate of whole animal cholesterol synthesis (nmol of water incorporated into sterols per h) and the proportion of synthesis that occurred in the liver. For the rates of whole-animal synthesis, the values are the mean ± SEM of data from 4 mice of each genotype fed the basal diet alone, and 5 mice of each genotype given ezetimibe. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
The dominating role of the liver in determining the rate of whole-body cholesterol synthesis, especially in the Lal−/− mice, is illustrated in Fig.3. In untreated Lal−/− mice, the liver accounted for ~71% of all sterol synthesis in the animal, and on ezetimibe, this figure was ~64%. This slightly lower contribution simply reflected the fact that whole-liver mass was reduced in the treated mutants (Fig.1B). In the untreated Lal+/+ mice, only ~18% of synthesis in the entire animal occurred in the liver but this fraction increased to ~45% with ezetimibe. The proportion of whole-body synthesis attributable to the small intestine is not depicted in Fig.3. Despite intestinal synthesis in the Lal+/+ mice being increased from 547 ± 69 to 1639 ± 66 nmol/h/organ with ezetimibe (Fig.2B), its contribution to whole-animal synthesis remained at 25%, the same as in the untreated Lal+/+ mice. This occurred because the increase in hepatic synthesis in the treated wildtypes (from 393 ± 55 to 2867 ± 304 nmol/h/organ) (Fig.2B) was so pronounced that it masked the accelerated rate of synthesis in their small intestine. Such overshadowing of the intestinal contribution was even more overt in the Lal−/− mice. This is clearly illustrated by comparing the intestinal synthesis rates in Fig. 2E with those for the liver in Fig. 2B. In the chow-fed Lal−/− mice, the fraction of whole-body synthesis contributed by the small intestine was just ~9%. This increased to only ~14% in the treated Lal−/− mice, even though their rate of intestinal synthesis, either per gram of tissue (Fig.2D) or per whole organ (Fig.2E), was appreciably enhanced with ezetimibe.
In contrast to the data for cholesterol synthesis, there were no significant genotypic or treatment-related changes in the rate of fatty acid synthesis in either the liver (Fig.2C) or small intestine (Fig.2F). For all four experimental groups, the rate of fatty acid synthesis in the liver markedly exceeded that in the intestine. There was a non-significant (p > 0.05) trend towards higher rates of hepatic fatty acid synthesis in both the Lal+/+ and Lal−/− mice given ezetimibe (Fig.2C).
3.3 Ezetimibe markedly reduced the hepatic and intestinal esterified cholesterol pools in the Lal−/− mice, leading to a contraction of their whole-body cholesterol content, along with a marked fall in plasma ALT activity
In the livers of the Lal−/− mice given ezetimibe, the concentration of esterified cholesterol (EC) fell to 37.2 mg/g from 55.6 ± 2.4 mg/g in the mutants on chow only (Fig.4A). When this fall in EC concentration was combined with the reduction in liver mass, their whole-liver EC content was half of that in the untreated Lal−/− mice (Fig.4B). This response was very similar to that observed in our initial publication on ezetimibe treatment in male Lal−/− mice [38]. Although not discernable in Fig.4A or 4B, EC accounted for only about 18% of the total cholesterol in the livers of the Lal+/+ mice, irrespective of treatment. In the small intestine of the Lal−/− mice, the EC concentration (Fig.4C) and content (Fig.4D) were markedly elevated, although not to the degree seen in the liver. However, with ezetimibe treatment, the intestinal concentration of EC in the mutants fell to 1.2 mg/g from 2.4 mg/g in their chow-fed counterparts. Together, these data showed that, by starting the Lal−/− mice on ezetimibe from weaning, there was a decisive blunting of the amount of EC sequestered in these organs at early adulthood. The same effect was not seen with tissue triglyceride levels. Although the data are not illustrated, in the Lal−/− mice on ezetimibe vs. chow only, the hepatic triglyceride concentrations equaled 14.8 ± 1.0 and 15.8 ± 0.9 mg/g, respectively. In their matching Lal+/+ controls, the hepatic triglyceride concentrations averaged 6 to 7 mg/g in both treated and untreated groups. For the Lal−/− mice on ezetimibe vs chow only, the intestinal triglyceride concentration was 16.4 ± 1.0 mg/g and 16.6 ± 1.4 mg/g, respectively. In their matching Lal+/+ controls, the intestinal triglyceride concentrations averaged 8 to 9 mg/g in both the treated and untreated groups.
Fig. 4.
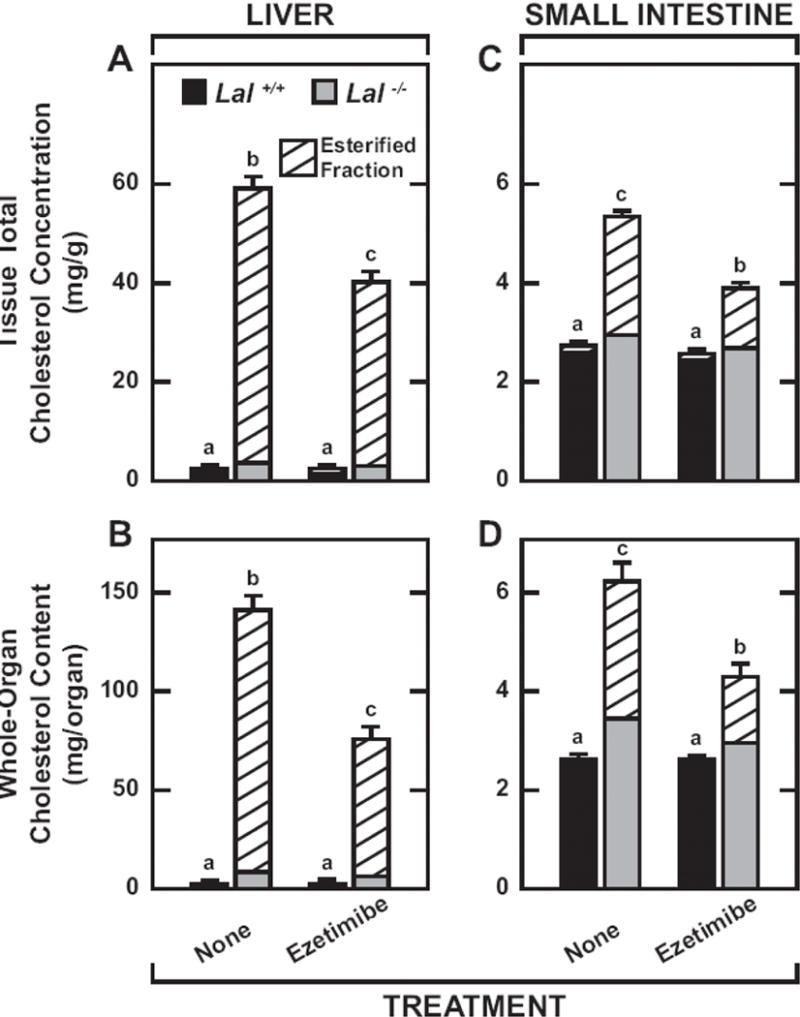
Proportion of hepatic and intestinal cholesterol present in esterified form in Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days. Those mice described in the legend for Fig. 1 that were not used for cholesterol synthesis measurements were used for determining several parameters including the proportion of hepatic and intestinal cholesterol present in esterified form as illustrated above. The concentrations of unesterified and esterified cholesterol were determined in aliquots of liver and the entire small intestine as described in Materials and Methods. The data were expressed in two ways. The total cholesterol concentrations (mg/ g wet weight of tissue) and the fraction of the total present in esterified form for the liver and small intestine are shown in A and C, respectively. The whole organ cholesterol contents and the proportion of each that was esterified in the liver and small intestine are illustrated in B and D, respectively. Values are the mean ± SEM of data from 6 mice in each group. Different letters (a-c) denote statistically significant (p<0.05) differences in the total cholesterol (ie esterified plus unesterified) concentrations (A and C) and contents (B and D) as determined by 2-way ANOVA with genotype and treatment as variables.
In LAL deficiency there is a marked rise in the EC content of many extrahepatic organs, not just the small intestine [20, 23]. Therefore, the significant reduction in whole-body cholesterol content in the ezetimibe-treated mutants (Fig.5A) raised the question of how much of this fall reflected a diminution of the EC content of organs other than the liver and small intestine. From the data in Fig.4A and 4B, it can be calculated that the reduction in whole-liver and whole-small intestine cholesterol content in the Lal−/− mice on ezetimibe together accounted for 92.4% of the contraction in whole-body cholesterol content (Fig.5A). The liver itself accounted for 89.8%. Consistent with these findings was the decisive fall in plasma ALT activity, an indicator of liver injury, in the mutants on ezetimibe (Fig.5B).
Fig. 5.
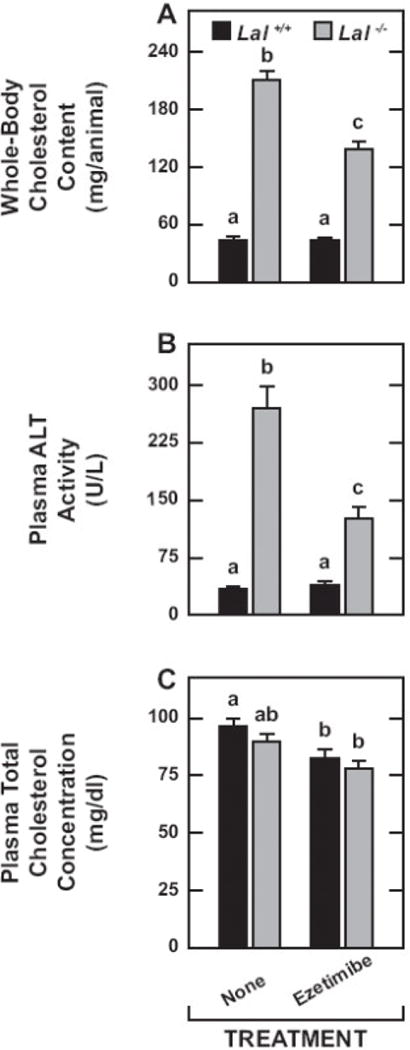
Whole-body cholesterol content, plasma ALT activity, and plasma cholesterol concentration in Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days of age. These parameters were determined in the same groups of mice used for the measurement of hepatic and intestinal esterified and unesterified cholesterol concentrations. The details of how each parameter was measured are given in Materials and Methods. Values are the mean ± SEM of data from 6 mice in each group. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
The plasma total cholesterol concentration data (Fig.5C) warrant particular comment. In contrast to the marked dyslipidemia frequently seen in CESD patients [26–28], LAL-deficient mice, depending considerably on their strain background, do not develop elevated plasma total cholesterol concentrations although there may be a shift in their lipoprotein composition [20, 38]. In the present study there was no genotypic difference in the plasma cholesterol levels either on the basal diet alone or containing ezetimibe (Fig. 5C). However, for mice of both genotypes, there was a modest reduction in the cholesterol concentrations with ezetimibe although this was statistically significant (p < 0.05) for only the Lal+/+ mice.
3.4 Hepatic and intestinal relative mRNA expression levels for key genes in the cholesterol biosynthetic pathway reflected changes found in sterol synthesis rates in vivo
The mRNA levels for Lipa confirmed the marked diminution in the expression of this gene in the livers (Fig.6A) and small intestines (Fig.7A) of the knockout mice. In the case of the liver, the mRNA levels for Hmgcr (Fig.6B) and Hmgcs (Fig.6C) faithfully reflected the cholesterol synthesis rates expressed per g of tissue (Fig.2A). There are dozens of enzymes in the cholesterol biosynthetic pathway, with changes in the mRNA expression levels for both Hmgcr and Hmgcs being invariably considered the best indicators of shifts in the activity of this pathway [7, 17, 58]. For Srebp2 (Fig. 6D), the genotypic and treatment-related shifts in the mRNA expression level broadly paralleled those in the synthesis rates. In the case of the mRNA level for Fas in the liver (Fig.6E), there were no clear changes as a function of either genotype or treatment which was also true of the fatty acid synthesis rates (Fig.2C). Hepatic mRNA levels for Ldlr (Fig.6F) were unchanged across all groups. The mRNA levels for integrin alpha X (CD11c) (Fig.6G) and tumor necrosis factor alpha (Tnfα) (Fig.6H) were significantly lower in the treated mutants. The data in Fig. 7 show that the changes in the mRNA expression levels for genes like Hmgcr (Fig.7B) and Srebp2 (Fig.7C) in the small intestine were more subdued than was the case for the liver (Fig.6B and 6D, respectively). When compared to the genotypic and treatment-related changes in intestinal cholesterol synthesis rates (Fig.2D), the shifts in the expression levels of Hmgcr and Srebp2 were more synchronous with treatment than with genotype. For Fas expression in the intestine (Fig.7D), there were some trends (p > 0.05) relating to ezetimibe treatment in both the Lal+/+ and Lal−/− mice that paralleled the shifts in their rates of fatty acid synthesis (Fig.2F). In the case of Npc1l1 (Fig.7E), no significant changes were detected whereas for Ldlr expression (Fig.7F) there was a significant (p < 0.05) increase with ezetimibe treatment for both the Lal+/+ and Lal−/− mice.
Fig. 6.
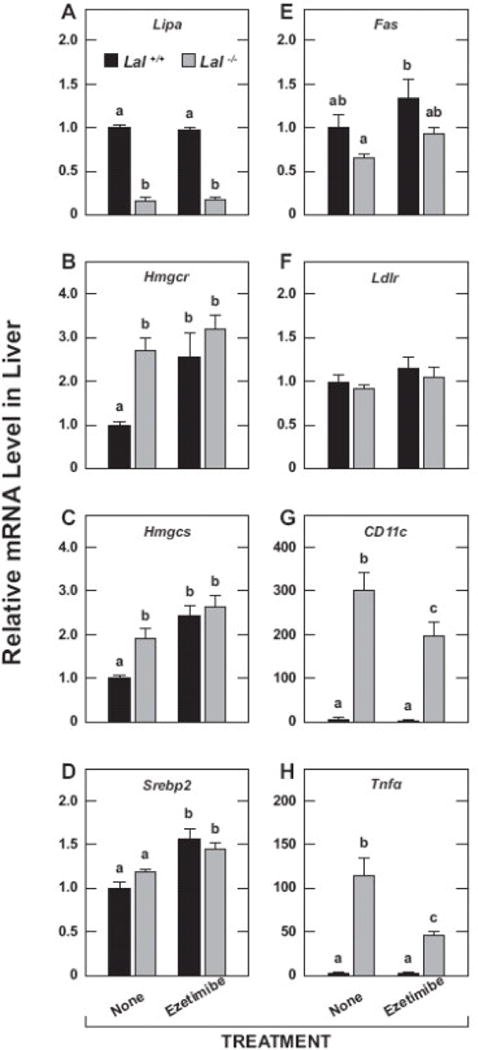
Relative expression level for various genes in the livers of Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days of age. These analyses were performed using aliquots of the same liver taken for the measurement of esterified and unesterified cholesterol concentrations (Fig. 4A, 4B). The mRNA levels were normalized to cyclophilin, and the values for each mouse were then expressed relative to those obtained for their matching Lal+/+ controls, which, in each case, were arbitrarily set at 1.0. Values are the mean ± SEM of data from 6 mice in each group. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
Fig. 7.
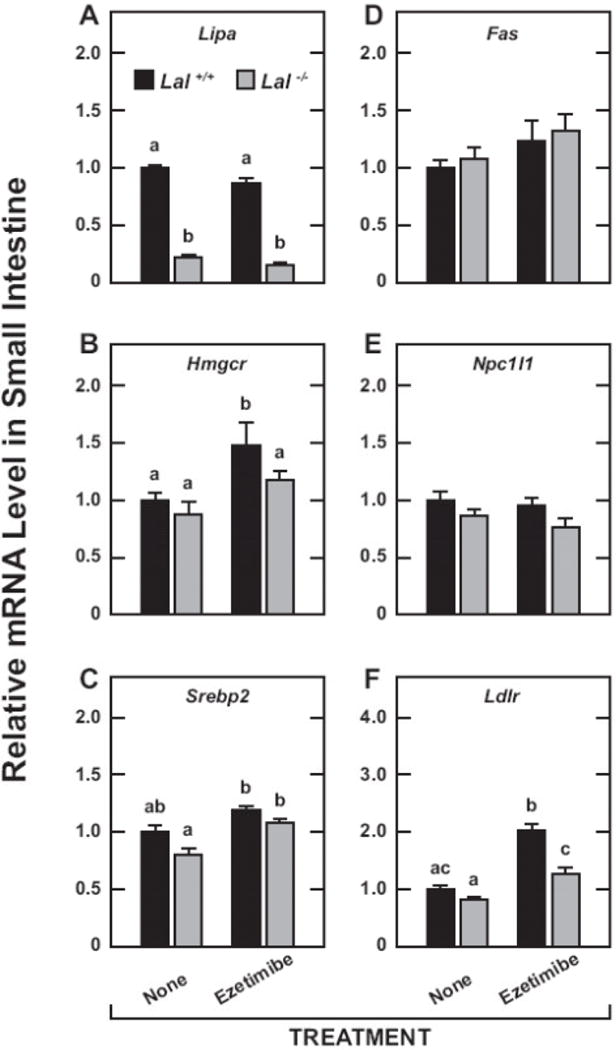
Relative expression level for various genes in the small intestines of Lal+/+ and Lal−/− mice given ezetimibe in their diet from 21 to 50 days of age. As described in Materials and Methods, these measurements were made using the mucosal scraping from the entire length of the small intestine. The mRNA levels were normalized and expressed in the same way as for the liver in Fig. 6. Values are the mean ± SEM of data from 4 mice in each group. Different letters (a-c) denote statistically significant (p<0.05) differences as determined by 2-way ANOVA with genotype and treatment as variables.
4. Discussion
The discovery and development of ezetimibe by Dr. Harry R. Davis Jr. and his colleagues represents the culmination of decades of research driven by a widely held belief that pharmacological control of intestinal cholesterol absorption was an important component of developing more efficacious strategies for treating dyslipidemia and lowering cardiovascular disease risk in the general population. Subsequent research conducted by this group of investigators into the mechanism of action of ezetimibe led to the discovery of the intestinal sterol transporter Niemann-Pick C1 Like 1 (NPC1L1), and the demonstration that this protein was the target of ezetimibe [45]. This publication contains a full schematic explanation of the sites and mechanism of action of ezetimibe in changing the enterohepatic flux of cholesterol. By binding to NPC1L1 primarily in jejunal enterocytes, ezetimibe inhibits the absorption of not only cholesterol, but also phytosterols and other non-cholesterol sterols. This latter effect has led ezetimibe to become the choice of treatment for sitosterolemia, a rare disease characterized by the retention of large amounts of non-cholesterol sterols in the plasma and tissues because of mutations in the genes that encode the production of ABCG5 and ABCG8, both of which facilitate the efflux of such sterols back into the intestinal lumen or the bile [46, 47]. The true prevalence of sitosterolemia is unknown but a 2008 publication noted a figure of 80–100 cases worldwide [48].
WD and CESD are also rare disorders although the prevalence of LAL deficiency is believed to be higher than figures suggest because patients are undiagnosed or misdiagnosed [26, 28, 49]. Improved detection rates, especially at earlier ages, and the availability of an ERT should potentially lessen the incidence of premature vascular disease in patients with diminished LAL function. There may, however, remain a subset of CESD patients who still require some form of adjunctive therapy for optimizing management of their dyslipidemia. In the literature on this subject, two case reports were found that focus on the use of ezetimibe, one as a monotherapy, the other in combination with lovastatin [50, 51]. Both involved patients in the second decade of life. Ezetimibe monotherapy improved the plasma lipid composition and reduced liver transaminase activities [51]. In the patient that had been on long-term lovastatin treatment, the co-administration of ezetimibe resulted in a further 16% reduction in the plasma LDL-cholesterol level [50]. Although, as noted earlier, the Lal−/− mice (FVB background) used in the present studies did not manifest elevated plasma total cholesterol concentrations, these were reduced in the mutants given ezetimibe but not to quite the same extent as seen in their Lal+/+ controls on treatment (Fig.5C).
The pathophysiological significance of the findings from the present studies resides partly in the fact that, like the CESD mouse model, patients with this disorder manifest accelerated rates of cholesterol synthesis which are believed to occur mainly in the liver [52, 53]. Presumably, intestinal cholesterol synthesis rates also rise with LAL deficiency in humans. The present findings for the small intestine in the LAL-deficient mouse, off and on ezetimibe therapy, are of particular importance given that changes in intestinal cholesterol handling invariably have a major downstream impact on cholesterol balance across the liver [1, 4, 6]. Several major points concerning the quantitative and qualitative differences in how ezetimibe affected cholesterol metabolism in the small intestines and livers of the mutant mice and their wild-type controls warrant discussion. In making these comparisons, the differences in the cellular make-up of these two organs in both the healthy and Lal-deficient animal have to be taken into account. One fundamental difference is that, while there is continual turnover of the absorptive cells in healthy intestinal mucosa [54], this is not the case with hepatocytes which represent the major cell type in normal liver tissue. Another consideration relates to the cholesterol esterifying enzymes in both organs. Enterocytes express SOAT2 activity whereas SOAT1 is localized to other cell types in the epithelium [55]. There is not complete agreement on which esterifying enzyme dominates in hepatocytes although the balance of evidence suggests that it is SOAT2 [55–57], while Kupffer cells express SOAT1 [57].
The first point of discussion has to do with the dramatic reduction in intestinal EC content in the mutants on treatment (Fig.4D). This was the product of a sharp fall in the EC concentration combined with a modest contraction in intestinal weight. At the dose of ezetimibe used, the absorption of cholesterol was probably almost fully blocked [7]. This would explain the compensatory increase in intestinal cholesterol synthesis, irrespective of genotype (Fig.2D and 2E). The intestinal mRNA data for the Ldlr (Fig.7F) imply there may have also been an increase in the uptake of LDL-cholesterol in the mutants but not nearly as much as was seen in their matching treated Lal+/+ controls. A marked increase in intestinal Ldlr has been described in ezetimibe-treated C57BL/6 mice maintained on a rodent chow diet [58]. Irrespective of whether this occurred in the treated Lal mutants, the perceived cellular cholesterol deficit caused by the sequestration of EC in the E/L compartment would have happened concurrently with the real cholesterol deficit arising from an essentially complete ezetimibe-mediated block of luminal cholesterol entering the absorptive cells. Despite the resulting compensatory increase in cholesterol synthesis, chylomicrons produced in the intestine of the treated mutants conceivably had a diminished EC content. If this did occur then it would have had a favorable impact in the livers of these mice because of a reduction in the amount of chylomicron remnant EC becoming sequestered in the E/L compartment following receptor-mediated uptake of these particles [59, 60]. The data in Fig 4A and 4B are consistent with this being the case.
While the reduction in hepatic EC content in the treated mutants is likely is attributable to an upstream effect of ezetimibe in the small intestine, the mechanism(s) accounting for the sharp fall in intestinal EC content are less clear. One possibility is that, in addition to a diminished rate of intestinal SOAT2-mediated esterification, EC formation by SOAT1 in other cell types within the mucosa of the treated mutants may have slowed. With the generally improved health of these mice, there may also have been a reduced level of infiltration of intestinal tissue by EC-laden macrophages originating from other organ systems.
A second discussion point centers on the question of why the expected compensatory increase in cholesterol synthesis with ezetimibe occurred in the small intestine of both the Lal+/+ and Lal−/− mice and in the livers of the wild types, but not in the livers of the mutants (Fig.2A and 2B). There is not a firm explanation for this but it may be that without full restoration of LAL function, the rate of cholesterol synthesis in those livers would continue to be maximally stimulated in response to the perceived cellular cholesterol deficit. The additional stimulus ordinarily coming from the reduced delivery of intestinal cholesterol to the liver would thus fail to have the same impact on hepatic sterol synthesis rates that it does in the livers of the treated Lal+/+ mice. Alternatively, the rate seen in the livers of the treated Lal−/− mice could reflect stimulation from both the prevailing level of EC sequestration and reduced chylomicron cholesterol delivery to the liver. Irrespective of which of these scenarios is correct, they raise the intriguing question of what happens to the liver cholesterol synthesis rate in CESD patients after an extended period of ERT. This could probably be explored using the techniques described here in LAL knockout mice given long-term ERT [61]. Irrespective of what the answers are to these questions, what matters more is that ezetimibe treatment in a CESD mouse model, when started long before maturity, clearly improved the health of the liver as judged by a decisive fall in the degree of hepatomegaly (Fig. 1B), a marked decrease in plasma ALT activity (Fig 5B), and clear reductions in the hepatic mRNA levels for CD11c (Fig.6G) and Tnfα (Fig.6H). The sharp decline in the level of hepatic EC entrapment in this model is also the main factor accounting for the significant contraction in the size of the whole-body cholesterol pool (Fig.5A).
The third point warranting discussion is the lack of effect of ezetimibe on the elevated hepatic and intestinal triglyceride levels in the Lal−/− mice. This is in contrast to the significant reductions in the triglyceride content of the liver and small intestine in Lal−/− mice given ERT [61]. In that investigation, the effect was much more pronounced in the liver. The lack of influence of ezetimibe on hepatic and intestinal triglyceride content in the present study was not unexpected given this agent selectively blocks the uptake of sterols, not fat in general [45]. This finding does however imply it is the entrapment of EC, more than that of triglyceride, that drives disease progression in LAL deficiency. The results of our earlier studies with Lal−/− mice treated with a low dose of a SOAT-2 selective inhibitor (PRD125) from the time of weaning are consistent with this interpretation [62].
A final point to be made concerns the potential relevance of the present findings to the quest for optimizing the management of CESD. With the advent of an effective ERT, and the presumption of an increased detection of individuals with this disorder, an expanding literature on this topic might be anticipated. One of the most searching questions surrounding the actions of ERT in hydrolyzing the vast pool of sequestered EC throughout the body, particularly in the liver, is the fate of all the UC that is generated once it exits the E/L compartment. Depending largely on the rate of its release relative to the operating capacity of the various pathways that this UC is channeled into, it is conceivable that there will be a transient, if not sustained increase in biliary cholesterol saturation. There is also the possibility that some of this UC will undergo re-esterification and subsequently become incorporated into nascent very low density lipoproteins. The most likely scenario however is an increased rate of delivery of the UC via the bile into the intestinal lumen where an unknown fraction of it will be reabsorbed and returned to the liver in chylomicron remnants. Clearly, in this instance, the co-administration of ezetimibe would be an effective strategy for preventing this from happening and maximizing the benefit of the ERT. Alternatively, a case could be made for blunting re-esterification of this surplus UC within the liver using the new generation of selective SOAT2 inhibitors [63]. All the resources are available for exploring these clinically relevant questions.
Acknowledgments
This work was supported entirely by US Public Health Service Grant R01HL009610. We are indebted to Dr. Gregory Grabowski and Dr. Hong Du for their generous gift of LAL heterozygous breeding stock. Ezetimibe was kindly provided by Dr. Harry R. Davis, Jr. at Merck & Co., Inc. We thank Monti Schneiderman and Stephen Ostermann for technical assistance.
Abbreviations
- ALT
alanine aminotransferase
- bw
body weight
- CESD
cholesteryl ester storage disease
- EC
esterified cholesterol
- E/L
endosomal/lysosomal
- ERT
enzyme replacement therapy
- Fas
fatty acid synthase
- Hmgcr
hydroxymethylglutaryl coenzyme A reductase
- Hmgcs
hydroxymethylglutaryl coenzyme A synthase
- LAL
lysosomal acid lipase
- Lipa
gene that encodes LAL
- Ldlr
low-density lipoprotein receptor
- Npc1l1
Niemann-Pick C1-Like 1
- Srebp2
sterol regulatory element-binding protein 2
- UC
unesterified cholesterol
- Soat
sterol O-acyltransferase
- VLDL
very low density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article: Ezetimibe (PubChem CID 150311)
References
- 1.Turley SD, Dietschy JM. The Metabolism and Excretion of Cholesterol by the Liver. In: Arias IM, Jakoby WB, Popper H, Schachter D, Shafritz DA, editors. The Liver: Biology and Pathobiology. Raven Press; New York: 1988. pp. 617–641. [Google Scholar]
- 2.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34:1637–1659. [PubMed] [Google Scholar]
- 3.Tso P. Intestinal Lipid Absorption. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1994. pp. 1867–1907. [Google Scholar]
- 4.Grundy SM. Absorption and metabolism of dietary cholesterol. Annu Rev Nutr. 1983;3:71–96. doi: 10.1146/annurev.nu.03.070183.000443. [DOI] [PubMed] [Google Scholar]
- 5.Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudel LL, Davis M, Sawyer J, Shah R, Wallace J. Primates highly responsive to dietary cholesterol up-regulate hepatic ACAT2, and less responsive primates do not. J Biol Chem. 2002;277:31401–31406. doi: 10.1074/jbc.M204106200. [DOI] [PubMed] [Google Scholar]
- 7.Repa JJ, Turley SD, Quan G, Dietschy JM. Delineation of molecular changes in intrahepatic cholesterol metabolism resulting from diminished cholesterol absorption. J Lipid Res. 2005;46:779–789. doi: 10.1194/jlr.M400475-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Valasek MA, Repa JJ, Quan G, Dietschy JM, Turley SD. Inhibiting intestinal NPC1L1 activity prevents diet-induced increase in biliary cholesterol in Golden Syrian hamsters. Am J Physiol Gastrointest Liver Physiol. 2008;295:G813–822. doi: 10.1152/ajpgi.90372.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMurry MP, Connor WE, Lin DS, Cerqueira MT, Connor SL. The absorption of cholesterol and the sterol balance in the Tarahumara Indians of Mexico fed cholesterol-free and high cholesterol diets. Am J Clin Nutr. 1985;41:1289–1298. doi: 10.1093/ajcn/41.6.1289. [DOI] [PubMed] [Google Scholar]
- 10.Sudhop T, Reber M, Tribble D, Sapre A, Taggart W, Gibbons P, Musliner T, von Bergmann K, Lutjohann D. Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J Lipid Res. 2009;50:2117–2123. doi: 10.1194/jlr.P900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Einarsson K, Ericsson S, Ewerth S, Reihner E, Rudling M, Stahlberg D, Angelin B. Bile acid sequestrants: mechanisms of action on bile acid and cholesterol metabolism. Eur J Clin Pharmacol. 1991;40(Suppl 1):S53–S58. [PubMed] [Google Scholar]
- 12.Parini P, Gustafsson U, Davis MA, Larsson L, Einarsson C, Wilson M, Rudling M, Tomoda H, Omura S, Sahlin S, Angelin B, Rudel LL, Eriksson M. Cholesterol synthesis inhibition elicits an integrated molecular response in human livers including decreased ACAT2. Arterioscler Thromb Vasc Biol. 2008;28:1200–1206. doi: 10.1161/ATVBAHA.107.157172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–E344. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- 15.Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, Turley SD. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem. 2000;275:39685–39692. doi: 10.1074/jbc.M007653200. [DOI] [PubMed] [Google Scholar]
- 16.Repa JJ, Buhman KK, Farese RV, Jr, Dietschy JM, Turley SD. ACAT2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: impact on hepatic cholesterol homeostasis. Hepatology. 2004;40:1088–1097. doi: 10.1002/hep.20439. [DOI] [PubMed] [Google Scholar]
- 17.Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–680. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. doi: 10.1146/annurev-physiol-012110-142233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res. 2001;42:489–500. [PubMed] [Google Scholar]
- 21.Grabowski GA, Du H, Charnas L. Lysosomal Acid Lipase Deficiencies: The Wolman Disease/Cholesteryl Ester Storage Disease Spectrum. 2015 http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374143. (Accessed June 2 2015).
- 22.Ghishan FK. Inborn errors of metabolism that lead to permanent liver injury. In: Boyer TD, Manns MP, Sanyal AJ, editors. Zakim and Boyer’s hepatology : a textbook of liver disease. Saunders/Elsevier; Philadelphia, PA: 2012. pp. 1155–1201. [Google Scholar]
- 23.Aqul A, Lopez AM, Posey KS, Taylor AM, Repa JJ, Burns DK, Turley SD. Hepatic entrapment of esterified cholesterol drives continual expansion of whole body sterol pool in lysosomal acid lipase-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2014;307:G836–847. doi: 10.1152/ajpgi.00243.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton BK, Balwani M, Feillet F, Baric I, Burrow TA, Camarena Grande C, Coker M, Consuelo-Sanchez A, Deegan P, Di Rocco M, Enns GM, Erbe R, Ezgu F, Ficicioglu C, Furuya KN, Kane J, Laukaitis C, Mengel E, Neilan EG, Nightingale S, Peters H, Scarpa M, Schwab KO, Smolka V, Valayannopoulos V, Wood M, Goodman Z, Yang Y, Eckert S, Rojas-Caro S, Quinn AG. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency. N Engl J Med. 2015;373:1010–1020. doi: 10.1056/NEJMoa1501365. [DOI] [PubMed] [Google Scholar]
- 25.Frampton JE. Sebelipase Alfa: A Review in Lysosomal Acid Lipase Deficiency. Am J Cardiovasc Drugs. 2016;16:461–468. doi: 10.1007/s40256-016-0203-2. [DOI] [PubMed] [Google Scholar]
- 26.Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol. 2013;24:332–338. doi: 10.1097/MOL.0b013e328361f6c6. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds T. Cholesteryl ester storage disease: a rare and possibly treatable cause of premature vascular disease and cirrhosis. J Clin Pathol. 2013;66:918–923. doi: 10.1136/jclinpath-2012-201302. [DOI] [PubMed] [Google Scholar]
- 28.Reiner Z, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Coric M, Calandra S, Hamilton J, Eagleton T, Ros E. Lysosomal acid lipase deficiency - An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235:21–30. doi: 10.1016/j.atherosclerosis.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6:447–470. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 30.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM. for the IMPROVE-IT investigators, Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 31.Kusters DM, Caceres M, Coll M, Cuffie C, Gagne C, Jacobson MS, Kwiterovich PO, Lee R, Lowe RS, Massaad R, McCrindle BW, Musliner TA, Triscari J, Kastelein JJ. Efficacy and safety of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia. J Pediatr. 2015;166:1377–1384. e1371–1373. doi: 10.1016/j.jpeds.2015.02.043. [DOI] [PubMed] [Google Scholar]
- 32.Yoo EG. Sitosterolemia: a review and update of pathophysiology, clinical spectrum, diagnosis, and management. Ann Pediatr Endocrinol Metab. 2016;21:7–14. doi: 10.6065/apem.2016.21.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Bari O, Neuschwander-Tetri BA, Liu M, Portincasa P, Wang DQ. Ezetimibe: its novel effects on the prevention and the treatment of cholesterol gallstones and nonalcoholic fatty liver disease. J Lipids. 2012;2012:302847. doi: 10.1155/2012/302847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labonte ED, Camarota LM, Rojas JC, Jandacek RJ, Gilham DE, Davies JP, Ioannou YA, Tso P, Hui DY, Howles PN. Reduced absorption of saturated fatty acids and resistance to diet-induced obesity and diabetes by ezetimibe-treated and Npc1l1−/− mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G776–G783. doi: 10.1152/ajpgi.90275.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng S, Hoos L, Cook J, Tetzloff G, Davis H, Jr, van Heek M, Hwa JJ. Ezetimibe improves high fat and cholesterol diet-induced non-alcoholic fatty liver disease in mice. Eur J Pharmacol. 2008;584:118–124. doi: 10.1016/j.ejphar.2008.01.045. [DOI] [PubMed] [Google Scholar]
- 36.Ushio M, Nishio Y, Sekine O, Nagai Y, Maeno Y, Ugi S, Yoshizaki T, Morino K, Kume S, Kashiwagi A, Maegawa H. Ezetimibe prevents hepatic steatosis induced by a high-fat but not a high-fructose diet. Am J Physiol Endocrinol Metab. 2013;305:E293–E304. doi: 10.1152/ajpendo.00442.2012. [DOI] [PubMed] [Google Scholar]
- 37.Loomba R, Sirlin CB, Ang B, Bettencourt R, Jain R, Salotti J, Soaft L, Hooker J, Kono Y, Bhatt A, Hernandez L, Nguyen P, Noureddin M, Haufe W, Hooker C, Yin M, Ehman R, Lin GY, Valasek MA, Brenner DA, Richards L. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial) Hepatology. 2015;61:1239–1250. doi: 10.1002/hep.27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chuang JC, Lopez AM, Posey KS, Turley SD. Ezetimibe markedly attenuates hepatic cholesterol accumulation and improves liver function in the lysosomal acid lipase-deficient mouse, a model for cholesteryl ester storage disease. Biochem Biophys Res Commun. 2014;443:1073–1077. doi: 10.1016/j.bbrc.2013.12.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sudhop T, Lütjohann D, Kodal A, Igel M, Tribble DL, Shah S, Perevozskaya I, von Bergmann K. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–1948. doi: 10.1161/01.cir.0000034044.95911.dc. [DOI] [PubMed] [Google Scholar]
- 40.Schwarz M, Russell DW, Dietschy JM, Turley SD. Marked reduction in bile acid synthesis in cholesterol 7 α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J Lipid Res. 1998;39:1833–1843. [PubMed] [Google Scholar]
- 41.Turley SD, Valasek MA, Repa JJ, Dietschy JM. Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1012–G1022. doi: 10.1152/ajpgi.00190.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 2005;42:886–893. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- 43.Chuang JC, Valasek MA, Lopez AM, Posey KS, Repa JJ, Turley SD. Sustained and selective suppression of intestinal cholesterol synthesis by Ro 48-8071, an inhibitor of 2,3-oxidosqualene:lanosterol cyclase, in the BALB/c mouse. Biochem Pharmacol. 2014;88:351–363. doi: 10.1016/j.bcp.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ. 2005;29:151–159. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- 45.Davis HR, Jr, Altmann SW. Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochim Biophys Acta. 2009;1791:679–683. doi: 10.1016/j.bbalip.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 47.Lee MH, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R, Srivastava AK, Salen G, Dean M, Patel SB. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet. 2001;27:79–83. doi: 10.1038/83799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kidambi S, Patel SB. Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis. J Clin Pathol. 2008;61:588–594. doi: 10.1136/jcp.2007.049775. [DOI] [PubMed] [Google Scholar]
- 49.Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230–1243. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 50.Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis. 2005;4:26. doi: 10.1186/1476-511X-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abello F, Guardamagna O, Baracco V, Bonardi R. P55 The treatment of colesteryl storage disease (CESD) by ezetimibe monotherapy. Atherosclerosis Supplements. 2010;11:28. [Google Scholar]
- 52.Ginsberg HN, Le NA, Short MP, Ramakrishnan R, Desnick RJ. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J Clin Invest. 1987;80:1692–1697. doi: 10.1172/JCI113259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cummings MH, Watts GF. Increased hepatic secretion of very-low-density lipoprotein apolipoprotein B-100 in cholesteryl ester storage disease. Clin Chem. 1995;41:111–114. [PubMed] [Google Scholar]
- 54.Lipkin M. Proliferation and differentiation of gastrointestinal cells in normal and disease states. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1981. pp. 145–167. [Google Scholar]
- 55.Lee RG, Willingham MC, Davis MA, Skinner KA, Rudel LL. Differential expression of ACAT1 and ACAT2 among cells within liver, intestine, kidney, and adrenal of nonhuman primates. J Lipid Res. 2000;41:1991–2001. [PubMed] [Google Scholar]
- 56.Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee CYG, Strom SC, Kashyap R, Fung JJ, Farese RV, Jr, Patoiseau JF, Delhon A, Chang TY. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem. 2000;275:28083–28092. doi: 10.1074/jbc.M003927200. [DOI] [PubMed] [Google Scholar]
- 57.Parini P, Davis MA, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, Rudel LL. ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. doi: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- 58.Engelking LJ, McFarlane MR, Li CK, Liang G. Blockade of cholesterol absorption by ezetimibe reveals a complex homeostatic network in enterocytes. J Lipid Res. 2012;53:1359–1368. doi: 10.1194/jlr.M027599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38:2173–2192. [PubMed] [Google Scholar]
- 60.Nguyen TM, Sawyer JK, Kelley KL, Davis MA, Rudel LL. Cholesterol esterification by ACAT2 is essential for efficient intestinal cholesterol absorption: evidence from thoracic lymph duct cannulation. J Lipid Res. 2012;53:95–104. doi: 10.1194/jlr.M018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Xu YH, Du H, Quinn B, Liou B, Stanton L, Inskeep V, Ran H, Jakubowitz P, Grilliot N, Grabowski GA. Reversal of advanced disease in lysosomal acid lipase deficient mice: A model for lysosomal acid lipase deficiency disease. Mol Genet Metab. 2014;112:229–241. doi: 10.1016/j.ymgme.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 62.Lopez AM, Chuang JC, Posey KS, Ohshiro T, Tomoda H, Rudel LL, Turley SD. PRD125, a potent and selective inhibitor of sterol O-acyltransferase 2 markedly reduces hepatic cholesteryl ester accumulation and improves liver function in lysosomal acid lipase-deficient mice. J Pharmacol Exp Ther. 2015;355:159–167. doi: 10.1124/jpet.115.227207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohshiro T, Ohtawa M, Nagamitsu T, Matsuda D, Yagyu H, Davis MA, Rudel LL, Ishibashi S, Tomoda H. New pyripyropene A derivatives, highly SOAT2-selective inhibitors, improve hypercholesterolemia and atherosclerosis in atherogenic mouse models. J Pharmacol Exp Ther. 2015;355:299–307. doi: 10.1124/jpet.115.227348. [DOI] [PMC free article] [PubMed] [Google Scholar]