

Abstract
TWIST1 is a transcription factor critical for development which can promote prostate cancer metastasis. During embryonic development, TWIST1 and HOXA9 are co-expressed in mouse prostate and then silenced post-natally. Here we report that TWIST1 and HOXA9 co-expression are re-activated in mouse and human primary prostate tumors and are further enriched in human metastases, correlating with survival. TWIST1 formed a complex with WDR5 and the lncRNA Hottip/HOTTIP, members of the MLL/COMPASS-like H3K4 methylases, which regulate chromatin in the Hox/HOX cluster during development. TWIST1 overexpression led to co-enrichment of TWIST1 and WDR5 as well increased H3K4me3 chromatin at the Hoxa9/HOXA9 promoter which was dependent on WDR5. Expression of WDR5 and Hottip/HOTTIP was also required for TWIST1-induced upregulation of HOXA9 and aggressive cellular phenotypes such as invasion and migration. Pharmacological inhibition of HOXA9 prevented TWIST1-induced aggressive prostate cancer cellular phenotypes in vitro and metastasis in vivo. This study demonstrates a novel mechanism by which TWIST1 regulates chromatin and gene expression by cooperating with the COMPASS-like complex to increase H3K4 trimethylation at target gene promoters. Our findings highlight a TWIST1-HOXA9 embryonic prostate developmental program that is reactivated during prostate cancer metastasis and is therapeutically targetable.
Keywords: EMT, TWIST1, HOXA9, MLL, metastasis
Introduction
Prostate cancer (CaP) is the most commonly diagnosed cancer for men, and leads to the second most cancer-related deaths in the United States (1). CaP natural history disease progression suggests that the largest therapeutic gains could be made by better understanding the progression of localized to metastatic disease (2).
Previous studies have implicated the epithelial-mesenchymal transition (EMT) transcription factor, TWIST1, in human prostate tumor pathogenesis correlating it with increased disease aggressiveness (3,4). TWIST1 can induce pro-metastatic behaviors in CaP cells (5) that is in part mediated by the homeobox protein HOXA9 (6).
Homeobox transcription factors, like HOXA9, are crucial to body plan organization during development, and are tightly regulated both spatially and temporally (7). The expression of genes in this cluster is regulated epigenetically that include critical alterations in chromatin methylation (8). HOX gene products also play a role in progression of cancer with HOXA9 and HOXB13 being the most commonly altered HOX genes in solid tumors (9). While the contribution of HOXA9 overexpression in leukemia, specifically AML, has been firmly established (10), the role of HOXA9 in CaP progression has not been well documented.
In normal tissue, the Hox/HOX cluster of genes is regulated at the chromatin level (11) by the complex of proteins associated with SET1 (COMPASS)-like complex that involves several mixed lineage leukemia (MLL) gene products (homolog of yeast Set1), the scaffolding protein, WDR5, and the long non-coding RNA (lncRNA), HOTTIP (12). COMPASS-like complex activates gene expression by methylation of histone 3 on lysine 4 (H3K4) (13). Although, first discovered in leukemias (14), KMT2 have since been found to be amongst the most frequently mutated genes in human cancer (15). KMT2D/MLL2 mutations distinguish the transition from localized to lethal metastatic castration resistant CaP (16). In addition, COMPASS-like H3K4 methyltransferase (HMT) complexes physically associate with androgen receptor (AR) and have been shown to be required for direct AR target gene expression and CaP growth (17).
In this study, we show that TWIST1 and HOXA9 were co-overexpressed in prostate cancer tumors and metastases. We demonstrated an interaction between TWIST1 and members of the COMPASS-like complex, WDR5 and Hottip/HOTTIP in regulation of Hoxa9/HOXA9 expression. Finally, we showed pharmacologic inhibition of HOXA9 could mitigate TWIST1-induced pro-metastatic behaviors in vitro and metastasis in vivo.
Materials and Methods
Plasmids, antibodies and reagents
pBABE-TWIST1-puro was used to construct the Twist1DQD mutant as described previously (5,18) using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA) and confirmed by sequencing. The following antibodies were used: Twist (Twist2C1a, sc-81417; Santa Cruz Biotechnology), E-cadherin (ab53033; Abcam), vimentin (ab92547; Abcam), ZO-1 (5406; Cell Signaling Technology), β-actin (A5316; Santa Cruz Biotechnology), WDR5 (ab56919; Abcam), H3K4me3 (ab8580; Abcam), H3K27me3 (ab6002; Abcam), and horseradish peroxidase-conjugated secondary antibodies (Invitrogen).
Cell lines and culture conditions
Cell-lines used in this study- PC3, K562, MOLM-13, HEK293 were obtained from American Type Culture Collection (ATCC, Manassas, VA). Myc-CaP cell line was a kind gift from Dr. John Isaacs (Johns Hopkins University). All cell lines were obtained between 2011 and 2014 and authenticated using short tandem repeat (STR) analysis. All the cell lines were expanded and frozen immediately after receipt. The cumulative culture length of the cells was less than 6 months after recovery. Early passage cells were used for all experiments and routinely tested for mycoplasma. Cells were maintained as described by ATCC.
Microarray data acquisition and analysis
Microarrays and bioinformatics analysis were performed previously (5). Details provided in Supplementary methods. The microarray data have been deposited to the Gene Expression Omnibus (GSE500002).
Retroviral experiments
The shRNA constructs against mouse Hoxa9, human HOXA9, mouse Wdr5 and human WDR5 were used according to the manufacturer's instructions (OriGene). Cells were transduced with pGFP-V-RS-based shRNA constructs as described above or with scrambled control vector for two successive times over a 36-hour period followed by selection with 1 mg/mL puromycin and passaged once until 80% confluent.
TALEN genetic knockout
TALEN genetic editing to knock out WDR5 in PC3 cells was accomplished using a TALEN-FastTALE kit (Allele Biotechnology) according to the manufacturer's instructions.
Patient tissue acquisition
Samples for IHC were obtained from patients who died of metastatic CRPC and who signed written informed consent for a rapid autopsy performed within 8 hours of death, under the aegis of the CaP Donor Program at the University of Washington. The institutional review board at the University of Washington approved the study. Bone metastases were formalin fixed, decalcified in 10% formic acid and embedded in paraffin.
Immunohistochemistry, Immunofluorescence and Western blotting
Immunohistochemistry, Immunofluorescence and Western blotting were conducted as described previously (19). Details in supplementary methods.
SYBR-green quantitative RT-PCR and CaP cDNA arrays
The iTaq Universal SybrGreen Master Mix (BioRad) was used according to the manufacturer's instructions and as described previously (6). Human normal prostate and CaP qPCR tissue arrays were purchased from OriGene.
Cell behavior assessment
Wound-healing migration assay: Two-dimensional migration assays were conducted using a scratch/wound model as described previously (6). Matrigel invasion assay: Invasion potential was assessed using Chemicon cell invasion assay kit (Millipore) as directed by the manufacturer. Soft agar colony formation assays were performed as previously described (20). Anoikis resistance was measured as described previously (21). Cell stiffness assay, Magnetic twisting cytometry (MTC) was used to measure mechanical properties of the cytoskeleton as described (22,23). Details in supplementary methods.
Protein co-immunoprecipitation and RNA-immunoprecipitation
As previously described (24). Details in supplementary methods.
Chromatin immunoprecipitation and ChIP-re-ChIP
Chromatin immunoprecipitation (ChIP) was conducted using a SimpleChIP Enzymatic IP Kit (Cell Signaling Technology) according to the manufacturer's instructions. Details in supplementary methods.
Experimental lung metastasis assay
Myc-CaP- cells stably overexpressing TWIST1 were treated with 10nm HXR9 or CXR9 for 24 hours before tail vein injection. 100 μl of PBS containing 5×105 cells were injected into athymic nude mice via the tail vein. After 4 weeks, the mice were sacrificed, necropsies were performed to score surface lung tumors and extra-thoracic metastases as previously described (6).
Statistics
Statistical analysis was carried out using GraphPad Prism v5.04 (GraphPad Software). Paired comparisons of the average were tested using the Mann–Whitney test. Paired comparisons of the frequency of an event were performed by contingency tables with the Fisher's exact test. Throughout this study: *, p< 0.05; **, p< 0.01; and ***, p<0.001.
Results
Bioinformatic analyses identified HOXA9 as putative downstream target responsible for TWIST1-induced pro-metastatic behaviors
We reported on TWIST1 structure-function studies and the effects of overexpression of mouse Twist1 mutants on the induction of TWIST1-dependent aggressive cellular pro-metastatic phenotypes in vitro and in vivo (5,6) (Figure 1A). Global gene expression analyses were performed on Myc-CaP cells expressing wild-type TWIST1, TWIST1-F191G, which does not induce a metastatic phenotype, and TWIST1-DQD, which induces a more pronounced metastatic phenotype (Figure 1B). Subtractive gene expression analysis identified genes whose expression was significantly altered in cells expressing wild-type TWIST1 but not in cells expressing TWIST1-F191G, and genes whose expression was significantly altered in cells expressing TWIST1-DQD but not in cells expressing wild-type TWIST1 (Figure 1C). Resultant gene sets were associated with increasing intensity of TWIST1-induced EMT phenotypes (Figure 1D).
Figure 1. Gene expression profiling of TWIST1 structure-function mutants revealed HOXA9 as a downstream target.

(A) Schematic of TWIST1 structure, T125D and S127D site-specific mutant, TWIST1-DQD, TWIST1-F191G. (B) Unsupervised hierarchal clustering heatmap visualization of gene expression differentially regulated by vector, TWIST1, TWIST1-DQD, and TWIST1-F191G. (C) Venn diagram represents significantly differentially expressed genes between Myc-CaP-TWIST1 and TWIST1 mutants. (D) GSEA analysis revealed that HOXA9 gene signatures were correlated with an increased EMT and metastatic phenotype.
Gene set enrichment analysis (GSEA) was performed on both sets of genes, and in both cases, gene sets representing target genes of constitutively active HOXA9 fusion proteins (25,26) were significantly overrepresented (Supplemental Table 3). This led us to hypothesize that HOXA9 activation was downstream of TWIST1 and was responsible for enforcing TWIST1-induced pro-metastatic phenotypes (Figure 1D).
TWIST1 and HOXA9 are co-expressed in the developing embryonic mouse prostate and reactivated in mouse prostate tumors
Limited mRNA expression data suggest that the posterior Hox cluster gene Hoxa9 is expressed transiently in the prostate following rodent birth and then expression is suppressed in adulthood (27). The role of TWIST1 and HOXA9 during prostate development is unknown. We observed by qRT-PCR of bulk fetal prostate tissue that Twist1 and Hoxa9 were both co-expressed with expression peaking during periods of early prostate bud invasion (E17.5 and E18.5, respectively), and then nadiring after birth at day P5 (Figure 2A). The adult prostate is derived from both the embryonic urogenital mesenchyme (UGM) and urogenital epithelium (UGE). To define the cell types expressing and sub-cellular localization of TWIST1 and HOXA9, we examined developmental protein expression in the mouse developing prostate using immunohistochemistry (IHC). Concordant with the temporal mRNA expression, TWIST1 and HOXA9 showed predominantly nuclear peak individual expression and co-expression during E17.5 in cells from the developing UGM, with highest co-expression among cells along the UGM-UGE interface (Figure 2B). In agreement with the mRNA expression data we observed a steady decrease in TWIST1 expression at P0 and P5 with no observable TWIST1 expression at time-points thereafter (Supplemental Figure 1). Overall, this observation suggests that HOXA9 may be a target of TWIST1 during mammalian prostate development.
Figure 2. TWIST1 and HOXA9 are co-expressed in the developing mouse prostate and in autochthonous prostate neoplastic lesions.

(A) Twist1 and Hoxa9 gene expression by qPCR in E15.5-E18.5 UGS and P0, P5 developing prostate (n=3-6 embryos-pups/time point/gene, Pearson correlation coefficient, R2= 0.82). Normalized to expression level at P35. (B) TWIST1 and HOXA9 IHC on serial sections of E17.5 UGS from wild-type mice in cells at the urogenital mesenchyme (UGM)-urogenital epithelium (UGE) interface. UGMi; section of the UGM closest to the UGE interface, UGMe; section of the UGM toward the edge. Graphs represent percentage positive cells (n=4 embryos). (C) TWIST1 and HOXA9 IHC on adjacent serial sections of prostate tissue from probasin (PB)-Cre; Apcflox/flox mice with prostate neoplasia (n=2). Arrows highlight TWIST and HOXA9 co-expression in the same cell-types.
TWIST1 expression is often aberrantly reactivated in human cancers including CaP (3,6,28), but data on expression of HOXA9 in CaP is lacking. We did not observe appreciable IHC staining for either TWIST1 or HOXA9 in prostate epithelial cells in tumors from the TRAMP (29), Hi-Myc (30) or Pten-/- (31) transgenic mouse models of CaP or the adult wild-type prostate (Supplemental Figure 2 and 5B). However, in the PB-Cre+;Apcflox/flox mouse model of CaP (32), we observed co-overexpression of TWIST1 and HOXA9 in neoplastic prostate epithelial cells (Figure 2C). Thus, we found that co-expression of TWIST1 and HOXA9 could be reactivated in at least one model of prostate epithelial tumorigenesis.
TWIST1 and HOXA9 are co-overexpressed in human CaP and correlated with poor patient survival
Analysis of publically available CaP patient gene expression data through Oncomine (www.oncomine.org) showed that HOXA9 trended towards overexpression overall in several different studies (Figure 3A; n=700 CaPs versus n=313 normal prostate, p=0.095) similar to TWIST1 (6). TWIST1 and HOXA9 were shown to be trending towards co-overexpression in primary CaP, 13% and 21%, respectively (n=131, p=0.12). Interestingly, co-amplification/overexpression cases were significantly enriched when examining metastases (42% alteration for TWIST1 and 32% for HOXA9, n=19, p=0.024 Fisher's exact test) [MKSCC Prostate Adenocarcinoma dataset (33)] using the cBio portal (34,35) (Figure 3B). We validated that HOXA9 was overexpressed in primary CaP (Figure 3C, n=91, p<0.001) and that TWIST1 and HOXA9 were both found to be overexpressed in primary CaP compared to normal prostate in another independent patient dataset (Origene TissueScan qPCR array) (Figure 3, C and D). Additionally, samples with high TWIST1 expression were disproportionately likely to have high HOXA9 expression (Figure 3 E). In a third independent patient dataset we found that TWIST1 and HOXA9 were co-amplified in metastatic CaP and HOXA9 amplification alone or when considered with TWIST1 amplification was correlated with worse overall patient survival (Michigan Prostate Adenocarcinoma dataset, Figure 3F and Supplemental Figure 3.; n=61, both p<0.024). Lastly, using CaP samples from a prospectively collected rapid autopsy series, we showed by IHC that TWIST1 and HOXA9 were co-localized in the nucleus of tumor cells in CaP primary tumors (n=3) and bone metastases (n=6 patients, Figure 3 G and H and Supplemental Figure 4 A and B). Collectively, multiple independent human CaP cohorts showed that TWIST1 and HOXA9 are dysregulated together at the genetic level and also co-overexpressed at the protein level in a subset of CaP, particularly metastatic samples that are correlated with poor patient survival.
Figure 3. TWIST1 and HOXA9 are co-overexpressed in metastatic CaP and correlate with poor survival.

(A) HOXA9 trended towards overexpression in several sets of prostate adenocarcinoma gene expression data analyzed using Oncomine (n=15 independent microarray datasets equaling 700 primary CaP samples and 313 normal prostates, p=0.095). (B) TWIST1 and HOXA9 trended towards co-amplification/overexpression in a subset of primary tumors from patients with prostate adenocarcinoma using the cBio portal (MKSCC Prostate Adenocarcinoma dataset, n=131 patient samples, p=0.12). (C, D) HOXA9 and TWIST expression in a commercially available cDNA array of CaP tissue samples compared to normal prostate tissue (n=91 CaP patient samples, Mann-Whitney t-test: **, p< 0.01; and ***, p<0.001). (E) TWIST1 and HOXA9 co-overexpression is enriched in prostate adenocarcinoma metastases (Fisher's exact test, n=19, p=0.024). (F) Amplification of HOXA9 is associated with shorter survival in patients with prostate adenocarcinoma, analyzed from cBio (Michigan Prostate Adenocarcinoma dataset, n=61 patients, p<0.024, Log rank test). (G-H) Representative example of TWIST1, HOXA9 and IgG/pan-cytokeratin on adjacent serial sections of a primary and bone metastasis from prostate adenocarcinoma patients. Arrows highlight TWIST and HOXA9 co-expression in the nuclei of tumor cells.
We have previously shown that HOXA9 was partially required for TWIST1-induced pro-metastatic behaviors in vitro (6). We found that stable overexpression of human HOXA9 alone in Myc-CaP and PC3 cells (Supplemental Figure 6, A and B) enforced a partial EMT, increased migratory potential (Supplemental Figure 6, C and D, all comparisons p<0.01), invasiveness (Supplemental Figure 6, E and F, p<0.05), anoikis resistance (Supplemental Figure 6, G and H, p<0.05) and radiation resistance (Supplemental Figure 6, K and L, p<0.05). These data suggest that HOXA9 alone is sufficient to induce many pro-metastatic behaviors in vitro and is partially required for TWIST1-induced pro-metastatic behaviors (6).
TWIST1 interacts with members of the MLL/COMPASS-like complex
Hoxa9 expression is activated during development by MLL/COMPASS-like-dependent methylation of promoter histones. It was recently shown that TWIST1 can lead to changes in the epigenetic landscape involving DNA as well as histone modifications (36). One possibility is that TWIST1 may modulate expression of Hoxa9 epigenetically through interaction with a COMPASS-like complex. In the MSKCC Prostate Adenocarcinoma data set (34,35) TWIST1 was co-overexpressed with a gene encoding a member of the COMPASS-like HMT complex, KMT2D (MLL2/ALR/MLL4) in CaP metastases (Figure 4A, n=19, p=0.049 by Fisher's exact test). TWIST1 has been shown to bind WDR5 under conditions of hypoxia-induced EMT (37). To test whether TWIST1 bound WDR5 in CaP cells and under normoxic conditions, we performed co-immunoprecipitation experiments on extracts from HEK293 embryonic kidney cells, Myc-CaP and PC3 CaP cells that stably overexpressed TWIST1 and also in K562 leukemia cells that have intrinsically high TWIST1 expression. Anti-TWIST1 antibody pulled down WDR5 in all cell lines, but PC3 (data not shown) and we also observed the reciprocal interaction when cell lysates were co-immunoprecipitated with anti-WDR5 antibody and probed for TWIST1 (Figure 4B). These results indicate TWIST1 and WDR5 interact with each other in cancer cells under normoxic conditions and this interaction may be in the context of the larger COMPASS-like HMT complex.
Figure 4. TWIST1 interacts with the components of the MLL/COMPASS-like complex WDR5 and Hottip/HOTTIP.

(A) Gene encoding MLL2, KMT2D, showed significant co-overexpression with TWIST1 in the MKSCC Prostate Adenocarcinoma data set in CaP metastases (Fisher's exact test, n=19, p=0.049). (B) TWIST1 and WDR5 pulldown by immunoprecipitation in HEK-293, Myc-CaP-TWIST1 and in K-562 cells compared to IgG control. (C-D) Expression of Hottip/HOTTIP in Myc-CaP-HOXA9 and PC3-HOXA9 cells compared to vector control (n=3-4, Mann-Whitney t-test: **, p< 0.01, +/-SEM). (E) Co-immunoprecipitation of HOTTIP with TWIST1 in K562, but not in (F) MOLM-13 cells (n≥2, Mann-Whitney t-test: **, p< 0.01, +/-SEM). (G) Co-immunoprecipitation of hottip/HOTTIP with TWIST1 in Myc-CaP-TWIST1 cells and (H) PC3-TWIST1 cells compared to isogenic vector control cells (n≥3, Mann-Whitney t-test: *, p< 0.05, +/-SEM).
We investigated how TWIST1 influences another component of the MLL/COMPASS-like complex that regulates the Hox cluster during development, Hottip, a lncRNA that directly binds to the complex and directs it to sites in the Hox cluster (24). We found that HOXA9 overexpression in Myc-CaP and PC3 cells led to an increase in Hottip/HOTTIP expression (Figure 4, C and D). In order to determine whether the TWIST1-WDR5 complex also contained Hottip/HOTTIP, we performed RNA immunoprecipitation (RIP) experiments using anti-TWIST1 antibody. In K562 leukemia cells which have high endogenous TWIST1 expression, HOTTIP co-precipitated with TWIST1 (Figure 4E, p<0.01), but there was no detectable enrichment in MOLM-13 cells (Figure 4F, p>0.05), which do not express TWIST1. When we performed RIP experiments using anti-TWIST1 antibody in lysates of Myc-CaP and PC3 cells overexpressing TWIST1, we found significant enrichment of Hottip/HOTTIP (Figure 4, G and H, p<0.05) as compared to vector controls. Overall, our results show that TWIST1 can interact with multiple members of the COMPASS-like HMT complex including the invariant component, WDR5, and the lncRNA, Hottip/HOTTIP. This physical interaction is highly suggestive that TWIST1-dependent upregulation of Hoxa9 expression may be mediated by increasing the amount of activating H3K4 histone methylation at the Hoxa9/HOXA9 promoter region.
Members of the MLL/COMPASS-like complex are required for TWIST1 mediated upregulation of Hoxa9 and induction of a metastatic phenotype
The active enzyme in the complex can be any one of several members of the MLL family (38), so genetic inhibition of any given member of the family may not have observable effects secondary to this redundancy. Therefore, we performed genetic knockdown of the invariant protein component WDR5 and the lncRNA Hottip/HOTTIP.
We showed that shRNA-mediated knockdown of Wdr5 (Supplemental Figure 7A) in Myc-CaP-TWIST1 cells led to a decrease in Hoxa9 expression (Figure 5A). Moreover, decreased Wdr5 led to decreased migration potential (Figure 5B; and Supplemental Figure 7B), invasiveness (Figure 5C), resistance to radiation (Supplemental Figure 5D) and anoikis resistance (Supplemental Figure 7E) as compared to scrambled shRNA control cells. We used single-cell biophysical technique to measure cytoskeletal stiffness, magnetic twisting cytometry (MTC) and observed that shRNA knockdown of Wdr5 decreased TWIST1-induced cellular stiffness (Supplemental Figure 7F). We did not observe increased anchorage independent growth as assayed by soft-agar clonogenicity (Supplemental Figure 7C) upon shRNA knockdown of Wdr5 in Myc-CaP-TWIST1 cells. Transcription activator-like effector nuclease (TALEN)-mediated knockout of WDR5 in PC3-TWIST1 cells (Supplemental Figure 7G) led to a decreased HOXA9 expression (Figure 5D). Additionally, loss of WDR5 resulted in decreased migration potential (Figure 5E and Supplemental Figure 6H), invasiveness (Figure 5F) and resistance to radiation (Supplemental Figure 6J) but not in soft-agar clonogenicity (Supplemental Figure 7I) or anoikis resistance (Supplemental Figure 7K).
Figure 5. WDR5 and Hottip/HOTTIP, are required for TWIST1-mediated expression of HOXA9 and induction of pro-metastatic cellular phenotypes.

(A) Wdr5 shRNA knockdown in Myc-CaP-TWIST1 cells reduced Hoxa9 expression as shown by qPCR. (B) Myc-CaP-TWIST1 cells with stable knockdown of Wdr5 showed decreased migration and (C) invasion through Matrigel compared to scrambled shRNA control. (D) WDR5 knockout by TALEN in PC3-TWIST1 cells decreased HOXA9 expression. (E) PC3-TWIST1 WDR5 knockout cells had decreased cell migration and (F) invasion through Matrigel cells compared to scrambled shRNA control. (G) Knockdown of Hottip/HOTTIP by siRNA in Myc-CaP-TWIST1 or (J) PC3-TWIST1 cells, respectively, led to decreased Hoxa9/HOXA9 mRNA. (H) Hottip or (K) HOTTIP knockdown led to decreased cell migration and (I) and (L) invasion through Matrigel in Myc-CaP-TWIST1 and PC3-TWIST1 cells, respectively, compared to scrambled control. All were n≥3, Mann-Whitney t-test: *, p< 0.05; **, p< 0.01; ***, p< 0.001, +/-SEM.
In parallel with our results from WDR5 knockdown/knockout, siRNA-mediated knockdown of Hottip (Supplemental Figure 8A) and HOTTIP (Supplemental Figure 8E) in TWIST1-overexpressing Myc-CaP and PC3 cells respectively, resulted in decreased expression of Hoxa9/HOXA9 (Figure 5, G and J). Furthermore, knockdown of Hottip/HOTTIP in Myc-CaP and PC3 cells overexpressing TWIST1, also led to a decrease in migration potential (Figure 5, H and K; and Supplemental Figure 8, B and F) and invasiveness (Figure 5, I and L) but not to an increase in MTC-determined cell stiffness (Supplemental Figure 8, C and G) or anoikis resistance (Supplemental Figure 8, D and H). All together, these results showed that members of the MLL/COMPASS-like complex, WDR5 and Hottip/HOTTIP, are partially required for HOXA9 expression downstream of TWIST1 and the consequent induction of several pro-metastatic cellular behaviors.
Expression of TWIST1 alters chromatin methylation at the Hoxa9/HOXA9 promoter region
TWIST1 and WDR5 bound directly to the Hoxa9/HOXA9 promoter as shown by chromatin immunoprecipitation (ChIP) using anti-TWIST1 and anti-WDR5 antibodies coupled with qPCR (ChIP-qPCR) in Myc-CaP and PC3 cells overexpressing TWIST1. Primers flanked E-box sequences, putative TWIST1 binding sites, in the mouse Hoxa9 (Figure 6A) as well as the human HOXA9 promoter region (Figure 6B). Enrichment of DNA fragments bound by TWIST1 in Myc-CaP and PC3 cells overexpressing TWIST1 are mapped to the Hoxa9/HOXA9 promoter region as shown by Figure 6, A and B; and Supplemental Figure 9B. WDR5 ChIP showed that WDR5 bound to the same regions of the Hoxa9/HOXA9 promoter in Myc-CaP and PC3 cells stably overexpressing TWIST1 (Supplemental Figure 9, A and C).
Figure 6. TWIST1 interacts with WDR5 to increase H3K4me3 chromatin configuration of the Hoxa9/HOXA9 promoter region.
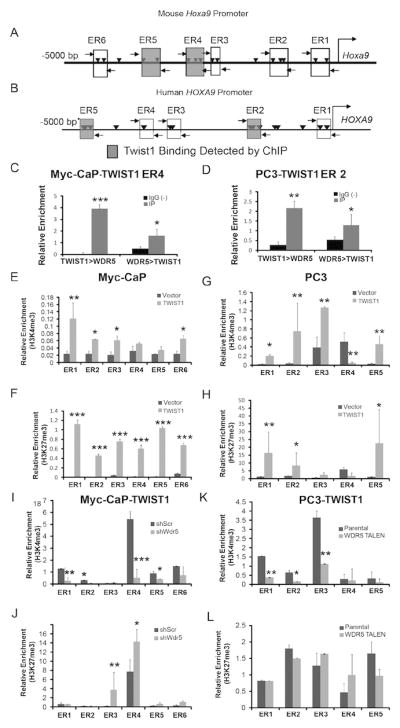
Schematic of the (A) mouse Hoxa9 and (B) human HOXA9 promoter. Rectangles represent E-box regions (ER) and shaded rectangles represent enrichment for TWIST1 binding. Arrowheads represent individual E-box motifs. Horizontal arrows represent ChIP primers. (C-D) TWIST1 and WDR5 bind together at the Hoxa9 promoter at ER4 by ChIP-re-ChIP in Myc-CaP-TWIST1 cells. (D) TWIST1 and WDR5 also bind together at the human HOXA9 promoter at ER2 by ChIP-re-ChIP in PC3-TWIST1 cells. ChIP with anti-H3K27me3 antibody showed increased H3K27me3 at several E-box regions in the Hoxa9/HOXA9 promoter region in (E) Myc-CaP-TWIST1 and (G) PC3-TWIST1 cells compared to vector control cells. Similarly, ChIP with an anti-H3K4me3 antibody showed increased H3K4me3 at E-box regions in the Hoxa9/HOXA9 promoter region of (F) Myc-CaP-TWIST1 and (H) PC3-TWIST1 cells compared to vector controls. (I) Wdr5 shRNA knockdown in Myc-CaP-TWIST1 cells decreased relative H3K4me3 enrichment observed with ChIP at the majority of E-box regions, but (J) there was still some increased H3K27me3 observed in ER3 and ER4 of the Hoxa9 promoter. (K) WDR5 knockout by TALEN in PC3-TWIST1 cells decreased H3K4me3 of the human HOXA9 promoter with no change on (L) H3K27me3. All ChIP results are normalized to an IgG-matched negative control and enrichment relative to that baseline reported on the y-axis. All were n≥3, Mann-Whitney t-test: *, p< 0.05; **, p< 0.01; ***, p< 0.001, +/-SEM.
TWIST1 and WDR5 bound together in a complex at the Hoxa9/HOXA9 promoter as shown by primary ChIP followed by secondary immunoprecipitation (re-ChIP) on TWIST1 overexpressing Myc-CaP and PC3 cells. In the ChIP re-ChIP technique, the soluble chromatin fractions derived from cross-linking are divided into two aliquots. The first aliquot was immunoprecipitated with anti-TWIST1 antibody, washed and the bound antibody-DNA complex was immunoprecipitated with anti-WDR5 antibody (TWIST1> WDR5). The second aliquot was treated identically, except that it was first precipitated with anti-WDR5 antibody followed by re-ChIP with anti-TWIST1 antibody (WDR5>TWIST1). The precipitated DNA was then amplified using primers flanking E-boxes in the mouse Hoxa9 and human HOXA9 promoter regions, respectively, as described above. The ChIP re-ChIP experiments showed enrichment of sequences in the Hoxa9/HOXA9 promoter bound by endogenous WDR5 with TWIST1 immunoprecipitates and of TWIST1 bound to WDR5 precipitates in both Myc-CaP and PC3 cells (Figure 6, C and D). This binding was specific as re-ChIP experiments using a non-specific antibody control did not immunoprecipitate TWIST1 and WDR5 at the same sites on the Hoxa9/HOXA9 promoter in CaP cells overexpressing TWIST1.
We next investigated the mechanism by which the TWIST1-WDR5 complex activated Hoxa9/HOXA9 expression. We found that the epigenetic activation marker, H3K4me3, was enriched in the E-box regions of the Hoxa9/HOXA9 promoter in Myc-CaP and PC3 cells overexpressing TWIST1 by H3K4me3 ChIP (Figure 6, E and G). Interestingly, we also saw enrichment of the repressive histone modification, trimethylation of lysine 27 of histone 3 (H3K27me3), in the Hoxa9/HOXA9 promoter region of TWIST1 stably overexpressing Myc-CaP and PC3 cells (Figure 6, F and H). When we used shRNA-mediated knockdown of Wdr5 or TALEN-mediated knockout of WDR5, we observed abrogated enrichment of the H3K4me3 activation marker in TWIST1 overexpressing Myc-CaP and PC3 cells at the Hoxa9/HOXA9 promoter region (Figure 6, I and K). We did not, however, observe abrogated enrichment of the repressive H3K27me3 marker in the Hoxa9/HOXA9 promoter region in these same cells with reduced or absent WDR5 (Figure 6, J and L). These results showed that TWIST1 binding to the Hoxa9/HOXA9 promoter led to increased H3K4 and H3K27 trimethylation and that WDR5 was required for TWIST1-induced H3K4me3 (activation) but not H3K27me3 (repression) modification of the Hoxa9/HOXA9 promoter region. In summary, our data indicated that TWIST1 and WDR5 bind as a complex to the E-box consensus sequences of the Hoxa9/HOXA9 promoter and promoted HOXA9 expression that was associated with enrichment of bivalent H3 chromatin markers.
Chemical inhibition of HOXA9 activity mitigates the pro-metastatic effects of TWIST1 expression
In order to determine whether this TWIST1-HOXA9 axis could be a potential clinical target, we investigated whether chemical inhibition of HOXA9 or its downstream effectors, would decrease the intensity of these pro-metastatic cellular phenotypes. The effects of two chemical inhibitors of HOXA9 activity were tested. UNC0646 is a small-molecule inhibitor of the methyltransferase G9a that has been shown to inhibit HOXA9 activity in leukemia cells (39) and HXR9 is a peptide inhibitor of HOXA9 that interferes with the interaction of HOXA9 with its cofactor PBX and has been shown to retard the growth of human meningioma (40).
IC50 values for each inhibitor were determined for several cell-lines including those stably overexpressing TWIST1 (Supplemental Figure 10), but in general we did not see an effect of either agent on cell viability. Treatment of Myc-CaP-TWIST1 and PC3-TWIST1 cells with 250 uM UNC0646 led to decreased migration (Supplemental Figure 11, A and C) and invasion (Supplemental Figure 11, B and D) compared to vehicle control. Similarly, treatment of Myc-CaP and PC3 cells stably overexpressing TWIST1 with 10 nM HXR9 led to decreased migration (Figure 7, A and B) and invasion (Figure 7, C and D) compared to a scrambled peptide control CXR9. Importantly, these inhibitory effects of UNC0646 and HXR9 were only observed in Myc-CaP and PC3 cells overexpressing TWIST1 and not in isogenic vector control cell lines. These contrasting effects on vector control versus TWIST1 overexpressing cells suggested that HOXA9 inhibition is very specific for TWIST1 overexpression.
Figure 7. Pharmacologic inhibition of HOXA9 activity mitigates the pro-metastatic effects of TWIST1 in vitro and in vivo.

(A) Treatment with 10 nM HXR9, a peptide inhibitor of HOXA9, led to decreased cell migration and (C) invasion compared to the control peptide, CXR9, in Myc-CaP-TWIST1 cells, but not in vector control cells. (B) Treatment with 10 nM HXR9 led to a decrease in cell migratory potential and (D) invasion through Matrigel compared to CXR9 treatment only in PC3-TWIST1 cells and not in PC3 vector control cells. (A)-(D) n≥3, Mann-Whitney t-test: *, p< 0.05; **, p< 0.01, +/-SEM. (E) A graph comparing number of athymic nude male mice with macroscopic lung tumors after tail vein injection of Myc-CaP-TWIST1 cells pre-treated with HXR9 or CXR9 (n=19-20 mice/arm, *p=0.018 by Fisher's one-sided exact test). (F) Graph comparing number of athymic nude male mice with presence of at least one extra-thoracic (ET) metastasis after tail vein injection of Myc-CaP-TWIST1 cells treated with HXR9 or CXR9 (n=19-20 mice/arm, p=0.039 by Fisher's one-sided exact test). (G) Model of a proposed TWIST1-WDR5-HOTTIP-KMT2D complex.
To investigate the functional consequences of HOXA9 inhibition in TWIST1 overexpressing cells in vivo, we injected Myc-CaP cells incubated with HXR9 or the control peptide, CXR9 into the tail veins of athymic nude mice. We have previously shown that TWIST1 overexpression significantly increased the ability of tail vein injected Myc-CaP cells to colonize the lungs and form macroscopic metastases as well as extra-thoracic metastases in distant subcutaneous tissues, abdominal organs and distant lymph nodes (6). Thus, TWIST1 allows cells injected into the venous circulation too not only colonize the lungs but also undergo the full metastatic pathway to produce extra-thoracic metastases. We found that TWIST1 overexpressing Myc-CaP cells lost the potential to form macroscopic lung tumors in vivo when treated with HXR9 (7/19 mice) as compared to control peptide, CXR9, treated cells (15/20 mice) (Figure 7E, p=0.018 by Fisher's exact one-sided test). This trend was also observed with the extra-thoracic metastases when Myc-CaP-TWIST1 cells were treated with HXR9 (2/19 mice) as compared to control CXR9 (8/20 mice) treated cells (Figure 7F, p=0.04 by Fisher's exact one-sided test). These results showed that HOXA9 promoted TWIST1-induced metastasis of CaP cells in vivo and that pharmacologic HOXA9 inhibition could be a strategy to target TWIST1-induced CaP metastasis.
Discussion
Prostate cancer co-opts embryonic developmental programs especially during neoplastic transformation and metastasis as indicated by a significant overlap in differentially expressed genes during prostate development and CaP progression (41). In this study, we show that HOXA9 expression correlates with TWIST1 expression during mouse prostate development. This expression was silenced post-natally in mice, however, co-expression of TWIST1 and HOXA9 in mouse and human CaP suggests that this developmental mechanism is reactivated during CaP progression. Extending our previous study which showed that TWIST1 can upregulate HOXA9 expression and that HOXA9 was partially required for TWIST1-mediated EMT phenotypes (6), here we demonstrate that HOXA9 is sufficient for the induction of pro-metastatic phenotypes alone in CaP cells. Importantly, we show that chemical inhibition of HOXA9 mitigates TWIST1-induction of these pro-metastatic cellular phenotypes in vitro and metastasis in vivo implicating HOXA9 as a potential therapeutic target in aggressive and metastatic CaPs.
We identify a novel role for TWIST1 in epigenetic regulation of the Hoxa9/HOXA9 locus via interaction with at least two members of the COMPASS-like complex, WDR5 and Hottip/HOTTIP (Figure 7G). In corroboration with a previous study from another group (37), we show that TWIST1 binds to WDR5, an invariant component of the COMPASS-like complexes. Importantly, we demonstrated the novel finding that TWIST1 also binds to the lncRNA Hottip/HOTTIP that recruits and directs the COMPASS-like complex to the Hox/HOX cluster (24). Furthermore, both WDR5 and Hottip/HOTTIP are required to observe the full potential of TWIST1-induced HOXA9 expression and acquisition of pro-metastatic properties in CaP cells. Overexpression of HOTTIP and subsequent HOX gene expression has been associated with poor prognosis and increased aggressiveness in other cancers (42–44). The common overexpression of the WDR5-HOTTIP complex across multiple types of cancer suggests that it is a key pathway in cancer progression and targeting this pathway successfully may have potential to benefit many cancer patients. Our new findings also extend the catalogue of protein targets for aggressive TWIST1 and MLL/COMPASS-driven CaP to include HOXA9. TWIST1 and HOXA9 were important for metastatic phenotypes in both AR-dependent and AR-independent models suggesting that this TWIST1-COMPASS-like-HOXA9 axis can function independently of the AR axis.
TWIST1 as a transcription factor, directly activates and represses the transcription of target genes, but there is evidence to suggest that TWIST1 also has broad epigenetic effects. TWIST1 can interact with SET8 leading to H4K20 monomethylation and target gene expression (45). TWIST1 led to a two-fold genome-wide increase in the number of TWIST1 target genes with bivalent chromatin configurations (36) rendering gene promoters poised for activation (46) and facilitating increased cellular plasticity that is characteristic of EMT (36). Herein, we have uncovered a mechanistic role for TWIST1 in directly targeting the COMPASS-like HMT complex to the Hoxa9/HOXA9 promoter resulting in alteration of chromatin methylation patterns in CaP cells. Overexpression of TWIST1 led to an increase in both H3K4 and H3K27 trimethlyation at the Hoxa9/HOXA9 promoter region, consistent with a bivalent chromatin configuration. Intriguingly, knockdown of WDR5 in the presence of TWIST1 abrogates the increase in H3K4 trimethylation, but not H3K27 trimethylation. These observations are consistent with TWIST1 cooperating with WDR5 and the whole COMPASS-like complex to increase H3K4 trimethylation at the Hoxa9/HOXA9 promoter region and thereby upregulate Hoxa9/HOXA9 expression. However, the presence of concurrent increased H3K27 trimethylation indicates the possible presence of additional chromatin regulatory mechanisms interacting with TWIST1, such as the Polycomb repressive complexes.
In conclusion, this study demonstrates a novel mechanistic role of TWIST1 in promoting CaP aggressiveness not only by direct transcriptional activation of Hoxa9/HOXA9 but also by epigenetic reprogramming of the Hoxa9/HOXA9 locus. In addition to its documented functions as a direct transcriptional activator and repressor, TWIST1 cooperates with the COMPASS-like HMT complex to directly increase H3K4me3 in the promoter region of Hoxa9/HOXA9. TWIST1 and HOXA9 appear to direct an embryonic developmental program for prostate organogenesis that is re-activated during CaP metastasis. Importantly, therapeutic targeting of HOXA9 is sufficient to abrogate TWIST1-induced CaP metastasis. Our findings are consistent with the concept that targeting epithelial plasticity programs in advanced CaP is an area that should be studied more pre-clinically (47) with an eye towards future clinical translation.
Supplementary Material
Acknowledgments
We thank the patients and their families who were willing to participate in the University of Washington CaP Donor Program and investigators Drs. Robert Vessella, Celestia Higano, Bruce Montgomery, Evan Yu, Peter Nelson, Heather Cheng, Paul Lange, and Martine Roudier.
Financial Support: This research was supported by the following funding: R. Malek is a CaP Foundation (PCF) Young Investigator. R.D. Williams was a Johns Hopkins Laboratory Radiation Oncology Training Fellow (NIH-T32CA121937). K. Taparra was funded by the NIH (F31CA189588). H.L. Wang is funded by a Uniting Against Lung Cancer Young Investigator Award. S An was funded by the NIH (P50CA103175, U54CA141868 and HL107361). P.T. Tran is funded by the Motta and Nesbitt Families, the DoD (W81XWH-11-1-0272), a Kimmel Translational Science Award (SKF-13-021), an ACS Scholar award (122688-RSG-12-196-01-TBG), American Lung Association (LCD-339465), Movember-PCF and the NIH (R01CA166348 & U01CA183031). L. True and C. Morrissey were funded by the Pacific Northwest CaP SPORE (P50CA97186) and the Richard M. Lucas Foundation.
Footnotes
Conflict of Interest: The authors have declared that no conflict of interest exists
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016 Feb;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA J Am Med Assoc. 1999 May 5;281(17):1591–7. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 3.Kwok WK, Ling MT, Lee TW, Lau TCM, Zhou C, Zhang X, et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005 Jun 15;65(12):5153–62. doi: 10.1158/0008-5472.CAN-04-3785. [DOI] [PubMed] [Google Scholar]
- 4.Shiota M, Yokomizo A, Tada Y, Inokuchi J, Kashiwagi E, Masubuchi D, et al. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene. 2010 Jan 14;29(2):237–50. doi: 10.1038/onc.2009.322. [DOI] [PubMed] [Google Scholar]
- 5.Gajula RP, Chettiar ST, Williams RD, Nugent K, Kato Y, Wang H, et al. Structure-function studies of the bHLH phosphorylation domain of TWIST1 in prostate cancer cells. Neoplasia N Y N. 2015 Jan;17(1):16–31. doi: 10.1016/j.neo.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gajula RP, Chettiar ST, Williams RD, Thiyagarajan S, Kato Y, Aziz K, et al. The Twist Box Domain is Required for Twist1-induced Prostate Cancer Metastasis. Mol Cancer Res MCR. 2013 Aug;27:1541–7786. doi: 10.1158/1541-7786.MCR-13-0218-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krumlauf R. Hox genes in vertebrate development. Cell. 1994 Jul 29;78(2):191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 8.Noordermeer D, Leleu M, Splinter E, Rougemont J, De Laat W, Duboule D. The dynamic architecture of Hox gene clusters. Science. 2011 Oct 14;334(6053):222–5. doi: 10.1126/science.1207194. [DOI] [PubMed] [Google Scholar]
- 9.Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med Berl Ger. 2014 Aug;92(8):811–23. doi: 10.1007/s00109-014-1181-y. [DOI] [PubMed] [Google Scholar]
- 10.Collins CT, Hess JL. Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene. 2016 Mar 3;35(9):1090–8. doi: 10.1038/onc.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soshnikova N, Duboule D. Epigenetic temporal control of mouse Hox genes in vivo. Science. 2009 Jun 5;324(5932):1320–3. doi: 10.1126/science.1171468. [DOI] [PubMed] [Google Scholar]
- 12.Li A, Yang Y, Gao C, Lu J, Jeong HW, Liu BH, et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J Clin Invest. 2013 Sep 24; doi: 10.1172/JCI62891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shilatifard A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annu Rev Biochem. 2012;81(1):65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Döhner K, Tobis K, Ulrich R, Fröhling S, Benner A, Schlenk RF, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol Off J Am Soc Clin Oncol. 2002 Aug 1;20(15):3254–61. doi: 10.1200/JCO.2002.09.088. [DOI] [PubMed] [Google Scholar]
- 15.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013 Oct 17;502(7471):333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012 Jul 12;487(7406):239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malik R, Khan AP, Asangani IA, Cieślik M, Prensner JR, Wang X, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015 Apr;21(4):344–52. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran PT, Shroff EH, Burns TF, Thiyagarajan S, Das ST, Zabuawala T, et al. Twist1 Suppresses Senescence Programs and Thereby Accelerates and Maintains Mutant Kras-Induced Lung Tumorigenesis. In: Grimes HL, editor. PLoS Genet. 5. Vol. 8. 2012. May 24, p. e1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J, et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PloS One. 2008 Jan;3(5):e2125. doi: 10.1371/journal.pone.0002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng J, See AP, Aziz K, Thiyagarajan S, Salih T, Gajula RP, et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol Ther. 2011 Oct 1;12(7):657–63. doi: 10.4161/cbt.12.7.17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene. 2002 Apr 4;21(15):2365–75. doi: 10.1038/sj.onc.1205300. [DOI] [PubMed] [Google Scholar]
- 22.An SS, Fabry B, Trepat X, Wang N, Fredberg JJ. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am J Respir Cell Mol Biol. 2006 Jul;35(1):55–64. doi: 10.1165/rcmb.2005-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Phys Rev Lett. 2001 Oct 1;87(14):148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- 24.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011 Apr 7;472(7341):120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeisig BB, Milne T, García-Cuéllar MP, Schreiner S, Martin ME, Fuchs U, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004 Jan;24(2):617–28. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda A, Goolsby C, Yaseen NR. NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 2006 Jul 1;66(13):6628–37. doi: 10.1158/0008-5472.CAN-06-0458. [DOI] [PubMed] [Google Scholar]
- 27.Huang L, Pu Y, Hepps D, Danielpour D, Prins GS. Posterior Hox gene expression and differential androgen regulation in the developing and adult rat prostate lobes. Endocrinology. 2007 Mar;148(3):1235–45. doi: 10.1210/en.2006-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiota M, Yokomizo A, Tada Y, Inokuchi J, Kashiwagi E, Masubuchi D, et al. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene. 2010 Jan 14;29(2):237–50. doi: 10.1038/onc.2009.322. [DOI] [PubMed] [Google Scholar]
- 29.Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol Ed John E Coligan Al. 2001 Nov; doi: 10.1002/0471142735.im2005s45. Chapter 20:Unit 20.5. [DOI] [PubMed] [Google Scholar]
- 30.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003 Sep;4(3):223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003 Sep;4(3):209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 32.Bruxvoort KJ, Charbonneau HM, Giambernardi TA, Goolsby JC, Qian CN, Zylstra CR, et al. Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Res. 2007 Mar 15;67(6):2490–6. doi: 10.1158/0008-5472.CAN-06-3028. [DOI] [PubMed] [Google Scholar]
- 33.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May 1;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 Apr 2;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malouf GG, Taube JH, Lu Y, Roysarkar T, Panjarian S, Estecio MR, et al. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013 Dec 24;14(12):R144. doi: 10.1186/gb-2013-14-12-r144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011 Sep 2;43(5):811–22. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Song JJ, Kingston RE. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem. 2008 Dec 12;283(50):35258–64. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehnertz B, Pabst C, Su L, Miller M, Liu F, Yi L, et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014 Feb 15;28(4):317–27. doi: 10.1101/gad.236794.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ando H, Natsume A, Senga T, Watanabe R, Ito I, Ohno M, et al. Peptide-based inhibition of the HOXA9/PBX interaction retards the growth of human meningioma. Cancer Chemother Pharmacol. 2014 Jan;73(1):53–60. doi: 10.1007/s00280-013-2316-5. [DOI] [PubMed] [Google Scholar]
- 41.Schaeffer E, Marchionni L, Huang Z, Simons B, Blackman A, Yu W, et al. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene. 2008 Dec 4;27(57):7180–91. doi: 10.1038/onc.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quagliata L, Matter MS, Piscuoglio S, Arabi L, Ruiz C, Procino A, et al. lncRNA HOTTIP / HOXA13 expression is associated with disease progression and predicts outcome in hepatocellular carcinoma patients. Hepatol Baltim Md. 2014 Mar;59(3):911–23. doi: 10.1002/hep.26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sang Y, Zhou F, Wang D, Bi X, Liu X, Hao Z, et al. Up-regulation of long non-coding HOTTIP functions as an oncogene by regulating HOXA13 in non-small cell lung cancer. Am J Transl Res. 2016 May 15;8(5):2022–32. [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z, Zhao X, Zhou Y, Liu Y, Zhou Q, Ye H, et al. The long non-coding RNA HOTTIP promotes progression and gemcitabine resistance by regulating HOXA13 in pancreatic cancer. J Transl Med. 2015 Mar 12;13 doi: 10.1186/s12967-015-0442-z. [Internet] [cited 2016 Jul 4] Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4372045/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, et al. SET8 promotes epithelial–mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012 Jan 4;31(1):110–23. doi: 10.1038/emboj.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006 Apr 21;125(2):315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 47.Ware KE, Somarelli JA, Schaeffer D, Li J, Zhang T, Park S, et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget. 2016 Jul;:7. doi: 10.18632/oncotarget.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.