

Hydrogels synthesized from crosslinking reactions between water soluble macromolecular precursors are widely used in a number of biomaterials applications, for example as drug or cell delivery vehicles and as cell culture scaffolds.[1,2] Many of the bio-click reactions used to form hydrogels allow direct encapsulation of biologics and cells, while maintaining their activity and viability, respectively.[3-7] Furthermore, many techniques exploit the ability to create multifunctional hydrogel systems with spatiotemporally controlled material properties,[8,9] biological functionalities,[7,10,11] and printed cell structures.[12,13] Along with this complexity, researchers have been further interested in methods to better characterize these complex systems, control their properties on demand, and temporally tune properties such as degradation or viscoelasticity.
With respect to temporally controlling hydrogel properties, many regenerative medicine applications that embed cells in hydrogels require degradation of the network structure to allow formation of focal adhesions, proliferation, migration, deposition of matrix components, and even to avoid fibrotic encapsulation of the implanted biomaterials. Hydrogels are routinely degraded by hydrolytic,[14] enzymatic,[15] or photolytic mechanisms,[8,16] and each mechanism provides specific advantages. With their high water content, hydrogels with hydrolytically cleavable crosslinks typically degrade through a uniform, bulk process; whereas hydrogels that are proteolytically cleavable often degrade through a local mechanism that depends on cell-secreted enzymes. More recently, hydrogels with photocleavable crosslinks have been developed that allow spatiotemporal control of the degradation process. In one example, Kloxin et al. demonstrated on demand control of network crosslinking density and elastic modulus, and used materials with photocleavable crosslinks to study the effects of mechanical properties on the reversibility of the fibroblast-to-myofibroblast transition in heart valve cells.[17] Since these early studies, spatiotemporal control of degradation has found numerous applications in guiding cellular proliferation, migration, and differentiation.[16,18-23] One limitation to many of the photodegradable groups used in biomaterial applications to date, however, is that the degradation relies on “one-photon one-event” reactions. A photolabile molecule, such as a nitrobenzyl or coumarin group, incorporated directly into the polymer backbone absorbs a photon and undergoes a cleavage reaction. This subsequently requires either long exposure times or high quantum yields.
With this in mind, we synthesized hydrogels that can be degraded via a radical addition-fragmentation chain transfer (AFCT) process, where a single photon initiates multiple events and amplifies the degradation process. Allyl sulfides have been used as efficient AFCT functionalities to introduce plasticity into crosslinked networks[24-26] and recently to reversibly photopattern biomolecules within a hydrogel.[10] To incorporate this moiety into biomaterial systems, we synthesized a symmetric allyl sulfide crosslinker flanked with azide functionalities for formation of a hydrogel network through a strain-promoted azide-alkyne cycloaddition (SPAAC). Subsequent exposure to light in the presence of a photoinitiator and a monofunctional thiol causes the crosslinked system to revert to soluble branched macromolecules. Upon exposure, photogenerated thiyl radicals rapidly propagate through thiol-ene addition reactions and chain transfer events (Scheme 1).[24,25,27] Allyl sulfide moieties in a crosslinking state participate in a reversible thiol-ene addition with non-crosslinking thiyl radicals, converting the allyl sulfides into a non-crosslinked state and generating a thiyl radical bound to the network (“network thiyl”). Alternating cycles of thiol-ene addition and chain transfer from a liberated network thiyl to a free thiol replace crosslinking allyl sulfides with non-crosslinking counterparts. Thiyl-thiol chain transfer events allow one absorbed photon to cleave multiple crosslinks, whereas current biocompatible photodegradation strategies typically rely on mechanisms where there is a maximum of one crosslink cleavage by one photon, with typical quantum yields being much lower.[28] Moreover, a lower concentration of photoinitator is required for radical mediated photodegradation of allyl sulfide-containing hydrogels, compared to traditional photodegradable hydrogels that have one or more photoactive constituents per crosslink.
Figure 1. Network structure of AFCT-based photodegradable hydrogels.
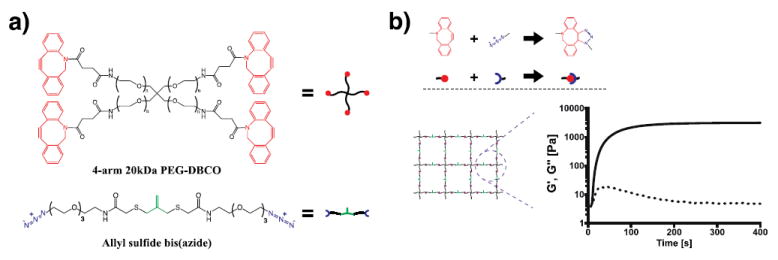
a) Chemical structures of cyclooctyne-terminated PEG macromer and azide crosslinker containing the allyl sulfide functionality. b) Upon mixing the two species, a hydrogel network is rapidly formed incorporating the allyl sulfide reactive groups. Gelation is monitored by shear rheology in situ, and the storage modulus (solid line) and loss modulus (dotted line) are tracked. The final elastic modulus is reached within 10 min.
Crosslinked hydrogel networks were formed through a strain promoted azide alkyne cycloaddition (SPAAC) reaction between a tetrafunctional poly(ethylene glycol) dibenzocyclooctyne (PEG-DBCO) and an allyl sulfide bis-(PEG3-azide) (Figure 1a). This bio-orthogonal “click” reaction proceeds rapidly at physiological conditions, and has been used in numerous studies as a cytocompatible crosslinking strategy.[16,20,29,30] Specifically, a 7.5 wt% solution of PEG-DBCO (15mM DBCO) and allyl sulfide crosslinker (16.5 mM azide) was polymerized in situ on a rheometer. Excess azide was chosen to ensure complete conversion of the DBCO functionalities and to avoid side reactions during the subsequent thiol-ene reactions, as strained alkynes are known to react with thiols in a Michael-type addition and also participate in thiol-yne radical additions.[31-37] The gel point was estimated by the cross-over of G’ and G”, which occurred in <30 s, while a final modulus of 3500±660 Pa was achieved in ~10 min. for this formulation (Figure 1b).
For subsequent photodegradation, the hydrogel was equilibrium swollen, and then placed in a solution containing varying concentrations of the photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP)[38,39] (1-8 mM) and a methoxy-PEG-thiol (mPEG-SH, Mn≈500 Da, 0-50 mM). Exposure to light generates photoinitiator radical species, which can add directly to the olefin of the allyl sulfide or undergo chain transfer to a free thiol, which in turn reacts with the allyl sulfide (Figure 2a). After 20 min of swelling, the gels were irradiated with 365 nm light (2-40 mW cm-2), and the degradation was tracked via changes in the shear storage and loss moduli with exposure time.
Figure 2. Light-triggered radical network degradation.
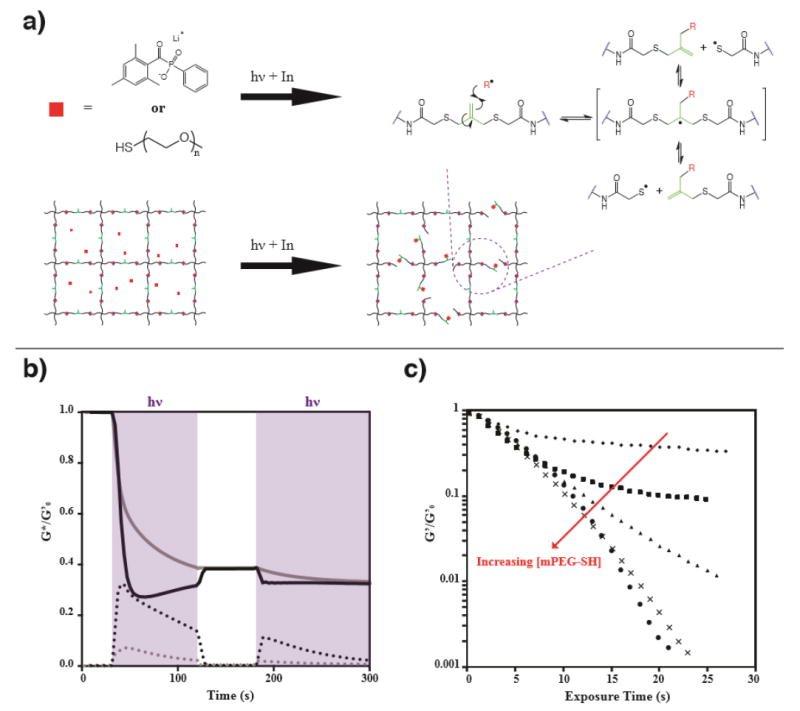
a) Photodegradation mechanism: in the presence of LAP and mPEG-SH, crosslinking molecules are fragmented by photoinitiator radicals or non-crosslinking monothiyl species when exposed to light. b) In the absence of free mPEG-SH, incomplete photodegradation is observed. The material is exposed to 365 nm light at 10 mW cm-2 at 30 s, and the light is shuttered for one minute at 120 s (light exposure is indicated by purple shading). Rheological traces were performed at a frequency of 1 rad/s (black) and 10 rad/s (grey), monitoring the storage (solid line) and loss (dotted line) moduli, with both normalized to the initial storage modulus. Evidence for rapid network reorganization is seen in the frequency dependence of the measurements during light exposure, while the curves converge when the light is shuttered (120 s – 180 s) and as the photoinitiator is depleted (less than 1% LAP remains after 300 s). c) Incorporation of mPEG-SH allows controlled photodegradation of the gel and tuning of the storage modulus. mPEG-SH was swollen into the network at 0 mM (◆), 5 mM (■), 15 mM (▲), 25 mM (×), and 50 mM (●). Reverse gelation occurs when mPEG-SH concentrations of 25 and 50 mM are used.
When the exchange solution contained only 4 mM LAP and no free thiol, significant photodegradation occurs, but not to an extent that results in reverse gelation (Figure 2b). In this condition, there are initially no free thiols present in the hydrogel system; the only sulfur atoms are in the thioethers of the crosslinker. Thus, photodegradation is likely due to the direct addition of the photoinitiator radical fragments to the olefin of the allyl sulfide crosslinker. According to Scheme S1, this reaction generates a new olefin and also a pendant thiyl radical. In the presence of thiyl radicals, the network is expected to reorganize rapidly as the system propagates through a number of thiol-ene additions, each consuming one thiyl radical and generating another.[24,27,40] Evidence for this network reorganization is shown in Figure 2b. The large increase in the loss modulus upon exposure to light indicates a significant shift in the viscoelastic properties of the network from an almost purely elastic material to one that is more fluid in nature. This behavior is typical of networks that are crosslinked by dynamic linkages, such as hydrazone bonds[41,42], host-guest interactions[43,12], electrostatics[44-46] and others (see reviews by C. Kloxin,[47] Rosales,[48] and Wang[49]), which display frequency-dependent mechanical properties. This is further emphasized by a near crossover of G’ and G”, indicating that the material is approaching the reverse gel point (i.e., behaving as soluble, highly branched polymer) at the given strain rate (1 rad/s).
An apparent anomaly is also seen in the 1 rad/s rheological trace, wherein G’ reaches a minimum after ~20 s of exposure and then increases before reaching a plateau (Figure 2b – black). This observation is likely due to a combination of photoinitiator consumption during light exposure and the generation of pendant network thiyls after allyl sulfide cleavage (Scheme S1). Initially, LAP is at its highest concentration (4 mM), leading to the highest rate of radical generation and allyl sulfide crosslink cleavage. The product of the allyl sulfide cleavage by LAP is a pendant thiyl radical. The pendant thiyl radicals have limited mobility, but are reactive towards other allyl sulfide species and can reform a crosslink upon addition to a network allyl sulfide molecule, which results in network reorganization, but not photodegradation.[24] Consequently, the measured G’ during the radical mediated network reorganization is lower than the value that would be obtained without radical generation, because crosslinked strands dissipate their potential energy upon crosslink reorganization. Here, we observe that the generation of network thiyls leads to a shift from a purely elastic network to a viscoelastic network with a frequency dependent storage and loss modulus. For comparison, the same experiment was performed at 10 rad/s and the results superimposed (Figure 2b - gray). At the higher sampling frequency, the hydrogel is less capable of dissipating the imposed force, and the storage modulus is a closer representation of what would be measured in the absence of radicals. To demonstrate the effect of network rearrangement on the storage modulus, the light was briefly shuttered at 120 s. The hydrogel subsequently reverted back to more purely elastic behavior, which led to an increase in G’ and a decrease in G”. At this point, the traces from the 1 rad/s and 10 rad/s experiments converge. Re-exposure of the sample at 180 s returns the gel to its previous viscoelastic storage modulus as it continues towards the final elastic modulus and the photoinitiator is completely consumed (photoinitiator half-life = 45s, Equation S1). Clearly, these reaction conditions result in rapid but incomplete photodegradation. Examination of the relevant functional groups – LAP (4 mM) and initial network thioethers (16.5 mM) – can help explain this. According to the Flory-Stockmayer equation,[50] the gel point for this hydrogel is estimated as ~ 61% (Equation S2), meaning that 39%, or 6.5 mM, of crosslinks would need to be cleaved to achieve reverse gelation. Theoretically, if every initiator fragment added directly to a crosslinking olefin, it would be possible to cause reverse gelation. However, proton abstraction from the increasing number of liberated pendant thiols may bias the system towards reactions that do not result in a net change in the crosslink density.
To increase the efficiency of the photodegradation reaction and allow complete network degradation, we studied the AFCT reaction in the presence of a free monothiol (mPEG-SH). In contrast to network-bound thiols, addition by a monofunctional thiol changes the overall network connectivity and effectively cleaves crosslinks. These replacement reactions can be favored by increasing the concentration of free mono-thiol relative to the concentration of thioethers initially present in the network. mPEG-SH was introduced at concentrations ranging from 5 to 50 mM, along with 4 mM LAP and irradiated with 365 nm light at 10 mW cm-2. As seen in Figure 2c, 25 and 50 mM of mPEG-SH resulted in reverse gelation, while concentrations of 0, 5, or 15 mM did not. If we assume similar reactivity of the alkyl thiols from mPEG-SH and the liberated network thiols, it is expected that the various thiolated molecules in Scheme 1 would be expected to approach an equilibrium defined by Equation 1:
Scheme 1. Amplification of photodegradation by chain transfer.
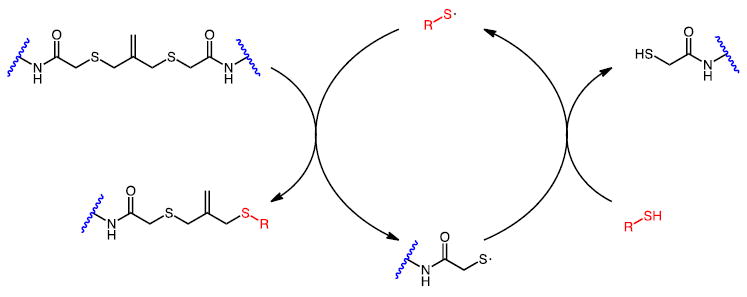
Addition-fragmentation chain transfer (AFCT) crosslinks exposed to a photogenerated mono-thiyl radical (red) transition from a crosslinked state to a non-crosslinked state and also regenerate a mono-thiyl radical capable of additional crosslink fragmentation reactions.
| (1) |
Figure S4 shows the expected equilibrium as a function of initial monothiol concentration. The predicted amount of initial free monothiol needed to cleave 6.5 mM crosslinks is 11 mM, and thus, we would expect complete degradation at 15 mM. Instead, we find a very weak gel with G’ approximately 1% of its initial value. It is possible that increasing the photoinitiator concentration would allow this equilibrium to be reached, but the competing reaction of initiator with olefin makes this analysis complex.
We also investigated the effect of varying photoinitiator concentration while keeping free thiol concentration constant at 25 mM and light intensity constant at 10 mW cm-2 (Figure 3a). As expected, the photoinitiator concentration had a strong effect on both the rate and extent of photodegradation. By increasing the photoinitiator concentration to 8 mM, complete reverse gelation was achieved after <13 seconds of irradiation, corresponding to a rate constant of kapp/I0 ×10-4 of 580 cm2 mW-1 s-1 and a photodegradation half-life of under 2 seconds. This result was compared to the rate of photodegradation of the widely used orthonitrobenzyl group and coumarin groups, and the AFCT mode of degradation was 70-2000 times faster than photodegradation methods based on alpha cleavage.[8,16,19,21,51-53] We then studied the effect of light intensity on the degradation rate (Figure 3b). The rate of photodegradation was easily tuned by setting the intensity of the illuminating light to 2, 10, or 40 mW cm-2. For 4 mM LAP and 25 mM mPEG-SH, a plot of kapp vs. I0 yields a straight line with a slope of 235 × 10-4 cm2 mW-1 s-1 (Figure 3c).
Figure 3. Photodegradation kinetics can be adjusted by changes in either the photoinitiator concentration or light intensity.
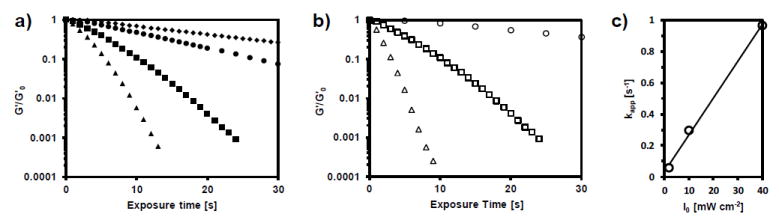
a) Effect of photoinitiator concentration. Hydrogels were swollen with 25 mM mPEG-SH and 1 mM (◆), 2 mM (●), 4 mM (■) or 8 mM (▲) LAP. b) Effect of light intensity. Hydrogels swollen with 4 mM LAP and 25 mM mPEG-SH and exposed to 2 (○), 10 (□) or 40 (∆) mW cm-2 365 nm light. c) Apparent rate constants for photodegradation are plotted as a function of light intensity, yielding a straight line with slope kapp/I0 ×104 of 235 cm2 mW-1 s-1.
AFCT-based photodegradable hydrogels also benefit from decreased light attenuation. There are two underlying causes to this effect. First, the molar absorptivity of our chosen photoinitiator, LAP, at 365 nm is 218 M-1 cm-1.[39] This is in contrast to the ortho-nitrobenzyl ester and coumarin photolabile groups which have molar absorptivities on the order of 3,000 to 7,000 M-1 cm-1 at 365 nm,[16,21,52] and while more transparent at longer wavelengths, the quantum yield and efficiency also decrease. The other contributing factor is that the concentration of photoactive species in this case can be lower because the radicals generated can propagate through numerous photocleavage events. In practice, 4 mM LAP was found to be sufficient for complete photodegradation, in comparison to 10-40 mM nitrobenzyl photodegradable groups commonly employed in photodegradable polymer strands. The combination of these factors allowed for photodegradation of much thicker hydrogel samples. To demonstrate the power of this effect, we created a 1 cm thick hydrogel with 4 mM LAP and subsequently swelled in mPEG-SH to a final concentration of 25 mM. The swollen hydrogen was then exposed to 365 nm light at 10 mW cm-2 along the 1 cm axis. For the LAP concentration employed, this 1 cm sample still allows ~13% transmission of the incident light at the bottom of the sample (Equation S3). As observed macroscopically in Figure 4a, this hydrogel rapidly erodes in ~ 1 min, which renders this hydrogel chemistry particularly useful for certain biomaterials applications. For example, in applications where one may wish to harvest selected cells or the entire cell population (e.g., for FACS or other analyses), light exposure in defined regions can allow cell capture in a manner akin to laser capture microdissection. In addition, as the field of mechanotransduction transitions from 2D to 3D culture systems, there is an ever increasing need for ways to expand and passage cells in 3D environments. One major roadblock in this approach is how to harvest cells from these 3D materials for futher expansion or characterization. Photodegradation is one attractive option, due to the spatial and temporal control that can be leveraged to release defined regions of cells at user-specified time points. However, rapid and spatially defined erosion is required, and current systems can be limited by relatively slow degradation kinetics and significant light attenuation. The aforementioned qualities of allyl sulfide crosslinked hydrogels give potential utility in this regard.
Figure 4. Allyl sulfide based photodegradable hydrogels can be used for large scale erosion and cytocompatible cell release.
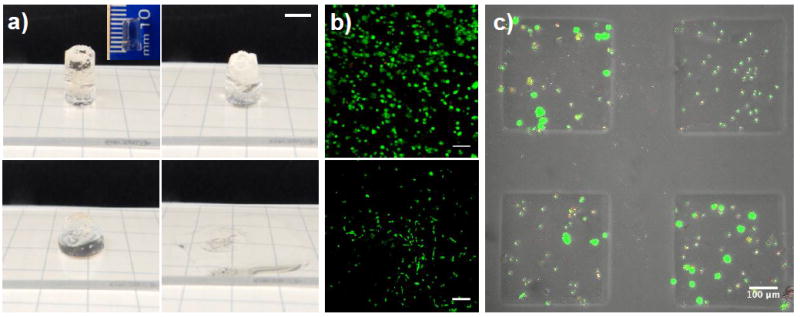
a) A 1 cm thick hydrogel containing 4 mM LAP and 25 mM mPEG-SH was irradiated with 10 mW cm-2 365 nm light, and completed eroded over the course of approximately 1 min. In this optically thick gel, 87 % of the incident light is attenuated through the sample (i.e., only 13% of the light reaches the bottom of the sample). Macroscopic images of the gel are shown at 0 s, 20 s, 44 s and 74 s (left to right, top to bottom). Scale bar 5 mm. b) Cells that were both encapsulated for 24 h and subsequently released remained highly viable. Top panel: hMSCs 24 h after encapsulation are 90% viable. Bottom panel: released cells remain viable and spread on glass coverslips over 24 h. Cells were stained with calcein AM (green, live) and ethidium homodimer (red, dead). Scale bars: 100 μm (top panel) and 300 μm (bottom panel). c) A 150 μm thick cell-laden hydrogel was selectively exposed to light (365 nm at 10 mW cm-2) for 1 min through a photomask to induce spatially controlled photodegradation and release a subset of the encapsulated cells. Cells remaining in hydrogel monoliths stained with with calcein AM (green) and ethidium homodimer (red). Scale bar = 100 μm.
To demonstrate some of these advantages, we encapsulated primary human mesenchymal stem cells (hMSCs) in 3 mm thick hydrogels at a density of 5×106 cells ml-1. For all of the cell culture experiments, an azide-functionalized RGD peptide was added to the gel formulation at 1 mM to provide cell-matrix interactions. Encapsulation using the SPAAC reaction is known to proceed with high cell viability;[16] indeed, hMSC viability was quantified as 90% and 74% after one and four days of encapsulation, respectively (Figure S2). This reduction in cell viability over time may be attributed, in part, to the inability of these encapsulated cells to remodel the PEG networks,[54] but the chemistry is readily modified to include protease degradable peptide linkers.
After one day of encapsulation, the cell laden hydrogels were swollen with mPEG-SH (50mM) and LAP (4 mM) for 1 h, followed by exposure to 10 mWcm-2 of 365 nm light for 1 min on a gelatin coated glass coverslip (Figure 4b). Under these exposure conditions, 2.4 mM photoinitiator is consumed, which is similar to photoinitiator concentrations that have been widely found to be cytocompatible for photoencapsulation and photopatterning.[3,39,55-57] The encapsulated cells were released from the photodegradable hydrogel and allowed to adhere to the underlying glass coverslip under standard culture conditions. hMSCs were viable upon release, and spread on the coverslip over 24 h, which demonstrates that these reaction conditions are mild enough to be useful for 3D cell culture and capture. Spatial control over cell release was also achieved under the same conditions by selective exposure of a 150 μm thick hydrogel through a chrome photomask (Figure 4c). In addition, we took advantage of the absorption spectrum of LAP, which extends up to 450 nm, to demonstrate that these hydrogels are capable of photodegradation under 405 nm light for cell release using a conventional microscope setup. hMSCs were seeded onto hydrogels at 1×104 cells cm-2 and using a confocal laser scanning microscope with a 405 nm laser (DAPI channel) at 60% power, a user defined area of the gel was degraded to release an adhered cell (Figure S3).
In conclusion, a photodegradable hydrogel system was synthesized incorporating an allyl sulfide functionality that allowed for a radical-initiated thiol-ene exchange reaction. By introduction of monothiols, the network connectivity and mechanical properties could be controlled on-demand by exposure to light. Under conditions that proved cytocompatible (4 mM LAP, 25 mM mPEG-SH, 10 mW cm-2 365 nm light), reverse gelation occurred in under 30 seconds and samples up to 1 cm thick could be eroded in ~1 min, representing a significant benefit over conventional photodegradable hydrogels in both respects. Importantly, both the SPAAC gel formation and photodegradation processes were designed to be compatible with biological systems, allowing new-found experiments to study cells in dynamic environments, and to readily capture cells from 3D laden systems. This new class of photodegradable hydrogels is unique in its mechanism, speed of degradation, and depths attainable, and provides access to experiments previously limited by light dose and attenuation.
Supplementary Material
Acknowledgments
This work was supported by the NSF DMR (1408955), the Howard Hughes Medical Institute, NSF GRFP (T.E.B. and I.A.M.) and NIH T32 GM-065103 (T.E.B.).
Contributor Information
Tobin E. Brown, Department of Chemical and Biological Engineering and the BioFrontiers Institute, University of Colorado Boulder, Jennie Smoly Caruthers Biotechnology Building, 3415 Colorado Ave, Boulder, CO, 80303.
Ian A. Marozas, Department of Chemical and Biological Engineering and the BioFrontiers Institute, University of Colorado Boulder, Jennie Smoly Caruthers Biotechnology Building, 3415 Colorado Ave, Boulder, CO, 80303.
Kristi S. Anseth, Department of Chemical and Biological Engineering and the BioFrontiers Institute, University of Colorado Boulder, Jennie Smoly Caruthers Biotechnology Building, 3415 Colorado Ave, Boulder, CO, 80303 Howard Hughes Medical Institute, University of Colorado Boulder, Jennie Smoly Caruthers Biotechnology Building, 3415 Colorado Ave, Boulder, CO, 80303.
References
- 1.Hoffman AS. Adv Drug Deliv Rev. 2012;64:18. [Google Scholar]
- 2.Annabi N, Tamayol A, Uquillas JA, Akbari M, Bertassoni LE, Cha C, Camci-Unal G, Dokmeci MR, Peppas NA, Khademhosseini A. Adv Mater Res. 2014;26:85. doi: 10.1002/adma.201303233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han LH, Tong X, Yang F. Adv Mater. 2014;26:1757. doi: 10.1002/adma.201304805. [DOI] [PubMed] [Google Scholar]
- 4.McCall JD, Anseth KS. Biomacromolecules. 2012;13:2410. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 6.Khetan S, Burdick JA. Biomaterials. 2010;31:8228. doi: 10.1016/j.biomaterials.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 7.DeForest CA, Tirrell DA. Nat Mater. 2015;14:523. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 8.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Nat Mater. 2013;12:458. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandavarapu NR, Azagarsamy MA, Anseth KS. Adv Mater. 2014;26:2521. doi: 10.1002/adma.201304847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP. Nat Mater. 2013;12:1072. doi: 10.1038/nmat3766. [DOI] [PubMed] [Google Scholar]
- 12.Highley CB, Rodell CB, Burdick JA. Adv Mater. 2015;27:5075. doi: 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- 13.Hinton TJ, Jallerat Q, Palchesko RN, Park JH, Grodzicki MS, Shue H-J, Ramadan MH, Hudson AR, Feinberg AW. Sci Adv. 2015;1:e1500758. doi: 10.1126/sciadv.1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metters AT, Anseth KS, Bowman CN. J Phys Chem B. 2001;105:8069. [Google Scholar]
- 15.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci U S A. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeForest CA, Anseth KS. Nat Chem. 2011;3:925. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kloxin AM, Benton JA, Anseth KS. Biomaterials. 2010;31:1. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang C, Tibbitt MW, Basta L, Anseth KS. Nat Mater. 2014;13:645. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azagarsamy MA, McKinnon DD, Alge DL, Anseth KS. ACS Macro Lett. 2014;3:515. doi: 10.1021/mz500230p. [DOI] [PubMed] [Google Scholar]
- 20.McKinnon DD, Brown TE, Kyburz KA, Kiyotake E, Anseth KS. Biomacromolecules. 2014;15:2808. doi: 10.1021/bm500731b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffin DR, Kasko AM. J Am Chem Soc. 2012;134:13103. doi: 10.1021/ja305280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norris SCP, Tseng P, Kasko AM. 2015 doi: 10.1021/acsbiomaterials.6b00237. [DOI] [Google Scholar]
- 23.Tsang KMC, Annabi N, Ercole F, Zhou K, Karst DJ, Li F, Haynes JM, Evans Ra, Thissen H, Khademhosseini A, Forsythe JS. Adv Funct Mater. 2015;25:977. doi: 10.1002/adfm.201403124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott TF, Schneider AD, Cook WD, Bowman CN. Science. 2005;308:1615. doi: 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- 25.Scott TF, Draughon RB, Bowman CN. Adv Mater. 2006;18:2128. [Google Scholar]
- 26.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans RA, Rizzardo E. Macromolecules. 2000;33:6722. [Google Scholar]
- 28.Tibbitt MW, Kloxin AM, Anseth KS. J Polym Sci A Polym Chem. 2013;51:1899. doi: 10.1002/pola.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekkebus R, Van Kasteren SI, Kulathu Y, Scholten A, Berlin I, Geurink PP, De Jong A, Goerdayal S, Neefjes J, Heck AJR, Komander D, Ovaa H. J Am Chem Soc. 2013;135:2867. doi: 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiu H-Y, Chan T-C, Ho C-M, Liu Y, Wong M-K, Che C-M. Chem - A Eur J. 2009;15:3839. doi: 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- 33.Shiu H-Y, Chong H-C, Leung Y-C, Wong M-K, Che C-M. Chem - A Eur J. 2010;16:3308. doi: 10.1002/chem.200903121. [DOI] [PubMed] [Google Scholar]
- 34.van Geel R, Pruijn GJM, van Delft FL, Boelens WC. Bioconjug Chem. 2012;23:392. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- 35.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Proc Natl Acad Sci U S A. 2010;107:1821. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fairbanks BD, Sims EA, Anseth KS, Bowman CN. Macromolecules. 2010;43:4113. [Google Scholar]
- 37.Fairbanks BD, Scott TF, Kloxin CJ, Anseth KS, Bowman CN. Macromolecules. 2009;42:211. doi: 10.1021/ma801903w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majima T, Schnabel W, Weber W. Die Makromol Chemie. 1991;192:2307. [Google Scholar]
- 39.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moad G, Rizzardo E, Thang SH. Polymer (Guildf) 2008;49:1079. [Google Scholar]
- 41.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2013;26:1. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Chem Mater. 2014;26:2382. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodell CB, Kaminski AL, Burdick JA. Biomacromolecules. 2013;14:4125. doi: 10.1021/bm401280z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee KY, Mooney DJ. Prog Polym Sci. 2012;37:106. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stowers RS, Allen SC, Suggs LJ. Proc Natl Acad Sci U S A. 2015;112:1953. doi: 10.1073/pnas.1421897112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif Sa, Weaver JC, Huebsch N, Mooney DJ. Nat Commun. 2015;6:1. doi: 10.1038/ncomms7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloxin CJ, Bowman CN. Chem Soc Rev. 2013;42:7161. doi: 10.1039/c3cs60046g. [DOI] [PubMed] [Google Scholar]
- 48.Rosales AM, Anseth KS. Nat Publ Gr. 2016;1:1. [Google Scholar]
- 49.Wang H, Heilshorn SC. Adv Mater. 2015:n/a. doi: 10.1002/adma.201501558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stockmayer WH. J Chem Phys. 1943;11:45. [Google Scholar]
- 51.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Adv Mater. 2010;22:3484. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffin DR, Kasko AM. ACS Macro Lett. 2012;1:1330. doi: 10.1021/mz300366s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeForest CA, Anseth KS. Angew Chemie Int Ed. 2012;51:1816. doi: 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hudalla GA, Eng TS, Murphy WL. Biomacromolecules. 2008;9:842. doi: 10.1021/bm701179s. [DOI] [PubMed] [Google Scholar]
- 55.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlara S, Khademhosseini A. Biomaterials. 2010;31:5536. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA. Sci Rep. 2016;6:21387. doi: 10.1038/srep21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong DY, Ranganath T, Kasko AM. PLoS One. 2015;10:1. doi: 10.1371/journal.pone.0139307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.