

Abstract
Genomic rearrangements are a hallmark of human cancers. Here, we identify the piggyBac transposable element derived 5 (PGBD5) gene as an active DNA transposase expressed in the majority of childhood solid tumors, including lethal rhabdoid tumors. Using assembly-based whole-genome DNA sequencing, we found previously undefined genomic rearrangements in human rhabdoid tumors. These rearrangements involved PGBD5-specific signal (PSS) sequences at their breakpoints, recurrently inactivating tumor suppressor genes. PGBD5 was physically associated with genomic PSS sequences that were also sufficient to mediate PGBD5-induced DNA rearrangements in rhabdoid tumor cells. Ectopic expression of PGBD5 in primary immortalized human cells was sufficient to promote cell transformation in vivo. This activity required specific catalytic residues in the PGBD5 transposase domain, as well as end-joining DNA repair, and induced structural rearrangements with PSS breakpoints. This defines PGBD5 as an oncogenic mutator and provides a plausible mechanism for site-specific DNA rearrangements in childhood and adult solid tumors.
Introduction
Whole-genome analyses have now produced near-comprehensive topographies of coding mutations for certain human cancers, enabling both detailed molecular studies of cancer pathogenesis and potential of precisely targeted therapies 1-5. For certain childhood cancers, recent studies have begun to reveal the essential functions of complex non-coding structural variants that can induce aberrant expression of cellular proto-oncogenes 6,7. However, for many aggressive childhood cancers including embryonal tumors, such studies have identified distinct cancer subtypes that have no discernible coding mutations 8-11. In addition, while for some cancers, defects in DNA damage repair have been suggested to explain their increased incidence at a relatively young age, the causes of complex genomic rearrangements in cancers of young children without apparent widespread genomic instability remain largely unknown.
Rhabdoid tumor is a prototypical example of this question. Rhabdoid tumors occur in the developing tissues of infants and children, leading to tumors with neuroectodermal, epithelial and mesenchymal components in the brain, liver, kidney and other organs 10,12,13. Rhabdoid tumors that cannot be cured with surgery are generally chemotherapy resistant and almost uniformly lethal 14. Rhabdoid tumors exhibit inactivating mutations of SMARCB1, generally as a result of genomic rearrangements of the 22q11.2 chromosomal locus 15. These mutations can be inherited as part of the rhabdoid tumor predisposition syndrome, but are not thought to involve chromosomal instability 13. While SMARCB1 mutations are sufficient to cause rhabdoid tumors in mice 16, human rhabdoid tumors have been observed to have multiple molecular subtypes and rearrangements of additional chromosomal loci that are poorly understood 9,10,17,18. These findings suggest that additional genetic elements and molecular mechanisms may contribute to the pathogenesis of rhabdoid tumors.
In humans, nearly half of the genome is comprised by sequences derived from transposons, including both autonomous and non-autonomous mobile genetic elements 19. The majority of human genes that encode enzymes that can mobilize transposons appear to be catalytically inactive, with the exception of L1 long interspersed repeated sequences (LINEs) that appear to induce structural genomic variation in human neurons and adenocarcinomas 20-22, Mariner transposase-derived SETMAR that functions in DNA repair 23, and Transib-like DNA transposase RAG1/2 that catalyzes somatic recombination of V(D)J receptor genes in lymphocytes 24. In particular, aberrant activity of RAG1/2 in lymphoblastic leukemias and lymphomas can induce the formation of chromosomal translocations that generate transforming fusion genes 25-27. The identity of and mechanisms by which similar genomic rearrangements may be formed in childhood solid tumors are unknown, but the existence of additional human recombinases that can induce somatic DNA rearrangements has long been hypothesized 28.
Recently, human PGBD5 and THAP9 have been found to catalyze transposition of synthetic DNA transposons in human cells 29,30. The physiologic functions of these activities are currently not known. PGBD5 is distinguished by its deep evolutionary conservation among vertebrates (∼500 million years) and developmentally restricted expression in tissues from which childhood embryonal tumors, including rhabdoid tumors, are thought to originate 30,31. PGBD5 is transcribed as a multi-intronic and non-chimeric transcript from a gene that encodes a full-length transposase that became immobilized on human chromosome 1 30,31. Genomic transposition activity of PGBD5 requires distinct aspartic acid residues in its transposase domain, and specific DNA sequences containing inverted terminal repeats with similarity to the lepidopteran Trichoplusia ni piggyBac transposons 30. These findings, combined with the recent evidence that PGBD5 can induce genomic rearrangements that inactivate the HPRT1 gene 32, prompted us to investigate whether PGBD5 may induce site-specific DNA rearrangements in human rhabdoid tumors that share developmental origin with cells that normally express PGBD5.
Results
Human rhabdoid tumors exhibit genomic rearrangements associated with PGBD5-specific signal sequence breakpoints
First, we analyzed the expression of PGBD5 in large, well-characterized cohorts of primary childhood and adult tumors (Supplementary Fig. 1a). We observed that PGBD5 is highly expressed a variety of childhood and adult solid tumors, including rhabdoid tumors, but not in acute lymphoblastic or myeloid leukemias (Supplementary Fig. 1a). The expression of PGBD5 in rhabdoid tumors was similar to that of embryonal tissues from which these tumors are thought to originate, and was not significantly associated with currently defined molecular subgroups or patient age at diagnosis (Supplementary Fig. 1a-f). To investigate potential PGBD5-induced genomic rearrangements in primary human rhabdoid tumors, we performed de novo structural variant analysis of whole-genome paired-end Illumina sequencing data for 31 individually-matched tumor versus normal paired blood specimens from children with extra-cranial rhabdoid tumors that are generally characterized by inactivating mutations of SMARCB1 10. By virtue of their repetitive nature, sequences derived from transposons present challenges to genome analysis. Thus, we reasoned that genome analysis approaches that do not rely on short-read alignment algorithms, such as the local assembly-based algorithm laSV and the tree-based sequence comparison algorithm SMuFin might reveal genomic rearrangements that otherwise might escape conventional algorithms 33,34.
Using this assembly-based approach, we observed recurrent rearrangements of the SMARCB1 gene on chromosome 22q11 in nearly all cases examined, consistent with the established pathogenic function of inactivating mutations of SMARCB1 in rhabdoid tumorigenesis (Fig. 1a). In addition, we observed previously unrecognized somatic deletions, inversions and translocations involving focal regions of chromosomes 1, 4, 5, 10, and 15 (median = 3 per tumor), which were recurrently altered in more than 20% of cases (Fig. 1a, Data S1). These results indicate that in addition to the pathognomonic mutations of SMARCB1, human rhabdoid tumors are characterized by additional distinct and recurrent genomic rearrangements.
Fig. 1. Human rhabdoid tumors exhibit genomic rearrangements associated with PGBD5-specific signal sequence breakpoints.
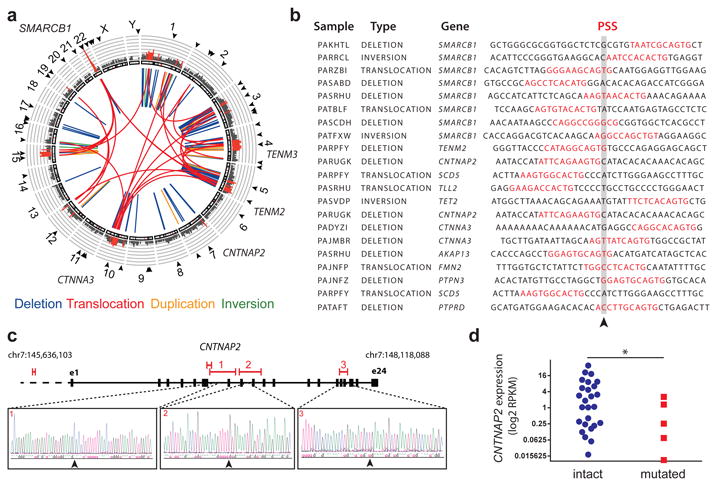
(a) Aggregate Circos plot of somatic structural variants identified in 31 human rhabdoid tumors using laSV, as marked for PSS-containing breakpoints (outer ring, arrowheads), recurrence (middle ring histogram, rearrangements occurring in ≥3 out of 31 samples and highlighted in red for rearrangements with recurrence frequency greater than 13%), and structural variant type (inner lines, as color-labeled). Recurrently rearranged genes are labeled. (b) Representation of 21 structural variant breakpoints in rhabdoid tumors identified to harbor PSS sequences (red) within 10 bp of the breakpoint junction (arrowhead). (c) Recurrent structural variants of CNTNAP2 (red) with gene structure (black) and Sanger sequencing of the rearrangement breakpoints. (d) CNTNAP2 mRNA expression in primary rhabdoid tumors as measured using RNA sequencing in CNTNAP2 mutant (red) as compared to CNTNAP2 intact (blue) specimens (* p = 0.017 by t-test for intact vs. mutant CNTNAP2).
To determine whether any of the observed genomic rearrangements may be related to PGBD5 DNA transposase or recombinase activity, we first used a forward genetic screen to identify PGBD5-specific signal (PSS) sequences that were specifically found at the breakpoints of PGBD5-induced deletions, inversions and translocations that caused inactivation of the HPRT1 gene in a thioguanine resistance assay 32. Using these PSS sequences as templates for supervised analysis of the somatic genomic rearrangements in primary human rhabdoid tumors, we identified specific PSS sequences associated with the breakpoints of genomic rearrangements in rhabdoid tumors (p = 1.1 × 10-10, hypergeometric test; Fig. 1b, Supplementary Fig. 2). By contrast, we observed no enrichment of the RAG1/2 recombination signal (RSS) sequences at the breakpoints of somatic rhabdoid tumor genomic rearrangements, in spite of their equal size to PSS sequences, consistent with the lack of RAG1/2 expression in rhabdoid tumors. Likewise, we did not find significant enrichment of PSS motifs at the breakpoints of structural variants and genomic rearrangements in breast carcinomas that lack PGBD5 expression, even though these breast carcinoma genomes were characterized by high rates of genomic instability (Data S1). PSS sequences observed in human rhabdoid tumors exhibited both similarities and differences to those found in the forward genetic screen (Supplementary Fig. 2), suggesting that context-dependent factors may control PGBD5 activity. In total, 580 (52%) out of 1121 somatic genomic rearrangements detected in rhabdoid tumors contained PSS sequences near their rearrangement breakpoints (Data S1).
Overall, the majority of the observed rearrangements were deletions and translocations (Fig. 1a, Supplementary Fig. 3a). Notably, we found recurrent PSS-containing genomic rearrangements affecting the CNTNAP2, TENM2, TENM3, and TET2 genes (Fig. 1a-c, Supplementary Fig. 3c, Data S1). Using allele-specific polymerase chain reaction (PCR) followed by Sanger DNA sequencing, we confirmed three of the observed intragenic CNTNAP2 deletions and rearrangement breakpoints (Fig. 1c). Likewise, we confirmed the somatic nature of mutations of CNTNAP2 and TENM3 by allele-specific PCR in matched tumor and normal primary patient specimens (Supplementary Fig. 3d-h).
CNTNAP2, a member of the neurexin family of signaling and adhesion molecules, has been previously found to function as a tumor suppressor gene in gliomas 35. Consistent with the potential pathogenic functions of the apparent CNTNAP2 rearrangements in rhabdoid tumors found in our analysis, CNTNAP2 has also been recently reported to be recurrently deleted in an independent cohort of rhabdoid tumor patients 18. By using comparative RNA sequencing gene expression analysis, we found that recurrent genomic rearrangements of CNTNAP2 in our cohort were indeed associated with significant reduction of its mRNA transcript expression in genomically rearranged primary cases as compared to those lacking CNTNAP2 rearrangements (p = 0.017, t-test; Fig. 1d). Additional mechanisms, including as of yet undetected mutations or silencing 35, may contribute to the loss of CNTNAP2 expression in apparently non-rearranged cases (Fig. 1d).
Interestingly, some of the observed genomic rearrangements with PSS-containing breakpoints in rhabdoid tumors involved SMARCB1 deletions (Fig. 1a-b, Data S1), suggesting that in a subset of rhabdoid tumors, PGBD5 activity itself may contribute to the somatic inactivation of SMARCB1 in rhabdoid tumorigenesis. Similarly, we observed recurrent interchromosomal translocations and complex structural variants containing breakpoints with the PSS motifs that involved SMARCB1 (Fig. 1b, Data S1), including chromosomal translocations, previously observed using cytogenetic methods 17. For example, we verified the t(5;22) translocation using allele-specific PCR followed by Sanger sequencing of the translocation breakpoint (Suppl. Fig. 3i-j). In all, these results indicate that human rhabdoid tumors exhibit recurrent complex genomic rearrangements that are defined by PSS breakpoint sequences specifically associated with PGBD5, at least some of which appear to be pathogenic and may be coupled with inactivating mutations of SMARCB1 itself.
PGBD5 is physically associated with human genomic PSS sequences that are sufficient to mediate DNA rearrangements in rhabdoid tumor cells
In prior studies, human PGBD5 has been found to localize to the cell nucleus 31. To test whether PGBD5 in rhabdoid tumor cells is physically associated with genomic PSS-containing sequences, as would be predicted for a DNA transposase that induces genomic rearrangements, we used chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) to determine the genomic localization of endogenous PGBD5 in human G401 rhabdoid tumor cells. We observed that human DNA regions bound by PGBD5 were significantly enriched for PSS motifs (p = 2.9 × 10-29, hypergeometric test), in contrast to the scrambled PSS sequences of identical composition, or the functionally unrelated RSS sequences of equal size that showed no significant enrichment (p = 0.28 and 1.0, respectively, hypergeometric test; Fig. 2a).
Fig. 2. PGBD5 is physically associated with human genomic PSS sequences that are sufficient to mediate DNA rearrangements in rhabdoid tumor cells.
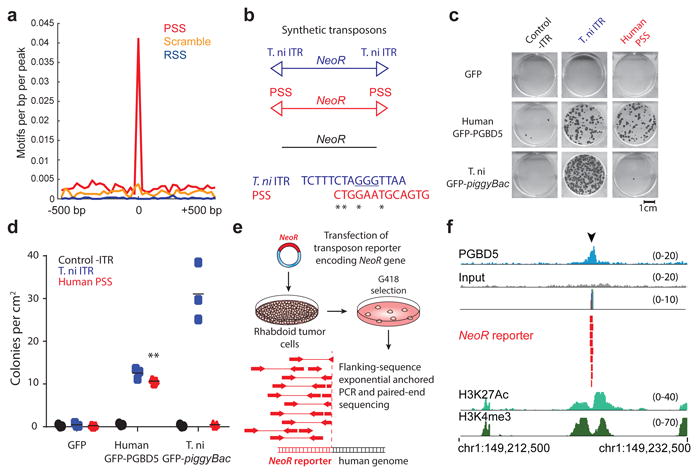
(a) Genomic distribution of PGBD5 protein in G401 rhabdoid tumor cells as a function of enrichment of PSS (red) as compared to scrambled PSS (orange) and RAG1 recombination signal sequence (RSS, blue) controls as measured using PGBD5 ChIP-seq (p = 2.9 × 10-29 for PSS, p = 0.28 for scrambled PSS, p = 1.0 for RSS by hypergeometric test). (b) Schematic of synthetic transposon substrates used for DNA transposition assays, including transposons with T. ni ITR marked by triangles in blue, transposons with PGBD5-specific signal sequence (PSS) marked by triangles in red and transposons lacking ITRs marked in black (top) and sequence alignment of T. ni ITR compared to human PSS (bottom). (c) Representative photographs of Crystal violet-stained colonies obtained upon G418 selection in the transposon reporter assay. (d) Genomic DNA transposition assay as measured using neomycin resistance clonogenic assays in HEK293 cells co-transfected with human GFP-PGBD5 or control GFP and T.ni GFP-PiggyBac, and transposon reporters encoding the neomycin resistance gene flanked by human PSS (red), as compared to control reporters lacking inverted terminal repeats (-ITR, black) and T. ni piggyBac ITR (blue). ** p = 5.0 × 10-5. Lepidopteran T. ni PiggyBac DNA transposase and its piggyBac ITR serve as specificity controls. Errors bars represent standard deviations of three independent experiments. (e) Schematic model of transposition reporter assay in G401 rhabdoid tumor cells followed by flanking sequence exponential anchored-polymerase chain reaction (FLEA-PCR) and Illumina paired-end sequencing. (f) Genomic integration of synthetic NeoR transposons (red) by endogenous PGBD5 in G401 rhabdoid tumor cells at PSS site (arrowhead), as shown in the ChIP-seq genome track of PGBD5 (blue), as compared to its sequencing input (gray), and H3K27Ac and H3K4me3 (bottom), consistent with the bound PGBD5 transposase protein complex.
To test the hypothesis that PGBD5 can act directly on human PSS-containing DNA sequences to mediate their genomic rearrangements, we used the previously established DNA transposition reporter assay 30. Human embryonic kidney (HEK) 293 cells were transiently transfected with plasmids expressing human GFP-PGBD5, hyperactive lepidopteran T. ni GFP-PiggyBac DNA transposase or control GFP, in the presence of reporter plasmids encoding the neomycin resistance gene (NeoR) flanked by a human PSS sequence, as identified from rhabdoid tumor rearrangement breakpoints (Suppl. Fig. 2-3, Data S1), lepidopteran piggyBac inverted terminal repeat (ITR) transposon sequence 30, or control plasmids lacking flanking transposon elements (Fig. 2b). Clonogenic assays of transfected cells in the presence of G418 to select neomycin resistant cells with genomic reporter integration demonstrated that GFP-PGBD5, but not control GFP, exhibited efficient activity on reporters containing terminal repeats with the human PSS sequences, but not control reporters lacking flanking transposon elements (p = 5.0 × 10-5, t-test; Fig. 2c & d). This activity was specific since the lepidopteran GFP-PiggyBac DNA transposase, which can efficiently mobilize its own piggyBac transposons, did not mobilize reporter plasmids containing human PSS sequences (Fig. 2c & d).
To determine whether endogenous PGBD5 can mediate genomic rearrangements in rhabdoid cells, we transiently transfected human G401 rhabdoid cells with the neomycin resistance gene transposon reporter plasmids, and determined their chromosomal integrations by using flanking sequence exponential anchored (FLEA) PCR to amplify and sequence specific segments of the human genome flanking transposon integration sites (Fig. 2e, Supplementary Fig. 4) 30. Similar assays in HEK293 cells that lack PGBD5 expression fail to induce measureable genomic integration of reporter transposons (Fig. 2c & d). In contrast, we observed that endogenous PGBD5 in G401 rhabdoid tumor cells was sufficient to mediate integrations of transposon-containing DNA into human genomic PSS-containing sites (Fig. 2f, Supplementary Tables 1 & 2). This activity was specifically observed for transposon reporters with intact transposons, but not those in which the essential 5′-GGGTTAACCC-3′ hairpin structure was mutated to 5′-ATATTAACCC-3′ (Supplementary Table 1) 30. Thus, PGBD5 physically associates with human genomic PSS sequences that are sufficient to mediate DNA rearrangements of synthetic reporters in rhabdoid tumor cells.
PGBD5 expression in genomically stable primary human cells is sufficient to induce malignant transformation in vitro and in vivo
Recurrent somatic genomic rearrangements in primary rhabdoid tumors associated with PGBD5-specific signal sequence breakpoints, their targeting of tumor suppressor genes, and specific activity as genomic rearrangement substrates raise the possibility that PGBD5 DNA transposase activity may be sufficient to induce tumorigenic mutations that contribute to malignant cell transformation. To determine if PGBD5 can act as a human cell transforming factor, we used established transformation assays of primary human foreskin BJ and retinal pigment epithelial (RPE) cells immortalized with telomerase 36. Primary RPE and BJ cells at passage 3-5 can be immortalized by the expression of human TERT telomerase in vitro, undergo growth arrest upon contact inhibition, and fail to form tumors upon transplantation in immunodeficient mice in vivo 36. Prior studies have established the essential requirements for their malignant transformation by the concomitant dysregulation of P53, RB, and RAS pathways 36. Thus, transformation of primary human RPE and BJ cells enables detailed studies of human PGBD5 genetic mechanisms that cannot be performed using mouse or other heterologous model systems.
To test whether PGBD5 has transforming activity in human cells, we used lentiviral transduction to express GFP-PGBD5 and control GFP transgenes in telomerase-immortalized RPE and BJ cells, at levels that are 1.1-5 and 1.5-8 fold higher as compared to primary rhabdoid tumor specimens and cell lines, respectively (Fig. 3a & b). We observed that GFP-PGBD5-expressing but not non-transduced or GFP-expressing RPE and BJ cells formed retractile colonies in monolayer cultures and exhibited anchorage-independent growth in semisolid cultures, a hallmark of cell transformation (Fig. 3c & d). When transplanted into immunodeficient mice, GFP-PGBD5-expressing RPE and BJ cells formed subcutaneous tumors with similar latency and penetrance to that seen in cells expressing both mutant HRAS and the SV40 large T antigen that dysregulates both P53 and RB pathways (LTA; Fig. 3f & g, Supplementary Figure 5). Importantly, both RPE and BJ cells transformed by GFP-PGBD5 had stable, diploid karyotypes when passaged in vitro (Supplementary Figure 6). By contrast, expression of the distantly related lepidopteran GFP-PiggyBac DNA transposase which exerts specific and efficient transposition activity on lepidopteran piggyBac transposon sequences (Fig. 2d), failed to transform human RPE cells (Fig. 3e), in spite of being equally expressed (Supplementary Fig. 7a). These results indicate that the PGBD5 transposase can specifically transform human cells in the absence of chromosomal instability both in vitro and in vivo.
Fig. 3. Ectopic expression of PGBD5 in human cells leads to oncogenic transformation both in vitro and in vivo.
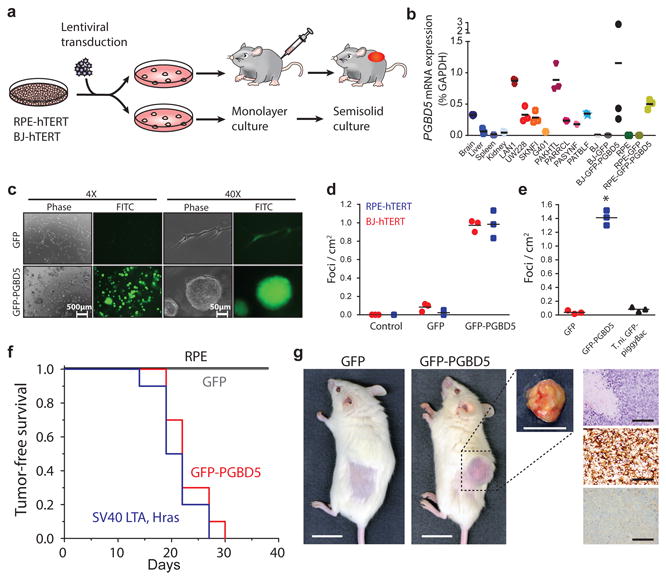
(a) Schematic for testing transforming activity of PGBD5. (b) Relative PGBD5 mRNA expression measured by quantitative RT-PCR in normal mouse tissues (brain, liver, spleen and kidney), as compared to human tumor cell lines (rhabdoid G401, neuroblastoma LAN1 and SK-N-FI, medulloblastoma UW-228 cells), primary human rhabdoid tumors (PAKHTL, PARRCL, PASYNF, PATBLF), and BJ and RPE cells stably transduced with GFP-PGBD5 and GFP. Error bars represent standard deviations of 3 independent measurements. (c) Representative images of GFP-PGBD5-transduced RPE cells grown in semisolid media after 10 days of culture, as compared to control GFP-transduced cells. (d) Number of refractile foci formed in monolayer cultures of RPE and BJ cells expressing GFP-PGBD5 or GFP, as compared to non-transduced cells (p = 3.6 × 10-5 and 3.9 × 10-4 for GFP-PGBD5 vs. GFP for BJ and RPE cells, respectively). (e) Expression of T. ni GFP-PiggyBac does not lead to the formation of anchorage independent foci in monolayer culture (* p = 3.49 × 10-5 for GFP-PGBD5 vs. T. ni GFP-PiggyBac). Error bars represent standard deviations of 3 independent experiments. (f) Kaplan-Meier analysis of tumor-free survival of mice with subcutaneous xenografts of RPE cells expressing GFP-PGBD5 or GFP control, as compared to non-transduced cells or cells expressing SV40 large T antigen (LTA) and HRAS (n = 10 mice per group, p < 0.0001 by log-rank test). (g) Representative photographs (from left) of mice with shaved flank harboring RPE xenografts (scale bar = 1 cm). Tumor excised from mouse harboring GFP-PGBD5 expressing tumor (scale bar = 1 cm). Photomicrograph of GFP-PGBD5 expressing tumor (top to bottom: hematoxylin and eosin stain, vimentin, and cytokeratin, scale bar = 1 mm).
PGBD5-induced cell transformation requires DNA transposase activity
To test whether the cell transforming activity of PGBD5 requires its transposase enzymatic activity, we used PGBD5 point mutants that are proficient or deficient in DNA transposition in reporter assays 30. Thus, we compared E373A and E365A PGBD5 mutants that retain wild-type transposition activity 30, to D168A, D194A, D386A or their double D194A/D386A (DM) and triple D168A/D194A/D386A (TM) mutants that occur on residues required for efficient DNA transposition in vitro, consistent with their evolutionary conservation and putative function as the DDD/E catalytic triad for the phosphodiester bond hydrolysis reaction 30. After confirming stable and equal expression of these PGBD5 mutants in RPE cells by Western immunoblotting (Fig. 4a), we assessed their transforming activity with contact inhibition assays in monolayer cultures and transplantation in immunodeficient mice. Whereas ectopic expression of wild-type GFP-PGBD5 induced efficient and fully penetrant cell transformation, neither D168A, nor D194A, nor DM or TM mutants deficient in transposition function in reporter assays induced contact inhibition in vitro or tumor formation in vivo (Fig. 4b & d). By contrast, transposition-proficient E373A and E365A mutants exhibited the same transforming activity as wild-type GFP-PGBD5 (Fig. 4b and 4d). Importantly, we confirmed that the catalytic mutants of GFP-PGBD5 on average retained their chromatin localization as compared to wild-type PGBD5, as assessed using ChIP-seq (Fig. 4c). Although the D386A mutant exhibited reduced transposition activity in reporter assays in vitro 30, its expression induced wild-type transforming activity in vivo (Fig. 4d). This suggests that the transforming activity of PGBD5 may involve non-canonical DNA transposition or recombination reactions, consistent with the dispensability of some catalytic residues for certain type of DNA transposase-induced DNA rearrangements 37,38. Thus, cell transformation induced by PGBD5 requires its nuclease activity.
Fig. 4. PGBD5 transposase activity is necessary to transform human cells.
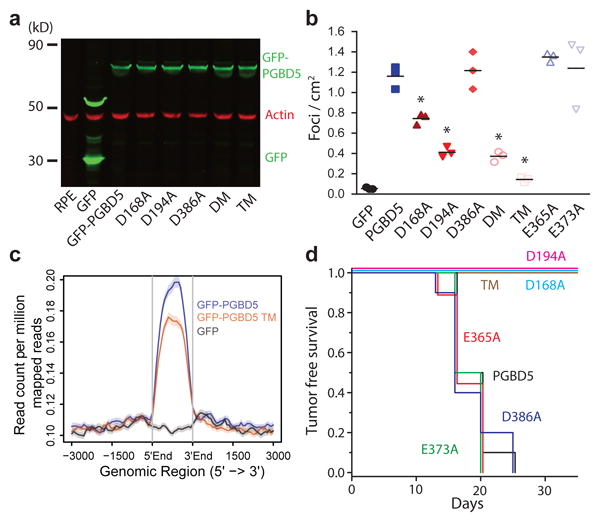
(a) Western blot of GFP in RPE cells expressing GFP-PGBD5, GFP-PGBD5 mutants, and GFP compared to RPE cells (DM = double mutant D194A/D386A; TM = triple mutant D168A/D194A/D386A). (b) Number of refractile foci formed in monolayer culture in RPE and BJ cells stably expressing GFP-PGBD5 or control GFP, as compared to non-transduced cells and cells expressing GFP-PGBD5 mutants (red = transposase deficient mutants, blue = transposase proficient mutants, * p = 2.1 × 10-4 for D168A vs. GFP-PGBD5, p = 2.7 × 10-6 for D194A vs. GFP-PGBD5, p = 1.8 × 10-6 for D194A/D386A vs. GFP-PGBD5, p = 2.4 × 10-7 for D168A/D194A/D386A vs. GFP-PGBD5). Error bars represent standard deviations of 3 independent experiments. (c) Composite plot of ChIP-seq of GFP-PGBD5 (green), as compared to the GFP-PGBD5 D168A/D194A/D386A catalytic TM mutant (orange) and GFP control (purple). (d) Kaplan-Meier analysis of tumor-free survival of mice with subcutaneous xenografts of RPE cells expressing GFP-PGBD5 as compared to cells expressing GFP-PGBD5 mutants (n = 10 per group, p < 0.0001 by log-rank test).
Transient expression of PGBD5 is sufficient for PGBD5-induced cell transformation
If PGBD5 can induce transforming genomic rearrangements, then transient exposure to PGBD5 should be sufficient to heritably transform human cells. To test this prediction, we generated doxycycline-inducible PGBD5-expressing RPE cells, and using Western immunoblotting confirmed lack of detectable expression of the enzyme in the absence of doxycycline and its induction upon exposure to doxycycline in vitro (Supplementary Fig. 7b). When transplanted into immunodeficient mice whose doxycycline chow treatment (–Dox) was stopped upon macroscopic signs of tumor formation (Fig. 5a, Supplementary Fig. 7c), the transduced cells retained essentially the same tumorigenicity as seen in continuously treated (+Dox) animals or in those transplanted with constitutively expressing GFP-PGBD5 cells (Supplementary Fig. 7c). Importantly, we confirmed the absence of measureable PGBD5 expression in tumors harvested from –Dox animals by Western immunoblotting (Fig. 5a, inset). Consistent with cell transformation by transient expression of PGBD5, both –Dox and +Dox tumors were indistinguishable histopathologically (Fig. 5b). To investigate the potential irreversibility and heritability of cell transformation induced by transient PGBD5 expression, we transplanted tumors harvested from –Dox and +Dox animals into secondary recipients, and observed that tumors were induced with the same latency and penetrance in both –Dox and +Dox animals (Fig. 5a). In agreement with this model of PGBD5-induced cell transformation, we observed that endogenous PGBD5 in established G401 and A204 rhabdoid tumor cells was dispensable for cell survival, as assessed using small hairpin RNA (shRNA) interference using two different shRNA vectors, as compared to control shRNA targeting GFP (Fig. 5c & d). Thus, transient expression of PGBD5 is sufficient to transform cells, as would be predicted from the ability of a catalytically active transposase to induce heritable cellular alterations.
Fig. 5. Transient PGBD5 transposase expression is sufficient to transform human cells.
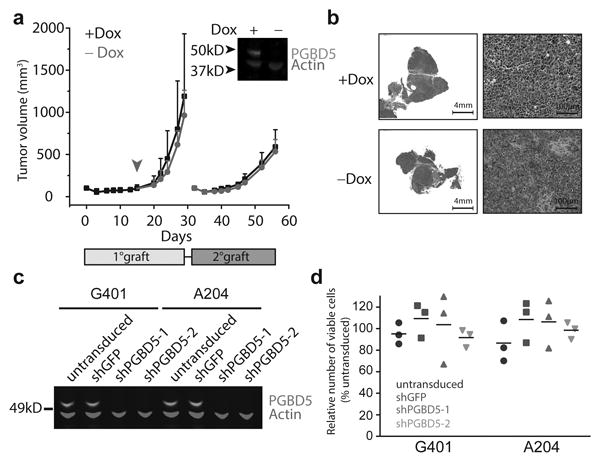
(a) Tumor volume of RPE cells as a function of time in primary (light gray box) and secondary (dark gray box) transplants, with PGBD5 expression induced using doxycycline (black), as indicated. RPE cells were treated with doxycycline in vitro for 10 days prior to transplantation. Arrowhead denotes withdrawal of doxycycline from the diet (red). Inset: Western blot of PGBD5 protein, as compared to actin control in cells derived from tumors after primary transplant. (b) Representative photomicrographs of hematoxylin and eosin stained tumor sections from doxycycline-inducible PGBD5-expressing RPE tumors after continuous (+Dox) and discontinuous (-Dox) doxycycline treatment. (c) Western blot of PGBD5 in G401 and A204 rhabdoid tumor cells upon depletion of PGBD5 using two independent shRNAs, as compared to non-transduced cells and control cells expressing shGFP. (d) Relative number of viable G401 and A204 cells upon 72 hours of PGBD5 shRNA depletion. Errors bars represent standard deviations of 3 independent experiments.
PGBD5-induced transformation requires DNA end-joining repair
If PGBD5-induced cell transformation involves transposase-mediated genomic rearrangements, then this process should depend on the repair of DNA double-strand breaks (DSBs) that are generated by the DNA recombination reactions 39. Genomic rearrangements induced by transposases of the DDD/E superfamily involve transesterification reactions that generate DSBs that are predominantly repaired by DNA non-homologous end-joining (NHEJ) in somatic cells 40, as is the case for human V(D)J rearrangements induced by the RAG1/2 recombinase 38. To test whether PGBD5-induced cell transformation requires NHEJ, we used isogenic RPE cells that are wild-type or deficient for the NHEJ cofactor PAXX, which stabilizes the NHEJ repair complex and is required for efficient DNA repair 41. In contrast to defects in other NHEJ components, such as LIG4, PAXX deficiency does not appreciably alter cell growth or viability but significantly reduces NHEJ efficiency without needing TP53 inactivation to survive 41. Thus, we generated RPE cells expressing doxycycline-inducible PGBD5 that were PAXX+/+ or PAXX-/-, and confirmed the induction of PGBD5 and lack of PAXX expression by Western immunoblotting (Fig. 6a). Doxycycline-induced expression of PGBD5 in PAXX-/- but not isogenic PAXX+/+ RPE cells caused the accumulation of DNA damage-associated γH2AX (Fig. 6b, Supplementary Figure 8b), apoptosis-associated cleavage of caspase 3 (Fig. 6c, Supplementary Figure 8a), and cell death (Supplementary Figure 8c). We confirmed the requirement of NHEJ for the repair of PGBD5-induced rearrangements using Ku80-deficient mouse embryonal fibroblasts (data not shown). Importantly, PGBD5-mediated induction of DNA damage and cell death in NHEJ-deficient PAXX-/- cells as compared to the isogenic NHEJ-proficient PAXX+/+ cells was nearly completely rescued by the mutation of D168A/D194A/D386A residues, which are required for transposase activity of PGBD5 (Fig. 6d). Thus, NHEJ DNA repair is required for the survival of cells expressing active PGBD5.
Fig. 6. DNA end-joining repair is required for survival of cells expressing active PGBD5.
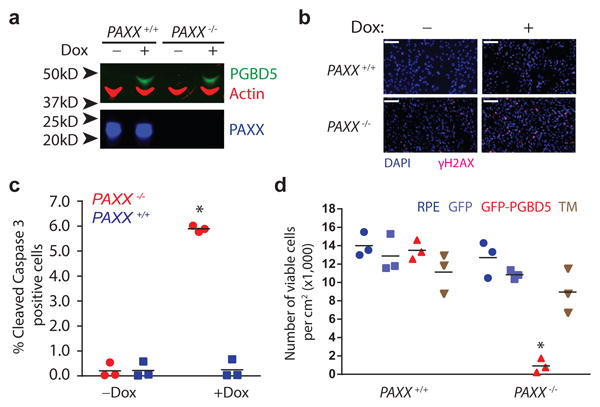
(a) Western blot of PGBD5 protein after 24 h of doxycycline (500 ng/ml) treatment of isogenic PAXX+/+ and PAXX-/- RPE cells stably expressing doxycycline-inducible PGBD5. (b) Representative photomicrograph of PAXX+/+ and PAXX-/- RPE cells after 48 h treatment with doxycycline (500 ng/ml) or vehicle control stained for DAPI (blue) and γH2AX (red). Scale bar = 100 μm. (c) Fraction of apoptotic cells as measured by cleaved caspase-3 staining and flow cytometric analysis of PAXX+/+ and PAXX-/- RPE cells after treatment with doxycycline or vehicle control. * p = 8.7 × 10-4 for PAXX+/+ vs. PAXX-/- with doxycycline. (d) Number of viable PAXX+/+ and PAXX-/- RPE cells per cm2 in monolayer culture as measured by Trypan blue staining after 48 h of expression of GFP-PGBD5, as compared to GFP-PGBD5 D168A/D194A/D386 mutant and GFP-expressing control cells. * p = 7.4 × 10-5 for PAXX-/- GFP-PGBD5 vs. GFP control. Error bars represent standard deviations of 3 independent experiments.
PGBD5-induced cell transformation involves site-specific genomic rearrangements associated with PGBD5-specific signal sequence breakpoints
The requirements for PGBD5 enzymatic transposase activity, cellular NHEJ DNA repair, and ability of transient PGBD5 expression to promote cell transformation are all consistent with the generation of heritable genomic rearrangements that mediate PGBD5-induced tumorigenesis. To determine the genetic basis of PGBD5-induced cell transformation, we sequenced whole genomes of PGBD5-induced tumors as well as control GFP-expressing and non-transduced RPE cells, using massively parallel paired-end Illumina sequencing at a coverage in excess of 80-fold for over 90% of the genome (Data S1). As for the rhabdoid tumor genome analysis, we used the assembly-based algorithm laSV as well as conventional techniques (Supplementary Table 3, Supplementary Figs. 9-11, Data S1) 33,34. This analysis led to the identification of distinct genomic rearrangements, specifically in PGBD5-induced tumor cell genomes as compared to control GFP and non-transduced RPE cells (Fig. 7a). The identified rearrangements were characterized by intra-chromosomal deletions with a median length of 183 bp, consistent with their apparent limited detectability by conventional genome analysis methods, as well as inversions, duplications and translocations (Supplementary Fig. 12a-c, Data S1). As with genomic rearrangements found in primary human tumors (Fig. 1), the analysis of genomic rearrangements found in PGBD5-transformed RPE cells detected significant enrichment of PSS motifs at the breakpoints of PGBD5-induced tumor structural variants (p = 7.2 × 10-3, hypergeometric test; Fig. 7b, Data S1). By contrast, breakpoints of structural variants in GFP control RPE cell genomes, presumably at least in part due to normal genetic variation, exhibited no enrichment for PSS motifs (p = 0.37). We independently verified these findings using the direct tree graph-based read comparative SMuFin analysis method (Supplementary Fig. 12a, Data S1). In addition, we validated five of these rearrangements using variant and wild-type allele-specific PCR followed by Sanger DNA sequencing of rearrangement breakpoints to confirm that they are specifically present in PGBD5-transformed but not control GFP-transduced RPE cells (Supplementary Fig. 12d-h). Additionally, we did not find genomic rearrangement breakpoints containing RSS sequences that are targeted by the RAG1/2 recombinase which is not expressed in RPE cells. We also did not find evidence of structural alterations of the annotated human MER75 and MER85 piggyBac-like transposable elements, in agreement with the distinct evolutionary history of human PGBD5 30. Notably, we found that the genomic rearrangements and structural variants observed in PGBD5-induced RPE tumors were significantly enriched for regulatory DNA elements important for normal human embryonal as opposed to adult tissue development (Fig. 7c, Supplementary Table 4).
Fig. 7. PGBD5-induced cell transformation involves site-specific genomic rearrangements associated with PGBD5-specific signal sequence breakpoints.
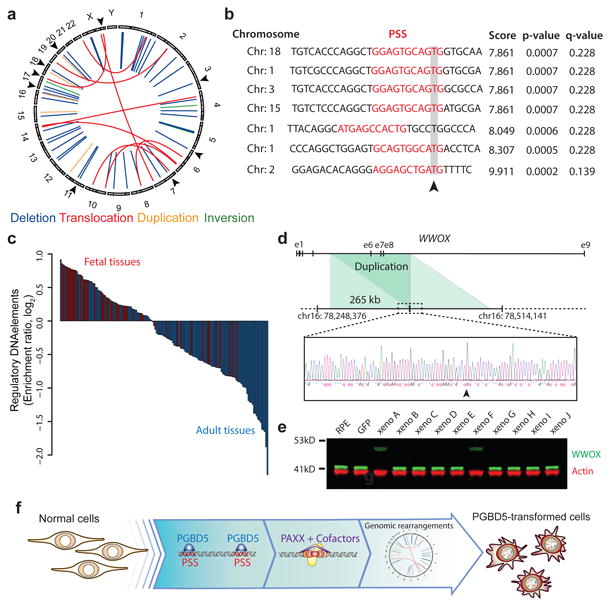
(a) Circos plot of structural variants discovered in RPE-GFP-PGBD5 tumor cells using assembly-based genome analysis. Black arrows on outer circle indicate the presence of PSS at variant breakpoints. (b) Representation of 7 breakpoints identified to harbor PSS sequences (red) within 10 bp of the breakpoint junction (arrowhead) of structural variants in PGBD5 expressing RPE cells. Genomic sequence is annotated 5′ to 3′ as presented in the reference genome (+) strand. (c) Waterfall plot of enrichment of ENCODE regulatory DNA elements with structural variants in fetal (red) as compared to adult tissues (blue) in PGBD5-transformed RPE cells (p = 5.7 × 10-8). (d) Schematic of the WWOX gene and its intragenic duplication in GFP-PGBD5-transformed RPE cells (top), with Sanger sequencing chromatogram of the rearrangement breakpoint (bottom). Arrowhead marks the breakpoint. (e) Western blot analysis of WWOX in 10 independent GFP-PGBD5-transformed RPE cell tumor xenografts, as compared to control GFP-transduced and non-transduced RPE cells. Actin serves as loading control. (f) Schematic model of the proposed mechanism of PGBD5-induced cell transformation, involving association of PGBD5 with genomic PSS sequences, their remodeling dependent on PAXX-meditated end-joining DNA repair, and generation of tumorigenic genomic rearrangements.
To identify genomic rearrangements that may be functionally responsible for PGBD5-induced cell transformation, we analyzed the recurrence of PGBD5-induced genomic rearrangements in 10 different RPE tumors from independent transduction experiments in individual mouse xenografts. We detected 59 PGBD5-induced structural variants per tumor, 42 (71%) of which were deletions, 36 (61%) affected regulatory intergenic elements, with 13 (22%) containing PSS motifs at their breakpoints (Data S1). In particular, we identified recurrent and clonal PSS-associated rearrangements of WWOX, including duplication of exons 6-8 (Fig. 7d). WWOX is a tumor suppressor gene that controls TP53 signaling 42. We confirmed the duplication of exons 6-8 of WWOX by PCR and Sanger DNA sequencing (Fig. 7d), and tested its functional consequence on WWOX protein expression by Western immunoblotting (Fig. 7e). Remarkably, this mutation resulted in low level expression of extended mutant form of WWOX protein, associated with loss of wild-type WWOX expression, consistent with the dominant negative or gain-of-function activity of mutant WWOX in RPE cell transformation. We observed this mutation in 2 out of 10 independent RPE tumors, consistent with its probable pathogenic function in PGBD5-induced cell transformation.
To determine its function in PGBD5-induced RPE cells transformation, we depleted endogenous WWOX and ectopically expressed wild-type WWOX in non-transformed wild-type and WWOX-mutant PGBD5-induced RPE cell tumors (Supplementary Fig. 13a & d). Consistent with the tumorigenic function of PGBD5-induced mutations of WWOX, we found that WWOX inactivation was necessary but not sufficient to maintain clonogenicity of PGBD5-transformed RPE tumor cells in vitro (Supplementary Fig. 13b-c & e-f). Thus, PGBD5-induced cell transformation involves site-specific genomic rearrangements that are associated with PGBD5-specific signal sequence breakpoints that recurrently target regulatory elements and tumor suppressor genes (Fig. 7f).
Discussion
We have now found that primary human rhabdoid tumor genomes exhibit signs of PGBD5-mediated DNA recombination, involving recurrent mutations of previously elusive rhabdoid tumor suppressor genes (Fig. 1). These genomic rearrangements involve breakpoints associated with the PGBD5-specific signal (PSS) sequences that are sufficient to mediate DNA rearrangements in rhabdoid tumor cell lines and physical recruitment of endogenous PGBD5 transposase (Fig. 2). The enzymatic activity of PGBD5 is both necessary and sufficient to promote similar genomic rearrangements in primary human cells, causing their malignant transformation (Figs. 3-7).
PGBD5-induced genomic rearrangements comprise a defined architecture, including characteristic deletions, inversions and complex rearrangements that appear distinct from those generated by other known mutational processes. We observe an imprecise relationship of PSS sequences with genomic rearrangement breakpoints, with evidence of incomplete ‘cut-and-paste’ DNA transposition, consistent with potentially aberrant targeting of PGBD5 nuclease activity. While our structure-function studies suggest that PGBD5 induces genomic rearrangements in conjunction with the canonical NHEJ apparatus, it is possible that PGBD5 activity can also promote other DSB repair pathways, such as alternative microhomology-mediated end joining (Supplementary Fig. 14). We confirmed that the putative catalytic aspartic acid mutants of PGBD5 on average maintain chromatin localization of wild-type PGBD5. It is also possible that these residues contribute to cell transformation due to their interaction with cellular cofactors or assembly of DNA regulatory complexes, or still yet unknown nuclease-independent functions that contribute to cell transformation.
PSS-associated genomic rearrangements induced by PGBD5 in rhabdoid tumors are reminiscent of McClintock's “mutable loci” induced upon DNA transposase mediated mutations of the Ds locus that controls position-effect variegation in maize 24,43. Insofar as nuclease substrate accessibility is controlled by chromatin structure and conformation, PGBD5-induced genomic rearrangements indeed may be coupled to developmental regulatory programs that control gene expression and specification of cell fate, as suggested by their strong association with developmental regulatory DNA elements in our analysis. The association of PGBD5-induced rearrangements may involve sequence-specific recognition of human genomic PSS sequences, or alternatively by their accessibility or the presence of cellular co-factors, as determined by cellular developmental states.
Importantly, the spectrum of PGBD5-induced genomic rearrangements and their PSS sequences identified in this study should provide a useful approach to the functional characterization of childhood tumor genomes and identification of cancer-causing genomic alterations. In the case of rhabdoid tumors, the association of SMARCB1 mutations with additional recurrent genomic lesions, such as structural alterations of CNTNAP2, TENM2 and TET2 genes that can regulate developmental and epigenetic cell fate specification, may lead to the identification of additional mechanisms of childhood cancer pathogenesis, including those that cooperate with the dysregulation of SWI/SNF/BAF-mediated nucleosome remodeling induced by SMARCB1 loss. Notably, the recurrence patterns of PGBD5-induced genomic rearrangements in rhabdoid tumors indicate that even for rare cancers, more comprehensive tumor genome analyses will be necessary to define the spectrum of causal genomic lesions and potential therapeutic targets. Our results also indicate that improved genome analysis methods, such as SMuFin and laSV used in our work 33,34, and confirmation of their sensitivity and specificity, will be needed to elucidate tumorigenic genome rearrangements. Similarly, given the existence of distinct molecular subtypes of rhabdoid tumors 9,10, it will be important to determine to what extent PGBD5-induced genome remodeling contributes to this phenotypic diversity.
In summary, PGBD5 defines a distinct class of oncogenic mutators that contribute to cell transformation not due to mutational activation but rather as a result of their aberrant induction and chromatin targeting to induce site-specific transforming genomic rearrangements. Our data identify PGBD5 as an endogenous human DNA transposase that is sufficient to fully transform primary immortalized human cells in the absence of chromosomal instability 36. Given the expression of PGBD5 in various childhood and adult solid tumors, either by virtue of its aberrant or co-opted tissue expression, we anticipate that PGBD5 may also contribute to their pathogenesis. Similarly, it will be important to investigate the functions of PGBD5 in normal vertebrate and mammalian development, given its ability to induce site-specific somatic genomic rearrangements in human cells. Finally, the functional requirement for cellular NHEJ DNA repair in PGBD5-induced cell transformation might foster rational therapeutic strategies for rhabdoid and other tumors involving endogenous DNA transposases.
Note Added in Proof
Since the work described in this paper was completed and submitted for publication, additional genome analysis of rhabdoid tumors was described, independently identifying recurrent mutations of CNTNAP2 and other loci in human rhabdoid tumors 44.
Online Methods
Reagents
All reagents were obtained from Sigma-Aldrich if not otherwise specified. Synthetic oligonucleotides were obtained from Eurofins (Eurofins MWG Operon, Huntsville, AL, USA), purified by HPLC, as listed in Supplementary Table 5. Antibodies are listed in Supplementary Table 6.
Plasmid constructs
Human PGBD5 cDNA (Refseq ID: NM_024554.3) was cloned into the lentiviral vector in frame with N-terminal GFP to generate pRecLV103-GFP-PGBD5 (GeneCopoeia, Rockville, MD, USA). pReceiver-Lv103 encoding GFP was used as a negative control in all experiments. Plasmid encoding the hyperactive T. ni piggyBac transposase, as originally cloned by Nancy Craig and colleagues 45, was obtained from System Biosciences (Mountain View, CA, USA), and subcloned into pReceiver-Lv103. The plasmids pBABE-neo-largeT, pBABE-puro-H-Ras, psPAX2, and pMD2.G were obtained from Addgene (Cambridge, MA, USA). Missense GFP-PGBD5 mutants were generated using site-directed mutagenesis according to the manufacturer's instructions (QuikChange Lightning, Agilent, Santa Clara, CA, USA), as described 30. Doxycycline-inducible pINDUCER21 vector was kind gift from Thomas Westbrook 46, and used to generate pINDUCER21-PGBD5 using Gateway cloning, according to the manufacturer's instructions (Fisher Scientific, Watham, MA, USA). Lentiviral shRNA and doxycycline-inducible WWOX expression vectors were a kind gift of Marcelo Aldaz 47. pLKO.1 shRNA vectors targeting PGBD5 (TRCN0000138412, TRCN0000135121) and control shGFP were obtained from the RNAi Consortium (Broad Institute, Cambridge, MA). The PB-EF1-IRES-NEO transposon reporter plasmid was used as described previously 30. pBS-EF1-IRES-NEO was created by cloning the EF1-IRES-NEO cassette from PB-EF1-IRES-NEO into pBluescript plasmid and modified by PCR mutagenesis to replace the T. ni piggyBac inverted terminal repeat with the PGBD5 signal sequence (CTGGAATGCAG). All newly generated plasmids are available from Addgene (https://www.addgene.org/Alex_Kentsis/).
Production and purification of anti-PGBD5 antibody
Synthetic peptide from human PGBD5 (Refseq ID: NM_024554.3) ELQLLSIVPGRDLQPSDSFTGPTRC was used to immunize mice (Lampire Biological Products, Ottsville, PA, USA). Hybridoma clones were screened using enzyme-linked immunosorbent assays (ELISA), and hybridoma supernatants were purified using Protein A affinity chromatography to generate the 10A8-11-7-P-5 Western blot antibody (Supplementary Table 6).
Lentivirus production and cell transduction
Lentivirus production was carried out as previously described 48. Briefly, HEK293T cells were transfected using TransIT-LT1 with 2:1:1 ratio of the lentiviral vector, and psPAX2 and pMD2.G packaging plasmids, according to manufacturer's instructions (Mirus, Madison, WI, USA). Virus supernatant was collected at 48 and 72 hours post-transfection, pooled, filtered and stored at -80 °C. RPE and BJ cells were transduced with virus particles at a multiplicity of infection (MOI) of 5 in the presence of 8 μg/ml hexadimethrine bromide. Transduced cells were selected for 2 days with puromycin hydrochloride (RPE cells at 10 μg/ml, BJ cells at 2 μg/ml) or G418 sulfate (2 mg/ml), depending on the vector-mediated resistance. For pINDUCER21 viruses, cells were transduced at a MOI of 1, and isolated using fluorescence-activated cell sorting (FACSAria III, BD Bioscience, San Jose, CA, USA). For inducible expression of WWOX, RPE cells were transduced with lentiviruses encoding tetOn-advanced-WWOX and selected with G418 sulfate (2 mg/ml) for 10 days. For shRNA depletion of WWOX, cells were transduced with lentiviruses encoding pGIPZ-shWWOX or pGIPZ-shScramble control and selected with puromycin hydrochloride (10 μg/ml) for 2 days.
Cell culture
Low-passage RPE and BJ cells, and human tumor cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). PAXX-/- RPE cells have been described previously 41. The identity of all cell lines was verified by STR analysis (Genetica DNA Laboratories, Burlington, NC, USA) and absence of Mycoplasma sp. contamination was determined using Lonza MycoAlert (Lonza Walkersville, Inc., Walkersville, MD, USA). Cell lines were cultured in 5% CO2 in a humidified atmosphere at 37 °C in Dulbecco's Modified Eagle medium with high glucose (DMEM-HG) supplemented with 10 % fetal bovine serum (FBS) and antibiotics (100 U / ml penicillin and 100 μg / ml streptomycin). Clonogenic assays of RPE cells were carried out in DMEM/F-12 medium. To assess the number of viable cells, cells were trypsinized, resuspended in media and sedimented at 500 g for 5 min. Cells were then resuspended in PBS and 10 μL mixed in a 1:1 ratio with 0.4 % Trypan Blue (Thermo Fisher) and counted using a hematocytometer (Hausser Scientific, Horsham, PA, USA).
Transposon reporter assay
The transposon reporter assay was performed using the pBS-EF1-IRES-NEO vector in HEK293 cells as described previously 30.
Quantitative RT-PCR
RNA was isolated using RNeasy Mini, according to manufacturer's instructions (Qiagen, Venlo, Netherlands). cDNA was synthesized using the SuperScript III First-Strand Synthesis System, according to the manufacturer's instructions (Invitrogen, Waltham, MA, USA). Quantitative real-time PCR was performed using the KAPA SYBR FAST PCR polymerase with 20 ng template and 200 nM primers, according to the manufacturer's instructions (Kapa Biosystems, Wilmington, MA, USA). PCR primers are listed in Supplementary Table 5. Ct values were calculated using ROX normalization using the ViiA 7 software (Applied Biosystems).
Western blotting
To analyze protein expression by Western immunoblotting, 1 million cells were suspended in 80 μl of lysis buffer (4% sodium dodecyl sulfate, 7% glycerol, 1.25% β-mercaptoethanol, 0.2 mg/ml Bromophenol Blue, 30 mM Tris-HCl, pH 6.8) and incubated at 95 °C for 10 minutes. Cell suspensions were lysed using Covaris S220 adaptive focused sonicator, according to the manufacturer's instructions (Covaris, Woburn, CA). Lysates were cleared by centrifugation at 16,000 g for 10 minutes at 4 °C. Clarified lysates (30 μl) were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electroeluted using the Immobilon FL PVDF membranes (Millipore, Billerica, MA, USA). Membranes were blocked using the Odyssey Blocking buffer (Li-Cor, Lincoln, Nebraska, USA), and blotted using antibodies listed in Supplementary Table 6. Blotted membranes were visualized using goat secondary antibodies conjugated to IRDye 800CW or IRDye 680RD and the Odyssey CLx fluorescence scanner, according to manufacturer's instructions (Li-Cor, Lincoln, Nebraska, USA).
Flow cytometry of cleaved Caspase-3
Cells were fixed using neutral-buffered formalin for 10 min on ice, washed with PBS, resuspended in 0.1% Triton-X100 in PBS, and incubated for 15 min at room temperature. Permeabilized cells were washed twice with PBS, and resuspended in 100 μl of Hank's balanced salt solution (HBSS) with 0.1% bovine serum albumin and 2 μl of Alexa Fluor 647-conjugated antibody against cleaved Caspase-3. Cells were incubated for 30 min at room temperature in the dark washed twice with PBS and stained with 1 μg/ml DAPI. Cells were analyzed on the Fortessa LSR as described before (BD Bioscience) 49,50.
Histological staining
Histologic processing and staining was done as described previously 51,52. In short, cell lines were plated on 8-well glass Millicell EZ chamber slides at 5000 cells/well, grown for 24 hours, and fixed using 4% paraformaldehyde for 10 min at room temperature (Millipore). Tumor xenograft tissue was fixed using 4% paraformaldehyde for 24 hours at room temperature. Tissues were embedded in paraffin using the ASP6025 tissue processor (Leica, Wetzlar, Germany), sectioned at 5 μm using the RM2265 microtome (Leica), and collected on SuperfrostPlus slides (Fisher Scientific). Tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems). Antigen retrieval was performed with Cell Conditioning 1 buffer (Ventana Medical Systems), and sections were blocked for 30 minutes with Background Buster solution (Innovex, Norwood, MA, USA). Primary antibodies were applied for 5 hours at 1 μg/ml (Supplementary Table 6). Secondary antibodies were applied for 60 minutes.
For immunohistochemistry staining, diaminobenzidine (DAB) detection was performed with the DAB detection kit according to manufacturer instruction (Ventana Medical Systems). Slides were counterstained with hematoxylin and a cover slip was mounted with Permount (Fisher Scientific).
For immunofluorescence staining, the detection was performed with Streptavidin-HRP D (Ventana Medical Systems), followed by incubation with Tyramide Alexa Fluor 647 prepared according to manufacturer instruction (Invitrogen). Slides were then counterstained with 5 μg/ml DAPI for 10 min and a cover slip was mounted with Mowiol (Sigma Aldrich).
Image acquisition
Bright-field images were acquired on an Axio Observer microscope (Carl Zeiss Microimaging, Oberkochen, Germany). Epifluorescence images were acquired using the EVOS FL microscope (Thermo Fisher). Slides were scanned using the Pannoramic 250 slide scanner and images analyzed using the Pannoramic Viewer (3DHistech, Budapest, Hungary).
Karyotype analysis
Five million cells were grown for 24 hours prior to harvesting. Cultures were treated with 0.005 μg/ml colcemid for 1 hr at 37 °C, resuspended in 75 mM KCl for 10 minutes at 37 °C and fixed in methanol : acetic acid (3:1). Cells were transferred onto slides and stained in 0.08 μg/ml DAPI in citric acid buffer for 3 minutes and mounted in Vectashield solution (Vector Labs). For each cell line, a minimum of 15 metaphases were counted.
Anchorage independence assay
One million RPE and BJ cells stably transduced with lentiviral vectors were expanded in 10 cm tissue culture plates until fully confluent. At confluence, cells were microscopically inspected for the occurrence of refractile colonies within the cell monolayer. For growth in semisolid medium, one million cells were resuspended in 2 ml of media mixed with 2 ml of Matrigel (BD Bioscience, Heidelberg, Germany). Cell suspensions were plated in 12-well tissue culture plates (200 μl per well). Semisolid suspensions were cultured for 10 days before scoring.
Xenografts
All mouse experiments were carried out in accordance with institutional animal protocols. Ten million RPE and BJ cells were suspended in 200 μl Matrigel (BD Bioscience, Heidelberg, Germany) and injected subcutaneously into the left flank of 6-week-old female NOD.Cg-Prkdc(scid)Il2rg(tm1Wjl)/SzJ mice (The Jackson Laboratory, Bar Harbor, Maine, USA). Tumor growth was monitored using caliper measurements, and tumor volume was calculated using the formula 3.14159 × length × width 2 / 6000. Mice were sacrificed by CO2 asphyxiation 35 days after transplantation or when tumor size exceeded 2,000 mm3. For secondary xenografts, primary xenografts were manually dissected and dissociated using 2 mg/ml collagenase in PBS for 30 min at 37 °C. Dissociated cell suspensions were filtered using 40 μm nylon mesh filters, and cryopreserved using 10% dimethyl sulfoxide (DMSO), 40% FBS and 50% DMEM-HG. For doxycycline treatment of mice, animals were fed 625 Doxycycline chow with weekly replacement (Harlan, Indianapolis, IN, USA). Photographs of mice and tumors were taken with a Nikon D3100 camera (Minato, Tokyo, Japan).
Analysis of published gene-expression arrays
The R2 visualization and analysis platform (http://r2.amc.nl) was used to re-analyze published HG-U133 Plus 2.0 microarray gene expression data from normal and tumor human tissues, with the analyzed data sets listed in the Supplementary Table 7.
Flanking sequence exponential anchored (FLEA) PCR
Transposon mapping using flanking sequence exponential anchored (FLEA) PCR were done as previously described 53.
Chromatin immunoprecipitation and sequencing (ChIP-seq)
ChIP was performed as previously described 54. Briefly, cells were fixed in 1% formalin in phosphate-buffered saline (PBS) for 10 minutes at room temperature. Glycine (125 mM final concentration) was added to the cells and cells were washed twice in ice-cold PBS and resuspended in sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1). Lysates were sonicated using the Covaris S220 adaptive focused sonicator to obtain 100-500 bp chromatin fragments (Covaris, Woburn, CA). Lysates containing sheared chromatin fragments were resuspended in 0.01 % SDS, 1.1 % Triton-X100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl. Rabbit anti-PGBD5 antibody was coupled to protein A and G Dynabeads according to the manufacture's protocol (Thermo Fisher Scientific, Waltham, MA). Lysates and antibody-coupled beads were incubated over night at 4 °C. Precipitates were washed sequentially with ice cold low salt washing solution (0.1 % SDS, 1 % Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high salt washing solution (0.1 % SDS, 1 % Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl washing solution (0.25 M LiCl, 1% IGEPAL CA-630, 1 % deoxycholic acid, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and Tris-buffered EDTA washing solution (1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and eluted in elution buffer (1 % SDS, 0.1 M NaHCO3). ChIP-seq libraries were generated using the NEBNext ChIP-seq library prep kit following the manufacturer's protocol (New England Biolabs, Ipswich, MA, USA). Libraries were sequenced on the Illumina HiSeq 2500 instruments, with 30 million 2 × 50 bp paired reads.
ChIP-seq analysis
Reads were trimmed for both quality and adapter sequences, with paired reads removed if either read length became less than twenty nucleotides. Bowtie2 (v2.2.2) with default parameters was used to align the reads to human reference assembly hg19, and PCR and optical duplicates were removed with Picard (http://picard.sourceforge.net). Genomic segments enriched for ChIP over input signal were classified using MACS (v1.4) with the default parameters, and genomic ‘blacklisted’ regions were subsequently filtered (http://www.broadinstitute.org/∼anshul/projects/encode/rawdata/blacklists/hg19-blacklist-README.pdf). Signal in enriched regions was then normalized to segment length and sequencing depth.
Whole-genome DNA sequencing
Genomic DNA was extracted using the PureLink Genomic DNA Mini Kit according to the manufacturer's instructions (Thermo Fisher Scientific). Genome sequencing libraries were constructed with the TruSeq Nano library kit following the manufacturer's protocol (Illumina, San Diego, CA, USA). Genomes were sequenced on the Illumina HiSeq X instruments, with 2 × 150 bp paired reads. For analysis of primary patient rhabdoid tumor genomes, sequencing files were downloaded from the TARGET Data Matrix, as previously described 10. Reads were aligned to the GRCh37 human reference using the Burrows-Wheeler Aligner (BWA aln and BWA MEM for GATK and laSV analyses, respectively) and processed using the best-practices pipeline that includes marking of duplicate reads with Picard tools (http://picard.sourceforge.net), and realignment around indels and base recalibration via Genome Analysis Toolkit (GATK) ver. 3.2.2 55,56.
Alignment-based mutational and structural variant analysis
MuTect v1.1.4 57, LoFreq v2.0.0 58 (SNVs only), Strelka v1.0.13 59 (both SNVs and indels), Pindel v0.2.5 and Scalpel v0.4 (indels only) were used with the default filtering criteria as implemented in each of the programs. Tri-allelic SNVs and common germline variants (>1 % MAF in 1000 Genomes Project release 3 or the Exome Aggregation Consortium server [http://exac.broadinstitute.org]), as well as a blacklist of recurrent artifactual calls seen in HapMap samples sequenced and analyzed with the same methodology were filtered out. The union of all SNV and indel calls was annotated using snpEff, snpSift 60 and GATK VariantAnnotator according to the annotation from ENSEMBL, COSMIC, 1000 Genomes, and ExAC 61,62. Copy number variant (CNV) were detected with BIC-seq2 63. DELLY v0.6.1 64, CREST v1.0 65, and BreakDancer v1.4.0 66 were used to detect structural variants (SVs). Bedtools pairtopair 67 was used to merge structural variants. Germline variants from the 1000 Genomes Project call set, Database of Genomic Variants and a blacklist of SVs seen in HapMap genomes were filtered out. SplazerS was used for the analysis of split reads 68, and SV breakpoints were annotated with coinciding BIC-seq2 CNV changepoints. SVs with split read support (tumor only), with at least one coinciding (within 500bp) CNV changepoint called by two or more tools or called by CREST are marked as higher confidence. The annotation with gene overlap (RefSeq, Cancer Gene Census) including prediction of potential effect on genes (e.g. disruptive/exonic, intronic, intergenic) and with annotated transposons was done using bedtools 67.
laSV
De novo assembly-based laSV was used with the following parameters: -s 15 -k 63 -p 3 33. Structural variants supported by less than 4 reads or with allele frequencies below 10% were filtered. Variant recurrence was measured in 100 kb bins using bedtools 67. Circos plots were generated using circos version 0.67-4 69. Scripts used in this analysis are openly available at: https://github.com/kentsisresearchgroup/Rhabdoid_PGBD5_MSK_paper.
SMuFin
SMuFin was used with default parameters as previously described 34. SMuFin results included single nucleotide variants (SNVs), as well as small (indels) and large structural variants (SVs). Large SVs were defined as structural variants identified with a single breakpoint, where the SV length exceeded the length of the underlying variant block called by SMuFin. Breakpoints supported by less than 4 reads were filtered. SV size was estimated assuming that SVs were caused by single genomic events.
Regulatory element analysis
Annotated regulatory elements compiled from both ENCODE and NIH Roadmap Epigenomics Consortium were downloaded from http://www.encodeproject.org/data/annotations/v2. The analysis focused on distal DNase I hypersensitivity sites, as distal sites have been shown to vary in a more cell type-specific manner, and DNase I sensitivity covers both active and poised regulatory elements. Cancer cell line data sets were removed, and the overlap of at least one base pair was calculated between breakpoints and DNase I hypersensitivity peaks in each cell type. In order to account for cell types with variable DNase I hypersensitive sites, the overlap count for each cell type was normalized to the total number of regulatory sites in that cell type.
PGBD5 signal sequence (PSS) analysis
The position weight matrix (PWM) for the PGBD5 signal sequence (PSS) and RAG1 recombination signal sequence (RSS) were generated as described 32. These PWMs were used to scan sequences around variant breakpoints (+/- 50 bp) for both PSS and RSS using the sequence motif match algorithm FIMO 70. Additionally, PGBD5 signal sequence motifs associated with structural variants were detected by analyzing 20 bp windows around variant breakpoints using MEME with default parameters 71. Matches with false discovery rate < 0.1 and within 15 bp from the variant breakpoints were retained and counted. All variants associated with PSS motif were manually verified. To construct the position-scrambled PSS, the perl rand function was used to generate 10 independent position-scrambled PWMs.
Statistical analysis
All experiments were performed a minimum of three times with a minimum of three independent measurements. For comparisons between two sample sets, statistical analysis of means was performed using two-tailed, unpaired Student's t-tests. Survival analysis was done using the Kaplan-Meier method, as assessed using a log-rank test. For gene expression analysis, statistical significance was assessed using paired t-tests. False discovery was assessed at the 0.05 level using the step-down Dunnett method, as extended to general parametric models 72,73. Significance of sequence motif enrichment was assessed using hypergeometric tests. For significance analysis of association of structural variants with regulatory elements, Welch's t-test was used. Calculations were performed using the R Project for Statistical Computing 74.
Supplementary Material
Acknowledgments
We are grateful to Alejandro Gutierrez, Marc Mansour, Daniel Bauer, Thomas Look, Hao Zhu, Cedric Feschotte, Michael Kharas, John Petrini and Maria Gil Mir for critical discussions, and John Gilbert for editorial advice. This work was supported by the NIH K08 CA160660, P30 CA008748, U54 OD020355, UL1 TR000457, P50 CA140146, Spanish Ministerio de Economía y Competitividad SAF2014-60293-R, Cancer Research UK, Wellcome Trust, Starr Cancer Consortium, Burroughs Wellcome Fund, Sarcoma Foundation of America, Matthew Larson Foundation, Josie Robertson Investigator Program, and Rita Allen Foundation. A.H. is supported by the Berliner Krebsgesellschaft e.V. and the Berlin Institute of Health. A.K. is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator.
Footnotes
Author Contributions: AGH study design and collection and interpretation of the data, RK ChIP-seq, whole genome sequencing and FLEA-PCR data analysis, JZ tumor genome sequencing data analysis with laSV, EJ in vitro transformation assays and vector design and cloning, CR in vitro transformation assays and vector design and cloning, AE in vitro transformation assays and vector design and cloning, ES in vitro transformation assays and vector design and cloning, ERF genome sequencing data analysis, SG genome sequencing data analysis, MP genome sequencing data analysis, ANB creation of PAXX deficient cells and study design, CEM genome sequencing data analysis, EDS mouse xenograft study design, MG statistical analysis of datasets, AKE genome sequencing data analysis, MS genome sequencing data analysis, KA genome sequencing data analysis, CRe genome sequencing data analysis, NDS genome sequencing data analysis, EP study design, CRA histological analysis of tumor samples, CWMR study design, HS study design, EM study design, SPJ creation of PAXX-deficient cells and study design, DT genome sequencing data analysis, ZW genome sequencing data analysis, SAA study design, and AK study design, data analysis and interpretation. AK and AGH wrote the manuscript with contributions from all authors.
Competing Financial Interests: There are no competing financial interests of any of the authors.
Data Availability and Accession Codes: Genome and chromatin immunoprecipitation sequencing data have been deposited to the NCBI Sequence Read Archive and Gene Expression Omnibus databases (Bioproject 320056 and DataSet GSE81160, respectively). Analyzed data are openly available at the Zenodo digital repository (http://dx.doi.org/10.5281/zenodo.50633).
References
- 1.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research N et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Futreal PA, et al. A census of human cancer genes. Nature reviews Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huether R, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature communications. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Northcott PA, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansour MR, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014 doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molenaar JJ, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 9.Johann PD, et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer cell. 2016;29:379–393. doi: 10.1016/j.ccell.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Chun HJ, et al. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer cell. 2016;29:394–406. doi: 10.1016/j.ccell.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones DT, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer HP, Thomsen H, Altmannsberger M, Bertram U. Malignant rhabdoid tumour of the kidney expressing neurofilament proteins. Immunohistochemical findings and histogenetic aspects. Pathology, research and practice. 1989;184:541–547. doi: 10.1016/S0344-0338(89)80149-9. [DOI] [PubMed] [Google Scholar]
- 13.Lee RS, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of clinical investigation. 2012 doi: 10.1172/JCI64400. In Press. doi:64400[pii]10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Heuvel-Eibrink MM, et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011;56:733–737. doi: 10.1002/pbc.22922. [DOI] [PubMed] [Google Scholar]
- 15.Versteege I, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 16.Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. doi:S153561080200185X. [pii] [DOI] [PubMed] [Google Scholar]
- 17.Rousseau-Merck MF, Fiette L, Klochendler-Yeivin A, Delattre O, Aurias A. Chromosome mechanisms and INI1 inactivation in human and mouse rhabdoid tumors. Cancer genetics and cytogenetics. 2005;157:127–133. doi: 10.1016/j.cancergencyto.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Takita J, et al. Genome-wide approach to identify second gene targets for malignant rhabdoid tumors using high-density oligonucleotide microarrays. Cancer science. 2014;105:258–264. doi: 10.1111/cas.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 20.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 21.Rodic N, et al. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1060–1064. doi: 10.1038/nm.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muotri AR, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 23.Shaheen M, Williamson E, Nickoloff J, Lee SH, Hromas R. Metnase/SETMAR: a domesticated primate transposase that enhances DNA repair, replication, and decatenation. Genetica. 2010;138:559–566. doi: 10.1007/s10709-010-9452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- 25.Navarro JM, et al. Site- and allele-specific polycomb dysregulation in T-cell leukaemia. Nature communications. 2015;6:6094. doi: 10.1038/ncomms7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papaemmanuil E, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nature genetics. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halper-Stromberg E, et al. Fine mapping of V(D)J recombinase mediated rearrangements in human lymphoid malignancies. BMC Genomics. 2013;14:565. doi: 10.1186/1471-2164-14-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreyer WJ, Gray WR, Hood L. The Genetics, Molecular, and Cellular Basis of Antibody Formation: Some Facts and a Unifying Hypothesis. Cold Spring Harb Symp Quant Biol. 1967;32:353–367. [Google Scholar]
- 29.Majumdar S, Singh A, Rio DC. The human THAP9 gene encodes an active P-element DNA transposase. Science. 2013;339:446–448. doi: 10.1126/science.1231789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henssen AG, et al. Genomic DNA transposition induced by human GBD5. eLife. 2015;4 doi: 10.7554/eLife.10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavelitz T, Gray LT, Padilla SL, Bailey AD, Weiner AM. GBD5: a neural-specific intron-containing piggyBac transposase domesticated over 500 million years ago and conserved from cephalochordates to humans. Mob DNA. 2013;4:23. doi: 10.1186/1759-8753-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henssen AG, et al. Forward genetic screen of human transposase genomic rearrangements. BMC Genomics. 2016;17:548. doi: 10.1186/s12864-016-2877-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhuang J, Weng Z. Local sequence assembly reveals a high-resolution profile of somatic structural variations in 97 cancer genomes. Nucleic acids research. 2015;43:8146–8156. doi: 10.1093/nar/gkv831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moncunill V, et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotechnol. 2014;32:1106–1112. doi: 10.1038/nbt.3027. [DOI] [PubMed] [Google Scholar]
- 35.Bralten LB, et al. The CASPR2 cell adhesion molecule functions as a tumor suppressor gene in glioma. Oncogene. 2010;29:6138–6148. doi: 10.1038/onc.2010.342. [DOI] [PubMed] [Google Scholar]
- 36.Hahn WC, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 37.Landree MA, Wibbenmeyer JA, Roth DB. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes & development. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu CP, Posey JE, Roth DB. Understanding how the V(D)J recombinase catalyzes transesterification: distinctions between DNA cleavage and transposition. Nucleic acids research. 2008;36:2864–2873. doi: 10.1093/nar/gkn128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 40.Mitra R, Fain-Thornton J, Craig L. piggyBac can bypass DNA synthesis during cut and paste transposition. The EMBO journal. 2008;27:1097–1109. doi: 10.1038/emboj.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochi T, et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015;347:185–188. doi: 10.1126/science.1261971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta. 2014;1846:188–200. doi: 10.1016/j.bbcan.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mc CB. The origin and behavior of mutable loci in maize. Proceedings of the National Academy of Sciences of the United States of America. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torchia J, et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer cell. 2016;30:891–908. doi: 10.1016/j.ccell.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, et al. piggyBac transposase tools for genome engineering. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2279–2287. doi: 10.1073/pnas.1305987110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westbrook TF, Stegmeier F, Elledge SJ. Dissecting cancer pathways and vulnerabilities with RNAi. Cold Spring Harb Symp Quant Biol. 2005;70:435–444. doi: 10.1101/sqb.2005.70.031. [DOI] [PubMed] [Google Scholar]
- 47.Ferguson BW, et al. The cancer gene WWOX behaves as an inhibitor of SMAD3 transcriptional activity via direct binding. BMC Cancer. 2013;13:593. doi: 10.1186/1471-2407-13-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kentsis A, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18:1118–1122. doi: 10.1038/nm.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fox R, Aubert M. Flow cytometric detection of activated caspase-3. Methods in molecular biology. 2008;414:47–56. doi: 10.1007/978-1-59745-339-4_5. [DOI] [PubMed] [Google Scholar]
- 50.Sordet O, et al. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood. 2002;100:4446–4453. doi: 10.1182/blood-2002-06-1778. [DOI] [PubMed] [Google Scholar]
- 51.Yarilin D, et al. Machine-based method for multiplex in situ molecular characterization of tissues by immunofluorescence detection. Scientific reports. 2015;5:9534. doi: 10.1038/srep09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujisawa S, Turkekul M, Barlas A, Fan N, Manova K. Double in situ detection of sonic hedgehog mRNA and pMAPK protein in examining the cell proliferation signaling pathway in mouse embryo. Methods in molecular biology. 2011;717:257–276. doi: 10.1007/978-1-61779-024-9_15. [DOI] [PubMed] [Google Scholar]
- 53.Henssen A, Carson JR, Kentsis A. Transposon mapping using flanking sequence exponential anchored (FLEA) PCR. Nature Protocol Exchange. 2015 doi: 10.1038/protex.2015.071. [DOI] [Google Scholar]
- 54.Krivtsov AV, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilm A, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic acids research. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 60.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narzisi G, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nature methods. 2014;11:1033–1036. doi: 10.1038/nmeth.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xi R, et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E1128–1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emde AK, et al. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics. 2012;28:619–627. doi: 10.1093/bioinformatics/bts019. [DOI] [PubMed] [Google Scholar]
- 69.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome research. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic acids research. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 73.Dunnett CW, Tamhane AC. Step-down multiple tests for comparing treatments with a control in unbalanced one-way layouts. Stat Med. 1991;10:939–947. doi: 10.1002/sim.4780100614. [DOI] [PubMed] [Google Scholar]
- 74.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. 2013 http://www.R-project.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.