

Abstract
Bithalamic gliomas are rare cancers diagnosed based on poorly defined radiologic criteria. Infiltrative astrocytomas account for most cases. While some previous studies reported dismal outcomes for patients with bithalamic gliomas irrespective of therapy and histologic grade, others described better prognoses even without anticancer therapy. Little is known about their molecular characteristics. We reviewed clinical, radiologic, and histologic features of patients with bithalamic gliomas treated at our institution over 15 years. Targeted sequencing of mutational hotspots in H3F3A, HIST1H3B, IDH1/2, and BRAF, and genome‐wide analysis of DNA methylation and copy number abnormalities was performed in available tumors. Eleven patients with bithalamic gliomas were identified. Their median age at diagnosis was 4.8 years (range: 1–15.7). Additional involvement of the brainstem, basal ganglia, and cerebral lobes occurred in 11, 9, and 3 cases, respectively. All patients presented with hydrocephalus. Two‐thirds of the patients had a histologic diagnosis of anaplastic astrocytoma. Despite aggressive therapy, our youngest patient, the only one diagnosed before 1 year of age, is the sole long‐term survivor. DNA methylation could be performed in seven tumors, all of which clustered with the RTK I ‘PDGFRA’ subgroup by unsupervised hierarchical analysis of methylation array against a previously published cohort of 59 pediatric high‐grade gliomas. Sequencing of hotspots mutations could be done in 10 tumors, none of which harbored H3F3A p.K27 and/or the respective DNA methylation signature, and any other hotspot mutations. Amplification of MDM4 (n = 2), PDGFRA (n = 2), and ID2 combined with MYCN (n = 1) were observed in 7 tumors available for analysis. In comparison with the previously published experience with unilateral high‐grade thalamic astrocytomas where H3F3A p.K27 was present in two‐thirds of cases, the absence of this molecular subgroup in bithalamic gliomas was striking. This finding suggests that unilateral and bithalamic high‐grade gliomas may represent two distinct molecular entities.
Keywords: bithalamic, children, high‐grade glioma, infiltrative, molecular characteristics
Introduction
Thalamic tumors account for approximately 5% of all central nervous system (CNS) cancers in children and consist mostly of astrocytomas with unilateral involvement 4, 17, 19, 23. Bithalamic gliomas represent a rare subset of poorly understood tumors diagnosed based on symmetrical involvement of both thalami 5, 7, 8, 14, 17, 19, 23. Although histologic confirmation is warranted for the diagnosis of bithalamic gliomas since their imaging characteristics overlap with those of other oncologic and non‐oncologic entities, 13 some studies have included patients with only radiologic diagnosis 5, 14, 23. Infiltrative low‐ and high‐grade astrocytomas account for most bithalamic gliomas 5, 8, 14, 17, 19, 23. The treatment and prognosis of patients with bithalamic gliomas remains controversial 5, 8, 14, 17, 19, 23. While some studies reported a dismal outcome for patients with bithalamic gliomas irrespective of therapy and histologic grade, 8, 19 others have reported long‐term survivors, including patients who underwent only tumor biopsy and/or decompression of hydrocephalus 5, 14, 17, 23.
Very little is known about the molecular characteristics of bithalamic gliomas since suitable tissue for detailed analysis is rarely obtained from small tumor biopsies 1, 27, 28. We conducted clinical, radiologic, and histologic review, and molecular analysis of a cohort of children with bithalamic gliomas. Unlike unilateral high‐grade thalamic astrocytomas, 11, 21, 25, 30 bithalamic gliomas lacked H3F3A p.K27M mutation and/or the respective DNA methylation characteristics, which suggests that both represent separate molecular entities.
Materials and Methods
Following institutional review board approval, we retrospectively reviewed the clinical and radiologic characteristics of all patients younger than 22 years with newly diagnosed bithalamic gliomas treated at our institution from March 1999 until August 1, 2014.
We defined bithalamic gliomas as tumors arising bilaterally and completely involving both thalami. Cases with a predominant unilateral involvement and partial spread to the contralateral side and those who developed a bithalamic appearance only at the time of progression were excluded from this analysis. Only patients with diffusely infiltrating gliomas were included in this study and the histologic diagnosis of pilocytic astrocytoma was an exclusion criterion.
Detailed clinical and therapy‐related data were collected for all patients. Brain MRIs at diagnosis of suspected cases were selected by a neuro‐oncologist (AB) and then independently reviewed by a neuro‐radiologist (SNH). A scoring system was used to assess the extent of gray matter involvement by T2‐weighted and/or FLAIR MRI sequences in the thalami, deep‐seated structures (ie, lentiform nucleus, caudate nucleus, insula, brainstem, and cerebellum), and cerebral lobes as previously described 6. A score of 1 was attributed to involvement of each unilateral structure, the brainstem, and cerebellum. Assessment of the presence of tumor mass was based on T1‐ and T2‐weighted/FLAIR signal characteristics independent of the presence of contrast enhancement. All cases underwent histologic review by a board‐certified neuro‐pathologist (BAO) according to the 2016 World Health Organization (WHO) classification. Immunoreactivity of H3K27M was tested on 4‐μm formalin fixed paraffin‐embedded (FFPE) sections as previously described using a polyclonal antibody (Millipore Sigma, Darmstadt, Germany, catalog number ABE419, 1:400) 28. Immunohistochemistry of p53 (Zeta Corporation Arcadia, CA, USA, clone DO‐7, 1:200), ATRX (Sigma, catalog number HPA001906, 1:600), and H3K27me3 (Cell Signaling Technology, Denver, MA, USA, C36B11, 1:200) were performed according to the manufacturer's specifications.
Molecular studies
Dual‐color fluorescence in situ hybridization (FISH) was performed on 4‐µm formalin‐FFPE tissue sections. Break‐apart and fusion probes for FGFR1 were derived from BAC clones RP11‐246A12 and RP11‐118H9 (BACPAC Resources, Oakland, CA). Probes were labeled with either AlexaFluor‐488 or AlexaFluor‐555 fluorochromes and nuclei were counterstained with DAPI (200 ng/mL; Vector Laboratories Inc., Burlingame, CA) for viewing on an Olympus BX51 fluorescence microscope equipped with a 100‐W mercury lamp; FITC, Rhodamine, and DAPI filters; 100X PlanApo (1.40) oil objective; and a Jai CV digital camera. Images were captured and processed using the Cytovision v7.3 software (Leica Biosystems, Inc., Buffalo Grove, IL, USA).
DNA was extracted from FFPE tissue using the Maxwell® 16 Plus LEV DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. DNA was quantified using the Qubit dsDNA BR assay kit (ThermoFisher Scientific, Grand Island, NY, USA).
Targeted sequencing of BRAF p.V600, H3F3A p.K27, H3F3A p.G34, HIST1H3B p.K27, IDH1 p.R132, and IDH2 p.R172 was performed as previously described 6, 29
Illumina infinium human 450 k bead array
Processing and acquisition of DNA methylation data were performed as previously described 6.
Analysis of DNA methylation data was performed using the open source statistical programming language R 18. Files with raw data generated by the iScan microarray scanner (Illumina, San Diego, CA) were read and processed using the minfi Bioconductor package as described in the Illumina GenomeStudio software (Illumina, San Diego, CA, USA) 2. Further filtering of the probes was done as previously described 25. In total, 438 370 probes were kept for clustering analysis. To determine the subgroup affiliation of our cohort by methylation array, we used previously published data of DNA methylation in pediatric high‐grade gliomas as a reference (n = 59; GSE36278) 25. Missing values were imputed using a k‐nearest neighbor algorithm 26. We selected the top 10 000 most variable methylation probes as measured by standard deviation across the combined dataset to conduct an unsupervised hierarchical clustering of bithalamic tumors and 59 reference samples. Samples were then clustered by applying the Ward's method and dissimilarity based on Euclidean distance. Methylation probes in the heatmap were reordered by hierarchical clustering using a similar approach.
We compared the methylation profile of tumors (n = 7) in the current manuscript to that of previously published unilateral thalamic high‐grade gliomas (n = 8) 25. We used the minfi package to obtain normalized M‐values for the methylation profile of each tumor 2. For each gene, we used Illumina annotation to identify all probes within the promoter region defined as the area within 1000 base pairs upstream of the transcription start site. For each gene and tumor, we then computed the average M‐value for this promoter region. Next, for each gene promoter region, we used the Welch t‐test to compare the average promoter M‐values of bithalamic (n = 7) and unilateral tumors (n= 8). To address multiple‐testing, we used a robust method to estimate the false discovery rate as a function of P‐value 16. To gain further biological insight from our study, we used the multi‐response permutation procedure (MRPP) to evaluate differential methylation of 9063 gene‐sets defined in the molecular signatures data base (MsigDB; http://software.broadinstitute.org/gsea/msigdb/) with statistical significance determined by 10 000 permutations of group label assignments 15.
Analysis of copy number abnormalities based on the 450 k Infinium methylation array was performed by using the conumee Bioconductor package 12. The combined intensities of all available CpG probes were normalized against 12 control samples from normal brain tissue using a linear‐regression approach. Probes were combined into genomic bins of 10 kb. Bins comprised of less than 10 probes were repeatedly merged with the adjacent bins. Subsequent analysis was performed with default setting in the conumee package. Detection of amplification and chromosomal gains and losses was performed by manual assessment of the respective loci for each individual profile following automatic scoring 6, 25.
A logistic regression model to estimate the probability of MGMT promoter methylation from the 450 k methylation array was performed as previously described 3.
Tumor and germline samples from one patient included in this study had already undergone whole exome sequencing as part of the Pediatric Cancer Genome Project 30.
Other statistical analyses
Descriptive statistical analyses were used to summarize the demographic and clinical characteristics of the patients. Overall survival (OS) was defined as the interval between date of diagnosis and death. Patients who were alive were censored at the time of last contact. The Wilcoxon rank sum test was used to assess the associations between categorical variables (ie, gender, presence of tumor mass, and contrast enhancement) and continuous variables (ie, age at diagnosis, interval from onset of symptoms and diagnosis, and number of gray matter areas affected). The Kruskal–Wallis test was used to test any associations between histologic diagnosis and the same continuous variables. Cox models were used to assess for associations between OS and continuous variables and two‐sided log‐rank tests were used for categorical variables.
Results
We identified 11 patients (5 [45%] females) with bithalamic gliomas (Table 1). The median age at diagnosis was 4.8 years (range: 1.0–15.7). Only one patient was younger than one 1 year at diagnosis. The median latency from onset of symptoms to diagnosis was 30 days (range: 1–120 days). Patient 4 had a 24‐h history precipitated by massive intra‐tumoral hemorrhage. Most patients presented with signs of increased intracranial pressure. Only two patients had complaints of new pyramidal signs. None of the patients had seizures at presentation.
Table 1.
Clinical, radiologic, and histologic characteristics of all patients.
| Patient no. | Age at diagnosis (yrs)/gender | Interval between onset of symptoms and diagnosis (days) | Race/ethnicity | No. of gray matter areas affected | Histologic diagnosis/surgery | Therapy | Survival (yrs) |
|---|---|---|---|---|---|---|---|
| 1 | 1.0/F | 120 | Caucasian | 3 | AA/biopsy | Chemotherapy | 7.2 + |
| 2 | 2.4/M | 14 | Caucasian | 3 | Glioblastoma/STR | Chemotherapy | 0.5 |
| 3 | 2.4/M | 21 | Caucasian | 5 | AA/biopsy | RT + chemotherapy | 0.8 |
| 4 | 3.3/M | 1 | Mixed race | 4 | HGG with small cell features/STR | RT + chemotherapy | 1.0 |
| 5 | 4.3/F | 60 | Hispanic | 6 | AA/biopsy | RT + chemotherapy | 0.75 |
| 6 | 4.8/M | 5 | African‐American | 6 | GBM/STR | RT + chemotherapy | 1.0 |
| 7 | 5.4/M | 7 | Caucasian | 6 | AA/biopsy | RT + chemotherapy | 1.2 |
| 8 | 9.8/F | 35 | Caucasian | 7 | AA/biopsy | RT + chemotherapy | 1.5 |
| 9 | 10.7/M | 30 | Caucasian | 6 | AA/biopsy | RT + chemotherapy | 1.2 |
| 10 | 14.9/F | 42 | African‐American | 7 | Diffuse astrocytoma WHO grade II/biopsy | RT + chemotherapy | 1.75 |
| 11 | 15.7/F | 30 | Caucasian | 5 | AA/biopsy | RT + chemotherapy | 1.0 |
Abbreviations: No. = number; yrs = years; F = female; M = male; AA = anaplastic astrocytoma; GBM = glioblastoma; HGG = high‐grade glioma; WHO = World Health Organization; STR = subtotal resection; RT = radiation therapy.
Figure 1A shows typical MRI characteristics observed in patient 11. All patients presented with hydrocephalus which was decompressed by placement of a ventriculo‐peritoneal shunt (n = 5) or third ventriculostomy (n = 5). Patient 4 had improvement of hydrocephalus following subtotal resection of the tumor. Tumor infiltration of the brainstem and basal ganglia were observed in 11 and 9 patients, respectively (Figure 1B). Patients 8, 10, and 11 had additional involvement of 2, 1, and 2 cerebral lobes at diagnosis, respectively. Distinct tumor masses were observed within the infiltrated thalami in 5 patients, 4 of which enhanced after contrast administration. Patients had a median of 6 (range: 3–7) gray matter areas involved at diagnosis.
Figure 1.
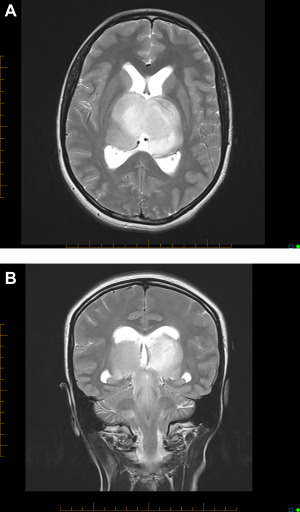
Axial T2‐weighted brain MRI of patient 11 showing symmetrical bithalamic involvement (A). This patient was also found to have hydrocephalus based on symptoms and enlargement of lateral ventricles with transependymal flow in their frontal horns. Coronal T2‐weighted brain MRI of same patient showing symmetrical bithalamic and midbrain involvement (B).
All patients underwent histologic confirmation (Table 1). The majority of samples analyzed consisted of at least 80% tumor cells. Two‐thirds (7 of 11) of the patients had a histologic diagnosis of anaplastic astrocytoma (WHO grade III). Three patients had a glioblastoma (WHO grade IV). Only one patient had a diffuse astrocytoma (WHO grade II; patient 10, Figure 2). All tumors consisted of infiltrative astrocytomas with round to oval nuclear morphology. The tumors lacked specific histologic findings except for one case which demonstrated a small‐cell phenotype (patient 4) and another that had scattered multinucleated giant cells (patient 9). Expression of p53 by immunohistochemistry could be assessed in 6 cases, one of which (patient 5) displayed a strong and diffuse pattern suggestive of a mutant TP53. While ATRX protein loss was observed in one (patient 4) of 5 cases, mutant H3K27M was not detected in 5 tumors with available tissue (Figure 2). H3K27me3 demonstrated heterogeneous expression in 5 tumors tested.
Figure 2.
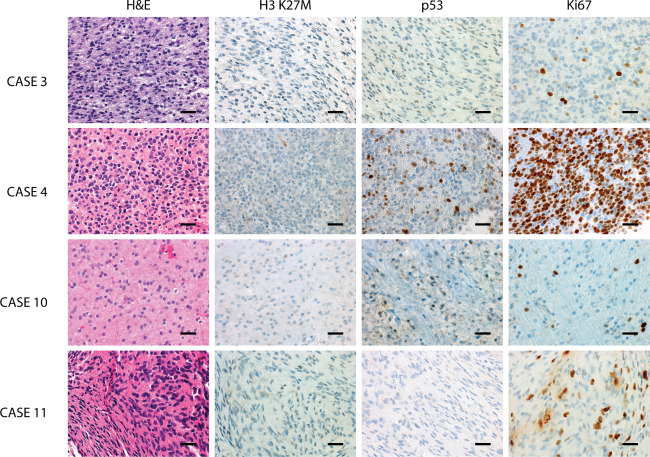
Representative histomorphology and immunophenotype of bithalamic gliomas demonstrating infiltrative growth and moderate cytologic pleomorphism. While tumors lacked immunoreactivity for H3K27M, p53 expression was mostly seen in a minority of tumor cells. The proliferative fraction, as assessed by Ki67/Mib‐1 immunolabeling, was variable from moderately to very high frequency. The scale bars represent 200 μm.
Molecular studies
Unsupervised hierarchical clustering of genome‐wide methylation was conducted in 7 (64%) tumors for which adequate DNA was available. By comparing with a previously published dataset of pediatric high‐grade gliomas, 25 our cohort clustered with the RTK I ‘PDGFRA’ subgroup (Figure 3 and Table 2). A subgroup representing either H3F3A p.K27‐ or p.G34‐mutated tumors was not observed. Only one tumor in the RTK I ‘PDGFRA’ subgroup displayed MGMT promoter methylation (Table 2).
Figure 3.
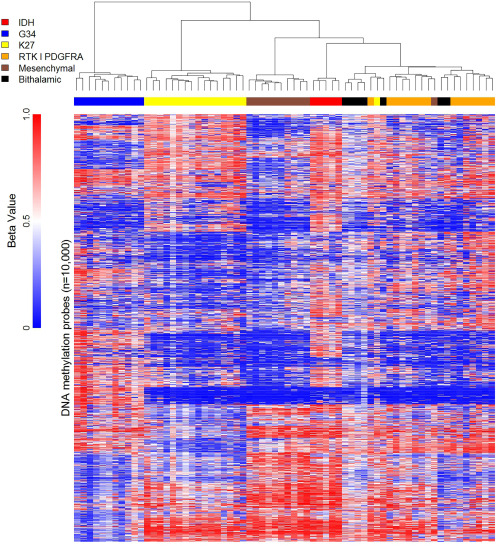
Heatmap representation of unsupervised hierarchical clustering analysis of 450 k methylation array profiles in 7 pediatric patients with bithalamic gliomas in comparison with a dataset of 59 previously published pediatric high‐grade gliomas.
Table 2.
Molecular characteristics of evaluable tumors.
| Patient no. | Methylation subgroup | MGMT promoter methylation | IDH1 p.R132H | H3F3A p.G34 | H3F3A p.K27 | HIST1H3B p.K27 | CDKN2A Loss | Other relevant molecular abnormalities |
|---|---|---|---|---|---|---|---|---|
| 1 | NA | NA | No | No | No | NA | NA | NA |
| 3 | RTK I ‘PDGFRA’ | Methylated | No | No | No | No | No | No |
| 4 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | PDGFRA amplification |
| 5 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | PDGFRA and EGFR amplification |
| 6* | NA | NA | No | No | No | No | No | PDGRFA (structural variation), EGFR G598V (LOH) and TP53 R175H (LOH) |
| 7 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | MDM4, ID2, and MYCN amplification |
| 8 | NA | NA | No | NA | No | NA | NA | NA |
| 9 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | No |
| 10 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | MDM4 amplification |
| 11 | RTK I ‘PDGFRA’ | Unmethylated | No | No | No | No | No | CDK6 amplification |
*Patient whose tumor previously underwent whole exome sequencing.
Abbreviations: No. = number; MGMT = methylguanine methyltransferase; NA = not available; LOH = loss of heterozygosity.
We observed 364 genes with differentially methylated promoter regions comparing unilateral and bithalamic tumors (estimated false discovery rate of 0.001) (Supporting Information Table 1). Thirty‐two of these genes exhibited greater methylation in bithalamic compared with unilateral tumors. The remaining 332 genes exhibited greater methylation in unilateral tumors. The top 100 genes sets that differed between the two groups are presented in Supporting Information Table 2.
Sequencing of hotspot mutations was available in 10 (91%) patients. No mutations were observed in H3F3A p.K27, H3F3A p.G34, HIST1H3B p.K27, IDH1 p.R132, IDH2 p.R172, and BRAF p.V600 (Table 2). EGFR and TP53 mutations were demonstrated in the tumor that had previously undergone whole exome sequencing 30 (Table 2).
Amplification of multiple oncogenes including MDM4 (n = 2), PDGFRA (n = 2), CDK6 (n = 1), EGFR (n = 1), and a combination of ID2 and MYCN (n = 1) was observed (Table 2). FGFR1 fusions were not detected using break‐apart FISH probes in 6 tumors with sufficient material. Further confirmation of copy number abnormalities by FISH was not possible due to limited amount of leftover tumor. The most common areas of large gains in 7 tumors were chromosome 7 (71%), 1q (57%), chromosome 2 (43%), 17q (43%), and chromosome 8, 9q, 12p, 13q, and 15q in 28.5% of tumors each. Likewise, the most common areas of large chromosomal losses were 4q (43%), 17p (43%), and 11q, 14q, and 16q in 28.5% of tumors each.
Outcome
Despite aggressive treatment with chemotherapy with or without radiation therapy (RT), (Table 1) only the patient diagnosed as an infant remains alive 7.2 years after diagnosis (Figure 4). Eight patients experienced local disease progression only. Patient 7 experienced massive leptomeningeal spread in his spine following local RT. The pattern of disease progression was not documented in one case. The median OS for all patients was 1 year (range: 0.5 to 7.2 + years). In this cohort, female patients were more likely to have longer intervals from onset of symptoms to diagnosis (median 42 vs. 10.5 days; P = 0.0065). None of the continuous and categorical variables analyzed showed any association with each other or with OS likely due to small sample size.
Figure 4.
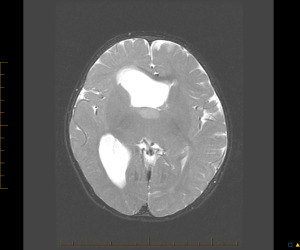
Axial T2‐weighted brain MRI of the only long‐term survivor (patient 1) at diagnosis. A tumor biopsy was obtained from the T2 hyperintense anterior portion of the right thalamus.
Discussion
We report for the first time detailed molecular characteristics of one of the largest cohorts to date of patients with bithalamic gliomas. Since this is a rare and poorly defined entity, previous studies contained a maximum of 15 affected subjects, even when reviewing the experience within entire countries 5, 8, 14, 17, 19, 23 (Table 3). The most remarkable molecular finding of our study was the complete lack of H3F3A p.K27 and/or the respective DNA methylation signature. The presence of H3F3A p.K27 and/or expression of the respective protein have been documented by sequencing or immunohistochemistry in approximately 70% of thalamic high‐grade astrocytomas 1, 9, 11, 20, 21, 22, 25, 27, 28 (Table 4). To our knowledge, molecular characteristics of only a few bithalamic high‐grade gliomas, one of which harbored the H3F3A p.K27 mutation, had been previously reported 1, 27, 28, 30. Since most patients with bithalamic gliomas undergo at most a biopsy, their representation in molecular studies would be assumed to be exceptional. Assuming that 70% of unilateral high‐grade thalamic astrocytomas harbor an H3F3A p. K27 mutation, 1, 9, 11, 20, 21, 22, 25, 27, 28 our results are substantially different despite the limited sample size (P = 5.9 × 10−6). The absence of other hotspot mutations including H3F3A p.G34 in our patients was not surprising because they predominantly occur in tumors arising in the cerebral cortex 21, 25, 30. In contrast to diffuse intrinsic pontine gliomas, another common pediatric midline CNS cancer, 11, 30 we did not observe hotspot HIST1H3B mutations in bithalamic gliomas.
Table 3.
Summary of previous series of patients with bithalamic gliomas.
| Author | Year of publication | Number of patients | Median age at diagnosis/range (years) | Histologic diagnosis | Upfront therapy | Outcome |
|---|---|---|---|---|---|---|
| Reardon et al | 1998 | 12 | NA/≤ 18 | LGG: 9; HGG: 3 | Not specified | 3‐year OS = 0% |
| Di Rocco et al | 2002 | 4 | 8/0.25–10 | DA grade II: 3; astrocytoma grade III: 1 | RT: 4 |
Three patients deceased with median survival of 10 months One patient alive after 8 months |
| Puget et al | 2007 | 9 | 9.6 (mean)/NA | PA: 1; LGG: 6; HGG: 2 | RT: 4 |
Four patients deceased Five long‐term survivors received no adjuvant therapy (mean F/U of 4.5 years) |
| Menon et al | 2010 | 7 | 17/5–29 | DA grade II 6; NC 1 | RT: 7 | All patients alive for a median F/U of 1 year (range; 1–5 years) |
| Steinbok et al | 2015 | 10 | 6.6 (mean)/1.5–14.7 | PA: 1; DA grade II: 3; AA: 2; GBM: 3; NC: 1 | Chemotherapy: 5; RT: 3 |
Seven patients deceased with median survival of 11.3 months Three long‐term survivors received no adjuvant therapy |
| Boesten et al | 2016 | 15 | 7.5/2–15.1 | PA: 2; DA grade II: 9; LGG: 1; NC: 3 | Chemotherapy: 3; RT: 3 |
10‐year EFS: 16% ± 13% 10‐year OS: 65% ± 13% |
Abbreviations: NA = not available; NC = not confirmed; LGGs = low‐grade glioma; HGG = high‐grade glioma; DA = diffuse astrocytoma; PA = pilocytic astrocytoma; AA = anaplastic astrocytoma; GBM = glioblastoma; RT = radiation therapy; EFS = event‐free survival; OS = overall survival; F/U = follow‐up.
Table 4.
Summary of H3F3A p.K27 mutations in unilateral high‐grade gliomas identified by sequencing and/or immunohistochemistry.
| Study | Number of patients | Median age at diagnosis (range; years) | % Tumors harboring H3F3A p.K27 |
|---|---|---|---|
| Schwartzentruber et al (2012) | 6 | 10.5 (6–14) | 50% |
| Sturm et al (2012) | 8 | 11 (6–23) | 100% |
| Venneti et al (2013) | 3 | 11.6 (7–21.4) | 100% |
| Aihara et al (2014) | 18 | 42 (17–78) | 55% |
| Fontebasso et al (2014) | 11 | 11 (6–15) | 82% |
| Venneti et al (2014) | 7 | 12 (7–21) | 71% |
| Feng et al (2015)* | 15 | 31.7 (mean; 22–53) | 66.7% |
| Solomon et al (2015) † | 23 | Adults and children | 65% |
| Ryall et al (2016) | 22 | 10.9 (4–17.6) | 50% |
| Total | 113 | 65% |
*Four patients included had a grade II glioma.
†One of 15 tumors harboring an H3F3A p.K27 was a grade 2 glioma. Histologic grade was not provided for 8 tumors.
Although our results showed differentially methylated genes comparing unilateral and bithalamic gliomas, (Supporting Information Tables 1 and 2) including those involved in DNA repair, cell‐cycle progression, and apoptosis, no solid conclusions can be drawn from such preliminary data.
Similar to other studies, 11, 21, 25, 30 we observed multiple other amplified oncogenes (ie, MDM4, MYCN, PDGFRA, and CDK6) in our patients. One tumor (patient 7) had concurrent amplification of MYCN and ID2, a finding recently reported in a subset of supratentorial high‐grade gliomas 24.
In contrast to two previous studies, 8, 14 our patients’ tumors commonly infiltrated adjacent structures, including the midbrain, basal ganglia, and in a few cases even cerebral lobes. The involvement of cerebral lobes in a minority of cases was reminiscent of gliomatosis cerebri, a condition where typical bithalamic involvement was seen in one‐fourth of patients 6. We confirmed that the risk of hydrocephalus is extremely high in bithalamic gliomas.
We acknowledge the bias of the current study since high‐grade gliomas accounted for 90% of cases in contrast to previous reports where low‐grade gliomas predominated 5, 8, 14, 17, 19. Sampling error of tiny biopsies in such extensive tumors probably accounts for some of this difference. We had already shown that even histologic low‐grade bithalamic gliomas portend a dismal prognosis 19. A few previous studies included patients without histologic confirmation, some of which were long‐term survivors despite not receiving any anticancer therapy 5, 14, 17, 23. We suspect that some of these patients with better outcomes might have a pilocytic astrocytoma. Pilocytic astrocytomas can display an infiltrative aspect with bithalamic involvement. When patients with thalamic gliomas undergo small biopsies, commonly the only means to differentiate tumors with better or worse prognosis is the presence of suggestive molecular features (eg, BRAF fusion) 10, 20. We removed patients with pilocytic astrocytomas from the current analysis because they represent a separate entity with a potentially better prognosis. Since the radiologic characteristics of bithalamic gliomas overlap with other oncologic and non‐oncologic entities, 13 we strongly recommend histologic confirmation before these patients are considered for any anticancer therapy.
The confusion in the definition of bithalamic gliomas accounts for the controversy in therapeutic approaches and outcomes reported for affected patients. While some studies described a terrible outcome for most patients despite treatment with RT with or without chemotherapy, 8, 19, 23 others reported several long‐term survivors 5, 14, 17, 23. Unfortunately, conventional methods may not be sufficient to distinguish bithalamic gliomas in clinical practice. The absence of immunoreactivity can rule out H3F3A p.K27 mutant diffuse midline gliomas. However, the heterogeneous immunolabeling of H3K27me3 stains was not useful in our diagnostic work‐up.
Until the imaging, histologic, and molecular characteristics of bithalamic gliomas are clearly defined, no consensus will emerge about their natural history, best therapeutic approaches, and prognosis of affected patients.
One of the limitations of this study was our inability to perform additional confirmatory or other exploratory molecular analyses caused by the lack of further tissue for analysis.
One previous study had speculated that bithalamic gliomas constitute a separate entity based on their unique clinical and radiologic characteristics 8. Although the number of tumors analyzed was limited, the complete absence of H3F3A p.K27‐mutated tumors among our patients is in stark contrast to other thalamic high‐grade gliomas. Therefore, our results suggest that bithalamic gliomas may represent a distinct entity compared with unilateral thalamic high‐grade gliomas.
Conflict of Interest
The authors have no conflicts of interest to disclose related to this manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. List of 364 genes with differentially methylated promoter regions comparing unilateral and bithalamic gliomas. Beta values ranged from 0 (no methylation) to 1 (complete methylation). FDR stands for false discovery rate.
Table S2. List of 100 gene sets with strongest differential methylation.
Acknowledgments
This work was supported by the United States National Institutes of Health Cancer Center Support (CORE) Grant P30 CA21765 and by the American Lebanese Syrian Associated Charities (ALSAC). We thank Geoffrey Neale and John Morris for assistance in performing the methylation studies. We thank Racquel Collins for her assistance in performing targeted sequencing.
References
- 1. Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S et al (2014) H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol 16:140––146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aryee MJ, Jaffe AE, Corrada‐Bravo H, Ladd‐Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363––1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP‐status. Acta Neuropathol 124:547––560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bilginer B, Narin F, Işıkay I, Oguz KK, Söylemezoglu F, Akalan N (2014) Thalamic tumors in children. Childs Nerv Syst 30:1493––1498. [DOI] [PubMed] [Google Scholar]
- 5. Boesten T, Gerber NU, Kandels D, Azizi AA, Schmidt R, Warmuth‐Metz M et al (2016) Management of primary thalamic low‐grade glioma in pediatric patients: results of the multicenter treatment studies HIT‐LGG 1996 and SIOP‐LGG 2004. Neuro Oncol Pract doi: 10.1093/nop/npw007 (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broniscer A, Chamdine O, Hwang S, Lin T, Pounds S, Onar‐Thomas A et al (2016) Gliomatosis cerebri in children shares molecular characteristics with other pediatric gliomas. Acta Neuropathol 131:299––307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colosimo C, di Lella GM, Tartaglione T, Riccardi R (2002) Neuroimaging of thalamic tumors in children. Childs Nerv Syst 18:426––439. [DOI] [PubMed] [Google Scholar]
- 8. Di Rocco C Iannelli A (2002) Bilateral thalamic tumors in children. Childs Nerv Syst 18:440–444. [DOI] [PubMed] [Google Scholar]
- 9. Feng J, Hao S, Pan C, Wang Y, Wu Z, Zhang J et al (2015) The H3.3 K27M mutation results in a poorer prognosis in brainstem gliomas than thalamic gliomas in adults. Human Pathol 46:1626––1632. [DOI] [PubMed] [Google Scholar]
- 10. Fontebasso AM, Bechet D, Jabado N (2013) Molecular biomarkers in pediatric glial tumors: a needed wind of change. Curr Opin Oncol 25:665––673. [DOI] [PubMed] [Google Scholar]
- 11. Fontebasso AM, Papillon‐Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO et al (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high‐grade astrocytoma. Nat Genet 46:462––466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hovestadt V, Zapatka M conumee: Enhanced copy‐number variation analysis using Illumina 450k methylation arrays. R package version 0.99.4. In: Bioconductor: Open Source Software for Bioinformatics. http://www.bioconductor.org/packages/release/bioc/html/conumee.html.
- 13. Khanna PC, Iyer RS, Chaturvedi A, Thapa MM, Chaturvedi A, Ishak GE, Shaw DW (2011) Imaging bithalamic pathology in the pediatric brain: demystifying a diagnostic conundrum. Am J Roentgenol 197:1449––1459. [DOI] [PubMed] [Google Scholar]
- 14. Menon G, Nair S, Sudhir J, Rao BR, Krishnakumar K (2010) Bilateral thalamic lesions. Br J Neurosurg 24:566––571. [DOI] [PubMed] [Google Scholar]
- 15. Nettleton D, Recknor J, Reecy JM (2008) Identification of differentially expressed gene categories in microarray studies suing nonparametric multivariate analysis. Bioinformatics 24:192––201. [DOI] [PubMed] [Google Scholar]
- 16. Pounds S, Cheng C (2006) Robust estimation of the false discovery rate. Bioinformatics 22:1979––1987. [DOI] [PubMed] [Google Scholar]
- 17. Puget S, Crimmins DW, Garnett MR, Grill J, Oliveira R, Boddaert N et al (2007) Thalamic tumors in children: a reappraisal. J Neurosurg 106:354––362. [DOI] [PubMed] [Google Scholar]
- 18. R Core Team (2015). R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing: Vienna, Austria. https://www.R-project.org/
- 19. Reardon DA, Gajjar A, Sanford RA, Heideman RL, Walter AW, Thompson SJ et al (1998) Bithalamic involvement predicts poor outcome among children with thalamic glial tumors. Pediatr Neurosurg 29:29–35. [DOI] [PubMed] [Google Scholar]
- 20. Ryall S, Krishnarty R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R et al (2016) Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun 4:93––103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226––231. [DOI] [PubMed] [Google Scholar]
- 22. Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ, Perry A (2015) Diffuse midline gliomas with histone H3‐K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 26:569––580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Steinbok P, Gopalakrishnan CV, Hengel AR, Vitali AM, Poskitt K, Hawkins C et al (2015) Pediatric thalamic tumors in the MRI era: a Canadian perspective. Childs Nerv Syst 32:269––280. [DOI] [PubMed] [Google Scholar]
- 24. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060––1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425––437. [DOI] [PubMed] [Google Scholar]
- 26. Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R et al (2001) Missing value estimation methods for DNA microarrays. Bioinformatics 17:520––525. [DOI] [PubMed] [Google Scholar]
- 27. Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A et al (2013) Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol 23:558––564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Venneti S, Santi M, Felicella MM, Yarilin D, Phillips JJ, Sullivan LM et al (2014) A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol 128:743––753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. List of 364 genes with differentially methylated promoter regions comparing unilateral and bithalamic gliomas. Beta values ranged from 0 (no methylation) to 1 (complete methylation). FDR stands for false discovery rate.
Table S2. List of 100 gene sets with strongest differential methylation.