Huai-Dong Song
Huai-Dong Song
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
a,b,
Chang-Chun Tu
Chang-Chun Tu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
c,b,
Guo-Wei Zhang
Guo-Wei Zhang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
a,b,
Sheng-Yue Wang
Sheng-Yue Wang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
d,b,
Kui Zheng
Kui Zheng
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Lian-Cheng Lei
Lian-Cheng Lei
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
c,
Qiu-Xia Chen
Qiu-Xia Chen
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Yu-Wei Gao
Yu-Wei Gao
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
c,
Hui-Qiong Zhou
Hui-Qiong Zhou
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Hua Xiang
Hua Xiang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
c,
Hua-Jun Zheng
Hua-Jun Zheng
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
d,
Shur-Wern Wang Chern
Shur-Wern Wang Chern
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
f,
Feng Cheng
Feng Cheng
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
a,
Chun-Ming Pan
Chun-Ming Pan
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
a,
Hua Xuan
Hua Xuan
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
c,g,
Sai-Juan Chen
Sai-Juan Chen
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
a,g,
Hui-Ming Luo
Hui-Ming Luo
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,b,
Duan-Hua Zhou
Duan-Hua Zhou
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,b,
Yu-Fei Liu
Yu-Fei Liu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Jian-Feng He
Jian-Feng He
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Peng-Zhe Qin
Peng-Zhe Qin
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Ling-Hui Li
Ling-Hui Li
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Yu-Qi Ren
Yu-Qi Ren
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
i,
Wen-Jia Liang
Wen-Jia Liang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Ye-Dong Yu
Ye-Dong Yu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
i,
Larry Anderson
Larry Anderson
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
f,
Ming Wang
Ming Wang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,g,
Rui-Heng Xu
Rui-Heng Xu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,g,
Xin-Wei Wu
Xin-Wei Wu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,b,
Huan-Ying Zheng
Huan-Ying Zheng
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,b,
Jin-Ding Chen
Jin-Ding Chen
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
j,b,
Guodong Liang
Guodong Liang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
k,
Yang Gao
Yang Gao
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Ming Liao
Ming Liao
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
j,
Ling Fang
Ling Fang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Li-Yun Jiang
Li-Yun Jiang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Hui Li
Hui Li
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,
Fang Chen
Fang Chen
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Biao Di
Biao Di
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Li-Juan He
Li-Juan He
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,
Jin-Yan Lin
Jin-Yan Lin
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
e,g,
Suxiang Tong
Suxiang Tong
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
f,g,
Xiangang Kong
Xiangang Kong
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
l,g,
Lin Du
Lin Du
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
h,g,
Pei Hao
Pei Hao
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
m,n,b,
Hua Tang
Hua Tang
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
o,b,
Andrea Bernini
Andrea Bernini
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
p,b,
Xiao-Jing Yu
Xiao-Jing Yu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
m,
Ottavia Spiga
Ottavia Spiga
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
p,
Zong-Ming Guo
Zong-Ming Guo
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
n,
Hai-Yan Pan
Hai-Yan Pan
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
n,
Wei-Zhong He
Wei-Zhong He
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
n,
Jean-Claude Manuguerra
Jean-Claude Manuguerra
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
q,
Arnaud Fontanet
Arnaud Fontanet
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
q,
Antoine Danchin
Antoine Danchin
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
q,
Neri Niccolai
Neri Niccolai
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
p,g,
Yi-Xue Li
Yi-Xue Li
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
m,n,g,
Chung-I Wu
Chung-I Wu
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
o,g,
Guo-Ping Zhao
Guo-Ping Zhao
aState Key Laboratory for Medical Genomics/Pôle Sino-Français de Recherche en Sciences du Vivant et Génomique, Ruijin Hospital Affiliated to Shanghai Second Medical University, 197 Rui Jin Road II, Shanghai 200025, China; cChangchun University of Agriculture and Animal Sciences, Changchun 130062, China; dChinese National Human Genome Center, 250 Bi Bo Road, Zhang Jiang High Tech Park, Shanghai 201203, China; eGuangdong Center for Disease Control and Prevention, 176 Xingangxi Road, Guangzhou 510300, Guangdong, China; fCenters for Disease Control and Prevention, 1600 Clifton Road, Atlanta, GA 30333; hGuangzhou Center for Disease Control and Prevention, 23 Third Zhongshan Road, Guangzhou 510080, Guangdong, China; iGuangdong Provincial Veterinary Station of Epidemic Prevention and Supervision, Guangzhou 510230, China; jCollege of Veterinary Medicine, South China Agriculture University, Guangzhou 510246, China; kNational Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 100052, China; lNational Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agriculture Sciences, Harbin 150001, China; mBioinformation Center/Institute of Plant Physiology and Ecology/Health Science Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; nShanghai Center for Bioinformation Technology, 100 Qinzhou Road, Shanghai 200235, China; oDepartment of Ecology and Evolution, University of Chicago, 1101 East 57th Street, Chicago, IL 60637; pBiomolecular Structure Research Center and Department of Molecular Biology, University of Siena, Via A. Fiorentina 1, I-53100 Siena, Italy; qInstitut Pasteur, 25, Rue du Docteur Roux, 75724 Paris Cedex 15, France; and rState Key Laboratory of Genetic Engineering/Department of Microbiology, School of Life Science, Fudan University, 220 Handan Road, Shanghai 200433, China
d,m,r,s



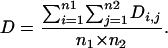
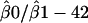 /2=28(days), which is equivalent to early December 2002.
/2=28(days), which is equivalent to early December 2002.

