

Abstract
Small interfering RNA (siRNA) is normally designed to silence preselected known genes. Such selections are inevitably prone to bias as a result of limited knowledge about the biological process, transcript identity, and functions. A library that contains all permutations of siRNA could avoid such problems. In this paper, it is shown that 5 × 107 siRNA-encoding plasmids can be constructed in a single tube by using vectors with two mutated RNA polymerase III promoters arranged in a convergent manner. Such a library was used to carry out genomewide screening of functional genes in a phenotype-driven manner. Multiple siRNAs that induce a significant increase of cell proliferation speed were identified.
Keywords: random targeting, screening, vector, dual promoters
The discovery of the RNA interference (RNAi) phenomena, especially small interfering RNA (siRNA), has dramatically simplified the manipulation of gene expression for functional genomics and drug target validation (1–3). Synthetic siRNAs were introduced and have been widely used for gene knockdown experiments (2, 3). siRNA-expressing plasmid or viral vectors were subsequently shown to be very effective in down-regulation of target mRNA and protein levels (4–10). The rapid development of this powerful method has made it possible to knock down almost any known gene in an applicable organism. The application of the method is, however, restricted by the required knowledge of the sequence of a gene to create siRNA to target it. In organisms where the transcriptome is largely known (such as human and mouse), the limitation on large-scale application is mostly reflected on the throughput level, which is further translated into limitations on cost and speed levels. As a consequence, most researchers are forced to shorten their gene list, either by knowledge-based prioritization or by using other first-line high-throughout methods such as microarrays to fit the research into individual cost structure and time frame. In important model species for which the transcriptome is largely unknown, parallel high-throughput applications are practically not possible. To overcome these limitations, the concept of siRNA libraries has been explored.
Here we present the construction of siRNA libraries that contain all permutations of siRNA sequences. For this purpose we created a plasmid vector system that contains two convergent RNA polymerase III (Pol III) promoters to drive the expression of both strands of the siRNA from a single randomized region in living cells. We further demonstrated the usefulness of this type of siRNA library for phenotype-driven genomewide screening in a cell proliferation model.
Materials and Methods
Construction and Validation of Vectors with Dual Pol III Promoters. The pBluescript vector with a human H1 promoter was a kind gift from Mauro D'Amato (Karolinska Institutet). The plasmid with a U6 promoter, pAV/U6, was a kind gift from David Engelke (University of Michigan, Ann Arbor) (4). First, the H1 promoter was engineered to replace the last 11 nucleotide pairs at its 3′ end with a BglII site by PCR (BglII versions of the promoters), and then cloned into the EcoRI site of the pBluescript II KS (+) vector (Stratagene) to form pMH. Second, the promoters were differently engineered to replace the last 11 nucleotide pairs at its 3′ end with a HindIII site by PCR (HindIII versions of the promoters). The HindIII tagged H1 promoter was cloned into the HindIII/SalI site of the pMH to form pDH, and the HindIII tagged U6 was cloned into the HindIII/SalI sites to form pBUH. The dual Pol III promoter cassette in pBUH was cleaved out by KpnI/XbaI and further cloned into the same sites of pcDNA3.1(-) (Invitrogen) with different manipulations on the CMV promoter to form pCUHa (with wild-type CMV promoter), pCUHb (with partially deleted CMV promoter), and pCUH (with CMV promoter completely deleted). The BglII/HindIII fragments between the two mutated promoters in pDH, pBUH, and pCUH were then removed and replaced by DNA duplexes formed by oligonucleotides having thegeneralstructureGATCTAAAAAX19TTTTTA,and AGCTTAAAAAY19TTTTTA where X19 and Y19 were complementary 19-mer sequences specific for Renilla and firefly luciferase. In this manner, we created siRNA-encoding vectors pDH_RL21, pDH_RL82, pDH_RL479, pBUH_82 (designed to target the nucleotide 21–39, 82–100, 479–497 regions of Renilla luciferase mRNA), and pCUHa_FL867, pCUHb_FL867, pCUH_FL867, and pCUHa_FL961, pCUHb_FL961, pCUH_FL961 (designed to target the nucleotide 867–885 and 961–979 regions of firefly luciferase mRNA). Sequences of siRNA were set to be the same as we previously reported (11). This cloning step will add AAAAA to the 3′ terminus of the promoters for two purposes: (i) this region will serve as the transcription termination site for the opposite RNA Pol III promoter; and (ii) the addition of AAAAA will rebuild the distance between the promoter and the transcription initiation site to make sure that the resulting RNA will start from the first X (or Y from the opposite direction). The activity of this modified H1 promoter was validated by cloning different hairpin siRNA coding sequences targeting the Renilla luciferase gene into BglII and HindIII sites of the modified vector pMH and measuring the inhibitory effect of these siRNAs against Renilla luciferase activity by dual luciferase assay (Promega).
Deletion of CMV Promoter. The CMV promoter in vector pcDNA3.1(-) was partially deleted by digestion with MluI and SnaBI (Promega) or entirely deleted by digestion with MluI and XhoI (Promega), followed by making blunt ends with the Klenow fragment of DNA polymerase (Promega) and self-ligation of the plasmid.
Preparation of Double-Stranded Random siRNA Library Sequence. Oligonucleotides containing N18C were synthesized, with a BglII site and AAAAA at their 5′ ends and TTTTT and a HindIII site at their 3′ ends (GAAGATCTAAAAAN18CTTTTTAAGCTTGGGCCGCCG). Double-stranded DNA was formed by annealing with a short oligonucleotide primer CGGCGGCCCAAGCTTAAAAAG that is complementary to its 3′ part sequence, followed by Klenow fill-in (Promega). The resulting DNA duplexes were purified with a QIAquick Nucleotide Removal Kit (Qiagen, Hilden, Germany), cleaved with BglII and HindIII, and ligated into vectors.
Plasmid Transfection and Luciferase Assay. Cell transfection and luciferase assay were done according to ref. 11.
Activity Comparison Between Chemically Synthesized siRNA and siRNA Encoded in pBUH. Chemically synthesized siRNA was purchased from Genset (Evry Cedex, France) to target CDK2 mRNA (BC018255) at GGC AGC CCT GGC TCA CCC T and CGG AGC TTG TTA TCG CAA A sites. Plasmids encoding siRNA targeting to the same sites were constructed by inserting the respective 19-mer duplex DNA into pBUH. The activities of synthetic siRNA and the plasmid-encoded siRNA were compared by using a system that was recently established that can precisely annotate the efficacy of a siRNA by using a reporter construct (12). The synthetic siRNA and plasmid were used to cotransfect HEK293 cells with the reporter construct harboring the respective targeting sites. Synthetic siRNAs were used a dose of 13 nM (final concentration), and plasmids were used at 200 ng per well in 24-well plates. Cells were transfected for 4 h before fresh medium was added. Cells were harvested after 24 h (for synthetic siRNA), or 72 h (for plasmid-encoded siRNA) for measuring silencing efficacy as described (12).
Confirmation of siRNA Production by the Vector. pCUH_FL867 was transfected into HEK293 cells at about 50% confluence. Total RNA was extracted from the cells 48 h after the transfection by using TRIzol reagent (Life Technologies/GIBCO). The product instructions were followed except that ethanol instead of 2-propanol was used for precipitation of RNA.
To prepare the probes for RNA-protection assay, 10 pmol of synthetic RNA oligonucleotide was labeled with 5 μlof[γ-32P]ATP by using a 5′ end labeling kit (Amersham Pharmacia Bioscience). The product instructions were followed except that the labeled product was dissolved in lysis/denaturation solution from an RPA kit (Ambion, Austin, TX). Both sense and antisense strands were labeled for the complementary strands detection.
A Direct Protect Lysate RPA kit (Ambion) was used to detect the expressed siRNA strand when the labeled siRNA sense strand was used as the probe to detect the expressed siRNA antisense strand, and the labeled siRNA antisense strand was used to detect the expressed siRNA sense strand. Thirty micrograms of total RNA was used for each reaction, and the product instructions were followed. The resulting RPA products were loaded on a denaturing 15% polyacrylamide gel, and subjected to electrophoresis in Tris/borate buffer at ≈200 V for ≈1 h. Then the gel was covered with plastic wrap, and the signal was detected by phosphorimaging on a Typhoon 9400 (Amersham Pharmacia Bioscience).
Verification of Silencing Function Under Stable Transfection. pCUH_FL867 (0.1 μg per flask) was transfected into MC3T3-E1 cells and the cells were maintained under the selection pressure of G418 for 3 weeks at a G418 concentration of 800 μg/ml for the first week and subsequently 400 μg/ml). The cells were then replated and transfected with the luciferase vectors as described above. Forty-eight hours after transfection the cells were harvested for luciferase activity measurement. pCUH_FL961 was used as a control in all experimental procedures.
Screening of siRNA That Can Induce Elevated Proliferation. A slow-growing cell line, MC3T3-E1 (mouse preosteoblast, CRL-2593, from the American Type Culture Collection) was used in these experiments. Cells were plated 12 h before transfection, in 12 T75 flasks at density of 2.4 × 106 per flask. The cells in each flask were transfected with 8 μg of siRNA library plasmid (equivalent to 5 × 107 plasmids) by Lipofectamine 2000 (Invitrogen). A plasmid was constructed in pCUH vector to encode a negative control siRNA duplex (sense: 5′-ACU ACC GUU GUU AUA GGU C-3′; antisense: 5′-GAC CUA UAA CAA CGG UAG U-3′) that targets to none of the known mammalian mRNA. This negative control plasmid was transfected into MC3T3-E1 cells under the same conditions as those of the siRNA library. G418 (Roche) was added to the cells at a concentration of 800 μg/ml to select stable cells 48 h after transfection. One week later, surviving cells were collected, pooled, counted, and then split into five flasks at the density of 2 × 106 per flask. The cells were split further into more flasks whenever the cells approached confluency. After 3 weeks of selection, the amount of G418 was reduced to 400 μg/ml. Cells were cultured for 60 days (≈30 generations). Afterward, total DNA was extracted from the cells and the siRNA-encoding cassette was amplified by PCR, cloned back into pCUH, and sequenced.
Cell Proliferation Test by MTS Assay. The cell proliferation rate was then measured by using an MTS Assay (CellTiter 96 aqueous one-solution cell proliferation assay, Promega; MTS being a tetrazolium compound) at daily time intervals. Cells were plated in 24-well plate at a density of ≈200 per well and then allowed to grow until being harvested 1–7 days afterward in the following way. For each well in the 24-well plate, culture media were discarded, and 250 μl of fresh culture medium was added. Then 50 μl of CellTiter 96 aqueous one-solution was added to each well. After 4 h of incubation, 100 μl of solution from each well was transferred into a well in 96-well plate. UV absorbance of the solution was measured at the wavelength of 490 nm. All MTS assays were done in triplicates.
Results
Design and Validation of siRNA-Encoding Vectors with Two Convergent Pol III Promoters. For making a vector system with two convergent promoters, it is critical to determine whether the wild-type Pol III promoters can be engineered to accommodate transcription terminator sequences without altering their transcriptional activity. Proper termination of transcription is essential in siRNA expression. For this purpose, we mutated the last five nucleotides of human H1 and U6 promoters into AAAAA to embed a TTTTT sequence on the minus strand for termination of transcription from the opposite direction. Additional BglII and HindIII restriction sites were engineered immediately ahead of the AAAAA region in the promoters for easy cloning (Fig. 1). The engineered promoters were compared with wild-type promoters for their activity to drive Pol III-mediated expression initiation of transcription from proper locations. The tests were done by inserting a normal hairpin siRNA-encoding DNA fragment in the plasmid and then assessing the inhibitory activity of the plasmid against its target gene, Renilla luciferase. Hairpin siRNA from the mutated promoters silenced the enzyme to the same degree as the wild-type promoters, suggesting that the mutated sequences still retained proper promoter activities when placed individually (data not shown).
Fig. 1.

Construction of siRNA-encoding vectors for gene silencing. (A) Human H1 promoter sequence with EcoRI and BglII sites tagged to the ends. (B) Comparison of the mutated and unmutated H1 promoters (only the last 29 nt was included; altered regions were underlined). The relevant restriction sites are shown in boldface. (C) Diagram of pDH [in pBluescript KS (+) II]. The two H1 promoters were placed in a convergent manner and 19-mer DNA identical to targeting site was placed between the two promoters. (D) Human U6 promoter sequences. (E) Comparison of the mutated and unmutated H1 promoters (only the last 26 nt was included; altered regions are underlined). The relevant restriction sites are shown in boldface. (F) Diagram of pCUH in pcDNA3.1(-). The CMV promoter of the pcDNA3.1(-) was deleted in pCUH.
Then we started to test vectors with two of the H1 promoters head-to-head as shown in Fig. 1C. The critical question was whether transcription in one direction would interfere with transcription from the other strand. To answer this question, two of the promoters, having BglII and HindIII sites, were cloned in opposite directions into a single plasmid with a 19-nt target sequence against Renilla luciferase sandwiched in between (pDH_RL82). siRNA chemically synthesized to target this sequence (nucleotides 82–100) was previously identified to be an effective siRNA for silencing the Renilla luciferase (11). The plasmid demonstrated excellent inhibitory activity against the intended target gene Renilla luciferase and repressed the enzymatic activity by >90% (Fig. 2A), suggesting efficient siRNA formation by transcription from both strands of the 19-mer sequence followed by efficient annealing of the two resulting RNA fragments. The dosing curve also showed that the inhibitory activities of vectors with the dual Pol III promoters are comparable to the efficiency of the classic hairpin siRNA-encoding vectors with a single Pol III promoter (Fig. 2). pBluescript II KS (+) and pcDNA3.1(-) were further tested as the backbone of the vectors (pDH series with pBluescript, and pCDH series with pcDNA3.1) (Fig. 3). Similar inhibition profiles induced with siRNA from both plasmids suggested that the siRNA expression properties of the mutated promoters are not plasmid-type dependent.
Fig. 2.

A plasmid vector with two convergent H1 promoters driving the expression of short RNA from a single 19-bp DNA can induce efficient gene silencing, at a level that is comparable to that induced by hairpin siRNA-encoding plasmids. CTL, vector control. (A) Dose responses of pDH-RL82 within a 0.3–2.7 μg per well dose range. (B) Dose responses of pHairpin_RL82 within a 0.3–2.7 μg per well dose range. All experiments were repeated at least two times with at least three replicas in each experiment.
Fig. 3.

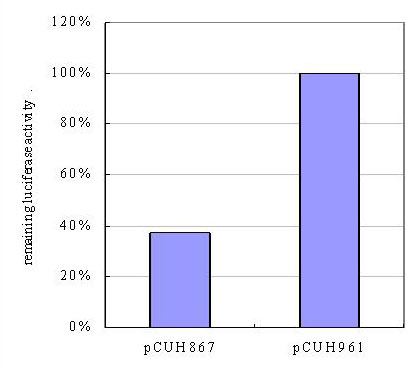
Inhibitory activities of different siRNA-encoding plasmids containing both H1 and U6 promoters. (A) siRNA-encoding plasmids with pcDNA3.1(-)as a plasmid backbone and U6/H1 promoters. pCUHa contains a CMV promoter upstream of the H1 promoter. In pCUHb, the CMV promoter was partially deleted, and, in pCUH, the CMV promoter was completely deleted. Positive siRNA encoded in vectors with CMV promoter deleted consistently showed about 30–40% improvement in gene-silencing efficiency. (B) With pBluescript as the backbone and U6/H1 promoters, pBUH_FL867 can silence the Renilla luciferase by ≈90% but the pBUH_FL961 showed no effect, in good agreement with results from chemically synthesized siRNA. (C) Similar inhibition profiles were observed for vectors with pBluescript and pcDNA3.1 backbones.
The presence of the two H1 promoters as long inverted repeats has raised a concern about plasmid instability and posed problems for downstream PCR amplification and sequencing (data not shown); therefore we further tested the combination of an H1 promoter and U6 promoter in a similar convergent manner (Fig. 1 D–F). To avoid possible interference of the CMV promoter with the transcriptional initiation of the H1 and U6 promoters when pcDNA3.1 was used, the CMV promoter was deleted from the pcDNA3.1 plasmid. Because U6 promoter prefers a G as the transcription start site, we used two previously identified siRNAs targeting the nucleotide 867–885 and 961–979 regions of firefly luciferase for the test because both siRNAs have a starting G in one strand (11). Vectors encoding siRNA targeting the nucleotide 867–885 region, including pCUHa with wild-type CMV promoter intact, pCUHb with CMV promoter partially deleted, and pCUH with CMV promoter completely deleted, all can induce efficient silencing of the target enzyme. Comparison suggested that deletion of the CMV promoter were found to enhance the inhibitory efficiency by 30–40% (Fig. 3C). pCUH was chosen to serve as the plasmid backbone for the construction of the siRNA libraries in the subsequent experiments. Vectors encoding siRNA targeting the nucleotide 961–979 region did not show silencing activities. The results were consistent with results obtained from experiments with chemically synthesized siRNA (11).
Activity Comparison Between Chemically Synthesized siRNA and Plasmid-Encoded siRNA. The efficacy of plasmid-encoded siRNA was compared with that of the chemically synthesized siRNA by using two siRNAs targeting CDK2 (cyclin-dependent kinase 2) sequences as an example. The assay was done by using an siRNA evaluation system recently developed in our lab in which a targeting site was conjugated into a reporter for precise measurement of siRNA activity (12). Sequences of a positive (cdk2A) and a negative (cdk2B) siRNA against endogenous cdk2 mRNA were used in this study. As shown in Fig. 4, siRNA encoded by plasmid pBUH has exactly the same activity profile as that of the synthetic siRNA. For the cdk2B siRNA, both types of reagent showed a silencing efficiency of 85%. The result further demonstrated the functionality of the vector system.
Fig. 4.

The functionality of the vectors for encoding siRNA was assessed by comparison with synthetic siRNA by using two siRNA sequences complementary to endogenous gene cdk2 by using a siRNA assessment system. Synthetic cdk2A siRNA does not have any silencing activity, and synthetic cdk2B siRNA has a potent silencing effect. The same efficacy was observed for siRNA encoded by pBUH plasmid.
Confirmation of Functionality of the siRNA Vector. pCUH867 was transfected into HEK293 cells in parallel with or without the addition of the luciferase plasmids so that the production of the two strands of the siRNA could be assessed simultaneously with the silencing effect. It was found that pCUH867 could silence the activity of the firefly luciferase by 85%. After total RNA was prepared from the cells, an RNase-protection assay was performed, using synthetic RNA oligonucleotides corresponding the two strands of the FL867 siRNA. As showed in Fig. 5, both the sense strand and the antisense strand of the FL867 siRNA were expressed in the cells by the pCUH867 plasmid. This finding is consistent with our expectations and the mounting information of natural antisense transcripts found in cells.
Fig. 5.

The production of the two strands of the siRNA by pCUH867 plasmid in HEK293 cells. For detecting the sense strand of the siRNA produced by the plasmid, total RNA from cells transfected with pCUH867 alone was used in RNase-protection assay (RPA) (lane 1). For detection of the antisense strand, total RNA from cells cotransfected with pCUH867 and the luciferase plasmids was used because in this case we can measure the presence of the antisense strand while measuring the silencing efficiency at the protein level without interference by the presence of the target sequence (lane 2). When a synthetic FL867 siRNA strand is spiked into total RNA from untransfected cells, RPA showed a protected product with the same size (lane 3), whereas the free probe moved significantly faster (lane 4).
pCUH867 and control pCUH961 were transfected into MC3T3-E1 cells in a stable transfection assay. After 3 weeks under the selection of G418, the surviving cells were plated and transfected with the luciferase plasmids. As shown in Fig. 8, which is published as supporting information on the PNAS web site, it was observed that firefly luciferase is specifically silenced in cells stably transfected with pCUH867 but not with pCUH961, suggesting that the siRNA expression cassette does perform in the same manner in a stable transfection as in a transient transfection.
Generation of a Fully Random siRNA-Encoding Plasmid Library. When the functionalities of the mutated promoters and plasmid constructs were verified, we further replaced the 19-bp gene-specific sequence by two versions of random sequences (N19 and N18C). Several considerations were included in the design of the library. Because statistics have shown that most active siRNAs have a G+C content of 35–55%, we biased the randomness of N according to different formula to achieve a median G+C content of ≈40–45% (Table 1). The cytidine in the N18C library was included for three different reasons. First, the U6 promoter, which drives the expression of RNA from the opposite stand, prefers a G as its transcription-starting site (13). Second, several lines of evidence suggested that the terminal nucleotides in siRNA do not contribute significantly to the target site recognition (14–16). Thus incorporation of a G·C base pair in this end will not alter the activity of the siRNA in the library, but it will reduce chemical complexity of the library by a factor of 4 without altering the fact that the library will contain all siRNA against any gene, which is favorable for applications of the library in screening. Third, recent evidence indicates that siRNA assembly into an RNA-induced silencing complex (RISC) is carried out in an asymmetric manner (17). The presence of a G·C base pair at one end would enable us to increase the proportion of effective siRNA in a directional manner, and this could assist future target gene identification from the siRNA sequence.
Table 1. Design and construction of the plasmid libraries encoding all permutations of siRNA.
| Determined nucleotide frequency, %
|
||||
|---|---|---|---|---|
| Library | G | C | A | T |
| (G15%C20%A33%T32%)18C | 13.5 | 20.4 | 22.4 | 43.7 |
| (G18%C20%A32%T30%)18C | 13.8 | 20.8 | 24.5 | 40.9 |
| (G22%C20%A38%T20%)18C | 20.1 | 23.7 | 36.7 | 19.5 |
| (G22%C20%A38%T20%)19 | 26.8 | 16.9 | 32.0 | 24.4 |
The design of the random siRNA-encoding DNA fragment was done according to the general formula of N18C, where N was set to generate a median G+C content of 35-45%. Because incorporation of different nucleotides appears to deviate significantly during individual chemical synthesis, several libraries have to be tested to arrive at this optimal nucleotide composition. Sequencing-determined nucleotide composition suggested that two of the libraries, (G22%C20%A38%T20%)18C and (G22%C20%A38%T20%)19, have G+C contents in the expected range.
The library was chemically synthesized as a single-stranded oligonucleotide with the randomized siRNA-encoding region embedded between two segments of known sequences (Fig. 6). After primer annealing and Klenow fill-in to form double-stranded DNA, the samples were cleaved with BglII and HindIII and then cloned into the same sites in pCUH. The sequence diversity of the library was verified by sequencing clones randomly chosen from the library. Because of the discrepancies between the designed nucleotide frequencies and the experimentally determined values, the synthesis of the library has been carried out multiple rounds to obtain libraries with 40–45% G+C content. The libraries (G22%C20%A38%T20%)19 and (G22%C20%A38%T20%)18C were both successful in this respect. Of 30–50 clones sequenced from each library, most clones were found to encode full-length 19-mer siRNA. Less than about 2–3% of clones contain a truncated sequence in the siRNA-encoding region that disables the generation of a siRNA, suggesting that the libraries are of good quality. One tradeoff for elevating A+T percentage to 60% is the increased occurrence of AAAA·TTTT or AAAAA·TTTTT sequence in the siRNA-encoding region (Table 2, which is published as supporting information on the PNAS web site). Pol III would terminate prematurely at such sequences to produce truncated RNA molecules that do not form siRNA.
Fig. 6.

Diagram of the structure of randomized siRNA library constructed in pCUH. Partially randomized single-stranded library oligonucleotides were synthesized according to the common formula of 5′-AGG GGA AGA TCT AAA AAN NNN NNN NNN NNN NNN NNC TTT TTA AGC TTG GGC CGC CG-3′ where N represents wobble bases with different G/A/T/C composition as listed in Table 1. The single-stranded library oligonucleotides were annealed to CGG CGG CCC AAG and then made double stranded by a fill-in reaction with Klenow fragment. The double-stranded fragments were cleaved with BglII and HindIII and cloned into the same sites in pCUH.
Screening of siRNA That Can Enhance Cell Proliferation by Using the Random siRNA Library. An siRNA library made in pCUH was used in a screening for functional siRNA that can enhance cell proliferation of murine MC3T3-E1 cells. Twelve flasks each containing 2.4 × 106 cells were used as the starting materials. The cells were transfected with 8 μg of the random siRNA library (representing ≈5 × 107 siRNA) per flask, and the cells were allowed to proliferate for ≈60 days (equivalent to ≈30 rounds of cell division) under the pressure of G418. One week after the selection started, surviving cells were collected, counted, and then replated in five flasks at the density of 2 × 106 cells per flask. Considering that some of the cells might have proliferated during the time, and some of the cells might still die later on, we estimate that this represents 1–3 × 106 independently transfected cells, but this is a rough estimation taking the slow proliferation of the MC3T3 cells into account. It may not apply to other cells lines with different proliferation speed.
It is anticipated that different siRNA plasmids will segregate into different cells after repeated cell division, thus each cell will have only a single or a small number of different plasmids. Plasmids were isolated from single cells, and sequencing of the plasmids indicated that each single cell contained only one or two siRNA sequences, suggesting that individual plasmids of the library do segregate into different cells as expected. At the same time, any cells harboring siRNA that offer advantages in the cell proliferation process will propagate more than others and eventually become over-represented in the final cell population.
After the 60-day period, we compared the proliferation rates of the cells transfected with the siRNA library and cells transfected with the single negative control plasmid. Indeed, we found that the cells that were transfected with the siRNA library proliferated faster than the cells that were transfected with the negative control plasmid (Fig. 7A). Then DNA was retrieved from the library-transfected cells, and sequences of siRNA-encoding region were obtained. When pCUH plasmids harboring the sequences were retransfected into the MC3T3-E1 cell line, some of the plasmids exhibited significant enhancement of cell proliferation (Fig. 7B). For example, plasmids A81 and B7 can elevate proliferation rate by about 100% over the 1-week measurement period. For example, it was interesting to note that siRNA B7 aligns nicely to the SET translocation gene in mouse. SET is a cell growth regulator that is implicated in leukemia (18). Recently, SET was shown to be a potent and specific inhibitor of protein phosphatase 2A, a family of major serine/threonine phosphatases involved in regulating cell proliferation and differentiation. No investigation has been performed to study the consequence of the direct disruption of the SET gene through antisense of RNA interference.
Fig. 7.

Proliferation enhancement by siRNA obtained from the screening of a random siRNA library. (A) A mixture of plasmids obtained from the screening was transfected into the cells (pink line), and the proliferation of cells was measured by MTS assay over 10 days. Cells transfected with a control plasmid were used as control (black line). (B) Individual siRNAs encoding plasmids isolated in the study (A66, A78, and B7) were transfected into cells and the proliferation of the cells was monitored for 7 days (pink for A66, green for A78, and blue for B7) in comparison with cells transfected with a control plasmid (black). The targeting site of B7 siRNA (TTT GAG GTG TCT GGG GGG C) aligns nicely with a sequence in translocation of SET, a gene implicated in leukemia.
Discussion
In a phenotype-driven screening, the size of a random siRNA library would be critical to obtaining reliable data. It should be extensive enough to completely cover all genes, but limited in size to facilitate the screening. The theoretical complexity of 19-mers, 2.75 × 1011, is achievable, but it would cause difficulties with respect to library maintenance and screening applications. What would then be an appropriate library size? To answer this question, we assessed data from studies on the effect of mutagenesis on siRNA activity and “off-target” hits of siRNA (14–16). For a typical siRNA, it appears that mutations on its terminal nucleotides provide minimal damage to its silencing activity (14). This observation was corroborated by the fact that some of the genes that share 15- to 16-nt identities with the siRNA can be efficiently modulated by the siRNA (15). Although internal mutations in the siRNA sequence can be detrimental to the ability of siRNA to degrade the target mRNA, it could transform the siRNA into an efficient translation inhibitor (16). A more concrete assessment was done by mutating the targeting site of an active siRNA. The experiment showed that mutations in the dinucleotides from either end of the targeting site do not affect the activity of the siRNA (Z.L., unpublished data). Taking all these aspects into consideration, it is conceivable that the minimal “specificity determinant sequence” of the siRNA could be just 15 nt in length. With this assessment, we propose that the effective complexity of a fully randomized 19-mer siRNA library could be only 1 × 109. Experimental data showed that our current library contains about 5 × 108 independent clones from 50 μg of starting plasmid vector, which means that the actual size of our libraries already covers 10–50% of the complexity of a fully random siRNA. Thus in essence, for any average mRNA of 2 kb from any organism where siRNA is applicable, there will be ≈200–1,000 siRNAs against it to be found in the libraries. If 1 in 3 siRNAs is active, for such a single mRNA, 70–300 effective siRNAs can exist in our libraries.
Pol III promoters are very well conserved on the sequence and functional levels (19). Even in species that are distantly related to humans, such as mouse, rat, and hamster, the human Pol III promoters can be used interchangeably for the expression of siRNA without any apparent loss of promoter activity. This fact makes it possible to apply a single siRNA library to a wide panel of mammalian species for comparative gene-screening purposes.
In summary, we have created and validated a vector system that can afford straightforward construction of siRNA libraries that contain a very large number of permutations of siRNA. The siRNA library generated here has the potential to be used in various high-throughput screening scenarios. By transfecting the library into cells under the selection pressure of antibiotics such as neomycin, single plasmids encoding distinct siRNAs can be segregated into single cells. The cells can then be sorted according to different phenotypes (or markers) (20). Such a screening process can easily be configured to run in a cycled manner. With this method, individual researchers can carry out genomewide screening of functional genes linked to a variety of phenotypes in a matter of days or weeks with minimal cost. It should be pointed out that the frequency of being stably transfected can differ hundreds of times between different cell lines when using a plasmid as the transfection vector. Thus it might be useful to consider retroviral or lentiviral vectors as alternatives. Preliminary results from our lab showed that the properties of the dual promoters also work in retroviral vectors.
Recently, two papers have been published to report complementary methods of constructing siRNA libraries by using enzymatically fragmented cDNA from a single mRNA or an mRNA pool (21, 22). Although they are very useful, the methods rely on the availability of the actual cDNA clones, and this can become a serious limiting factor for many researchers. Although this shortcoming is partially overcome by using cellular mRNA-derived cDNA as the source (22), the libraries generated from cellular mRNA would be heavily biased against genes expressed in a particular cell type, especially those highly expressed genes. In this respect, a strictly normalized, fully randomized, universal siRNA library would have much wider applicability in many cell types in a broad panel of organisms.
Supplementary Material
Acknowledgments
We thank Qiaomo Li for her technical help and Liam Good for comments on the manuscript. This work was supported by a National High Technology Research and Development Program (863 Program) (China) research grant (to Z.L.) and an International Collaboration Grant from 863 program (to Z.L. and M.C.) and in part by a Wallenberg Consortium North (Sweden) grant (to C.W.), as well as Grant 30400080 from the Natural Science Foundation of China.
Author contributions: M.C., H.-Y.Z., C.W., and Z.L. designed research; M.C., L.Z., H.-Y.Z., X.X., B.W., Q.D., and B.L. performed research; and C.W. and Z.L. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: siRNA, small interfering RNA; Pol III, RNA polymerase III.
References
- 1.Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E. & Mello, C. C. (1998) Nature 391, 806-811. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir, S. M., Harborthm, J., Lendeckel, W., Yalcin, A., Weber, K. & Tuschl, T. (2001) Nature 411, 494-498. [DOI] [PubMed] [Google Scholar]
- 3.McManus, M. T. & Sharp, P. A. (2002) Nat. Rev. Genet. 3, 737-747. [DOI] [PubMed] [Google Scholar]
- 4.Brummelkamp, T. R., Bernards, R. & Agami, R. (2002) Science 296, 550-553. [DOI] [PubMed] [Google Scholar]
- 5.Paul, C. P., Good, P. D., Winer, I. & Engelke, D. R. (2002) Nat. Biotechnol. 20, 505-508. [DOI] [PubMed] [Google Scholar]
- 6.Sui, G., Soohoo, C., Affar, B., Gay, F., Shi, Y., Forrester, W. C. & Shi, Y. (2002) Proc. Natl. Acad. Sci. USA 99, 5515-5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paddison, P. J., Caudy, A. A., Bernstein, E., Hannon, G. J. & Conklin, D. S. (2002) Genes Dev. 16, 948-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brummelkamp, T. R., Bernards, R. & Agami, R. (2002) Cancer Cell 2, 243-247. [DOI] [PubMed] [Google Scholar]
- 9.Rubinson, D. A., Dillon, C. P., Kwiatkowski, A. V., Sievers, C., Yang, L., Kopinja, J., Rooney, D. L., Ihrig, M. M., McManus, M. T., Gertler, F. B., et al,. (2003) Nat. Genet. 33, 401-406. [DOI] [PubMed] [Google Scholar]
- 10.Lee, N. S., Dohjima, T., Bauer, G., Li, H., Li, M. J., Ehsani, A., Salvaterra, P. & Rossi, J. (2002) Nat. Biotechnol. 20, 500-505. [DOI] [PubMed] [Google Scholar]
- 11.Xu, Y., Zhang, H. Y., Thormeyer, D., Larsson, O., Du, Q., Elmén, J., Wahlestedt, C. & Liang, Z. (2003) Biochem. Biophys. Res. Commun. 306, 712-717. [DOI] [PubMed] [Google Scholar]
- 12.Du, Q., Thonberg, H., Zhang, H. Y., Wahlestedt, C. & Liang, Z. (2004) Biochem. Biophys. Res. Commun. 325, 243-249. [DOI] [PubMed] [Google Scholar]
- 13.Tuschl, T. (2002) Nat. Biotechnol. 20, 446-448. [DOI] [PubMed] [Google Scholar]
- 14.Amarzguioui, M., Holen, T., Babaie, E. & Prydz, H. (2003) Nucleic Acids Res. 31, 589-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson, A. L., Bartz, S. R., Schelter, J., Kobayashi, S. V., Burchard, J., Mao, M., Li, B., Cavet, G. & Linsley, P. S. (2003) Nat. Biotechnol. 21, 635-637. [DOI] [PubMed] [Google Scholar]
- 16.Saxena, S., Jónsson, Z. O. & Dutta, A. (2003) J. Biol. Chem. 278, 44312-44319. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz, D. S., Hutvagner, G., Du, T., Xu, Z., Aronin, N. & Zamore, P. D. (2003) Cell 115, 199-208. [DOI] [PubMed] [Google Scholar]
- 18.Adachi, Y., Pavlakis, G. N. & Copeland T. D. (1994) J. Biol. Chem. 269, 2258-2262. [PubMed] [Google Scholar]
- 19.Myslinski, E., Ame, J. C., Krol, A. & Carbon, P. (2001) Nucleic Acids Res. 29, 2502-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suyama, E., Kawasaki, H., Nakajima, M. & Taira, K. (2003) Proc. Natl. Acad. Sci. USA 100, 5616-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen, G., Wehrman, T. S., Myers, J. W. & Blau, H. M. (2004) Nat Genet. 36, 183-189. [DOI] [PubMed] [Google Scholar]
- 22.Shirane, D., Sugao, K., Namiki, S., Tanabe, M., Iino, M. & Hirose, K. (2004) Nat. Genet. 36, 190-196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}