

Abstract
We report an efficient method for detecting functional RNAs. The approach, which combines comparative sequence analysis and structure prediction, already has yielded excellent results for a small number of aligned sequences and is suitable for large-scale genomic screens. It consists of two basic components: (i) a measure for RNA secondary structure conservation based on computing a consensus secondary structure, and (ii) a measure for thermodynamic stability, which, in the spirit of a z score, is normalized with respect to both sequence length and base composition but can be calculated without sampling from shuffled sequences. Functional RNA secondary structures can be identified in multiple sequence alignments with high sensitivity and high specificity. We demonstrate that this approach is not only much more accurate than previous methods but also significantly faster. The method is implemented in the program rnaz, which can be downloaded from www.tbi.univie.ac.at/~wash/RNAz. We screened all alignments of length n ≥ 50 in the Comparative Regulatory Genomics database, which compiles conserved noncoding elements in upstream regions of orthologous genes from human, mouse, rat, Fugu, and zebrafish. We recovered all of the known noncoding RNAs and cis-acting elements with high significance and found compelling evidence for many other conserved RNA secondary structures not described so far to our knowledge.
Keywords: comparative genomics, conserved RNA secondary structure
Traditionally, the role of RNA in the cell was considered mostly in the context of protein gene expression, limiting RNA to its function as mRNA, tRNA, and rRNA. The discovery of a diverse array of transcripts that are not translated to proteins but rather function as RNAs has changed this view profoundly (1–3). Noncoding RNAs (ncRNAs) are involved in a large variety of processes, including gene regulation (4), maturation of mRNAs, rRNAs, and tRNAs, or X-chromosome inactivation in mammals (5). In fact, a large fraction of the mouse transcriptome consists of ncRNAs (6), and about half of the transcripts from human chromosomes 21 and 22 are noncoding (7, 8). Structured RNA motifs furthermore function as cis-acting regulatory elements within protein-coding genes. Also in this context, new intriguing mechanisms are being discovered (9).
Hence, a comprehensive understanding of cellular processes is impossible without considering RNAs as key players. Efficient identification of functional RNAs (ncRNAs as well as cis-acting elements) in genomic sequences is, therefore, one of the major goals of current bioinformatics. Notwithstanding its utmost biological relevance, de novo prediction is still a largely unsolved issue. Unlike protein-coding genes, functional RNAs lack in their primary sequence common statistical signals that could be exploited for reliable detection algorithms. Many functional RNAs, however, depend on a defined secondary structure. In particular, evolutionary conservation of secondary structures serves as compelling evidence for biologically relevant RNA function. Comparative studies therefore seem to be the most promising approach. To date, complete genomic sequences of related species have been sequenced for almost all genetic model organisms as, for example, bacteria (10, 11), yeasts (12), nematodes (13, 14), and even mammals (15–17). Several studies (18–21) have identified a large collection of evolutionary conserved noncoding elements in mammalian (or, more generally, vertebrate) genomes, and it must be expected that a significant fraction of them are functional RNAs.
Possible candidates, however, have been identified only sporadically so far (19, 21), simply because there are no reliable tools to scan multiple sequence alignments for functional RNAs. The most widely used program qrna (22), which has been successfully used to identify ncRNAs in bacteria (23) and yeast (24), is not suitable for screens of large genomes. qrna is limited to pairwise alignments, and its reliability is low, especially if the evolutionary distance of the two sequences lies outside of the optimal range. An alternative approach, ddbrna (25), suffers from similar problems and so far has not been used in a real-life application. msari (26), on the other hand, gains its drastically enhanced accuracy from the large amount of information contained in large multiple sequence alignments of 10–15 sequences with high sequence diversity. At present, however, data sets of this kind are not available at a genomewide scale, at least for multicellular organisms.
In this article we address the problem by using an alternative approach: we combine a measure for thermodynamic stability with a measure for structure conservation. Using a combination of both scores we are able to efficiently detect functional RNAs in multiple sequence alignments of only a few sequences. Our method is substantially more accurate than qrna or ddbrna and performs better on pairwise alignments than msari does on alignments with 15 sequences. On the large, diverse alignments used for testing msari in ref. 26, our rnaz program achieved 100% sensitivity at 100% specificity.
Methods
Minimum Free Energy (MFE) RNA Folding. For MFE RNA folding we used the c libraries of the Vienna RNA package version 1.5 (27). We used rnafold for folding single sequences and rnaalifold (28) for consensus folding of aligned sequences. The same folding parameters were used for both algorithms to ensure that the obtained MFE values were comparable. For the covariation part of rnaalifold we used default parameters. Gaps were removed for single sequence folding.
Calculation of z Scores Using Support Vector Machine (SVM) Regression. To calculate z scores by regression analysis we used the following procedure: we generated synthetic sequences of different length and base composition. The length of the test sequences ranged from 50 to 400 nt in steps of 50. To quantify base composition, we used the GC/AT, A/T, and G/C ratios of the sequences and chose values for all ratios ranging from 0.25 to 0.75 in steps of 0.05. This process resulted in 10,648 points in a four-dimensional space of the independent variables. For each of the points we calculated the mean and standard deviation of the MFE of 1,000 random sequences, representing the dependent variables in our regression.
We used the SVM library libsvm (www.csie.ntu.edu.tw/~cjlin/libsvm) to train two regression models for mean and standard deviation. Input data for the SVM were scaled to mean of 0 and standard deviation of 1.
We chose the ν variant of regression and a radial basis function kernel. We optimized the parameters and found ν = 0.5, γ = 1, and C = 5 to yield the best results. Finally, we obtained two models for the mean and standard deviation we used for z-score calculation.
The traditional sampling of z scores depends on the randomization of the native sequence by shuffling the positions. In this context it was pointed out by Workman and Krogh (30) that a correct randomization procedure should conserve dinucleotide content because of the energy contributions of stacked base pairs in the energy model. In principle, the regression model could be extended to use dinucleotide frequencies. The good results with the simple model, however, allow us to neglect this effect.
Generation of the Test Alignments. Sequences for the test alignments were taken from the Rfam database (31) with the exception of the signal-recognition particle (SRP) RNA and RNaseP test sets, which were taken from other sources (32, 33) to use the same data as previous studies (22, 26). We used the procedure as described (34) to generate test sets consisting of a reasonable number of nonredundant alignments of different sizes, with a defined range of mean pairwise identities and in which all sequences were approximately equally represented.
Randomization of the Test Alignments. The program shufflealn.pl (34) was used to generate the randomized controls for alignments with up to n = 6 sequences. In brief, this program implements a randomization algorithm that takes care not to introduce randomization artifacts and produces random alignments of the same length, the same base composition, the same overall conservation, the same local conservation pattern, and the same gap pattern at the input alignment. For the large alignments (n = 10) we used the same procedure as in ref. 26: we completely shuffled all columns and realigned the alignment afterward by using clustalw.
SVM Classification. A binary classification SVM, again using libsvm, was trained to classify alignments as RNA or another sequence. Input parameters are the mean of the MFE z scores of the individual sequences in the alignments (without gaps), the structure conservation index (SCI) of the alignment, the mean pairwise identity, and the number of sequences in the alignment. For the final calibration of the SVM in the current implementation of rnaz we used all classes of ncRNA with the exception of tmRNAs and U70 small nucleolar RNAs (snoRNAs). For the tests on known families presented in this article, we generated models from all classes, leaving out one class at a time. In all cases, we used alignments with mean pairwise identities between ≈50% and 100% and two to six sequences per alignment. For each native alignment we included one randomized version in the training set. All parameters were scaled linearly from -1 to 1. We used a radial basis function-kernel and the parameters γ = 2 and C = 32 to train the models. The probability estimation option was used to obtain a model with probability information.
Test of Other Programs. We used qrna version 1.2b and ddbrna as available from the author (version of July 2004, www.tigem.it/Research/DiBernardoPersonalWebPage.htm). For the tests shown in Table 2, we chose the cutoffs log-odd = 15 for qrna and K = 1.5 for ddbrna, respectively. For rnaz we used a cutoff of P = 0.9 and customized models for the SVM that excluded both SRP RNA and RNaseP from the training set.
Table 2. Detection performance (sensitivity/specificity) for SRP RNA and RNAseP alignments with mean pairwise identities between 60% and 90%.
| No. of sequences in alignment
|
|||
|---|---|---|---|
| Program | 2 | 3 | 10 |
| qrna | 42.9/92.9 | — | — |
| ddbrna | 45.4/98.5 | 58.0/94.5 | — |
| msari | — | — | ≈ 56/100 |
| rnaz | 87.8/99.5 | 94.1/99.6 | 100/100 |
Results
The SCI. In a recent article (34) we demonstrated that the program rnaalifold [which originally was developed for prediction of secondary structure in aligned sequences (28)] also can be used for detection of evolutionarily conserved secondary structure. rnaalifold implements a consensus folding algorithm generalizing the standard dynamic programming algorithms for RNA secondary structure prediction algorithms (35) by adding sequence covariation terms to the folding energy model (36, 37). More precisely, a consensus MFE is computed for an alignment that is composed of an energy term averaging the energy contributions of the single sequences and a covariance term rewarding compensatory and consistent mutations (28). As this consensus MFE is difficult to interpret in absolute terms, we previously used a time-consuming random sampling method to assess its significance (34). This approach would require massive computational effort even for small-sized genomes and it does not seem practicable for large genomes as, for example, the human genome.
A much more efficient normalization can be achieved, however, by comparing the consensus MFE with the MFEs of each individual sequence in the alignment. To this end, we folded the alignment and calculated the consensus MFE EA of the alignment by using rnaalifold. If the sequences in the alignment fold into a conserved common structure, the average 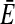 of the individual MFEs will be close to the MFE of the alignment,
of the individual MFEs will be close to the MFE of the alignment, 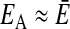 . Otherwise, the MFE of the alignment will be much higher (indicating a less stable structure) than the average of the individual sequences,
. Otherwise, the MFE of the alignment will be much higher (indicating a less stable structure) than the average of the individual sequences, 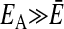 . We therefore define the SCI as
. We therefore define the SCI as
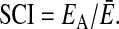 |
A SCI close to zero indicates that rnaalifold does not find a consensus structure, whereas a set of perfectly conserved structures has SCI ≈1. A SCI > 1 indicates a perfectly conserved secondary structure, which is, in addition, supported by compensatory and/or consistent mutations, which contribute a covariance score to EA.
A Normalized Measure for Thermodynamic Stability. It is widely believed that MFE predictions cannot be used for detection of functional RNAs after an in-depth study on the subject (38). Although thermodynamic stability is not significant alone, it still can be used as a valuable diagnostic feature because functional RNAs are indeed more stable than random sequences to some degree (34, 38). This effect is particularly dramatic in the case of microRNA precursors (39).
The significance of a calculated MFE value m is assessed by comparison with a large sample of random sequences. This approach was introduced 16 years ago (40) and is still widely used today (18, 19, 39). Typically, the normalized z score z = (m - μ)/σ is used, where μ and σ are the mean and standard deviations, respectively, of a large number of random sequences of the same length and same base or dinucleotide composition.
The parameters μ and σ are, by construction, functions of length and base composition. In the case of RNA molecules we found that they can be computed very accurately from a relatively simple regression model, which we obtained by means of a standard implementation of a SVM algorithm. SVMs are a set of related supervised learning methods with a solid mathematical foundation, applicable to both classification and regression. SVMs nonlinearly map their n-dimensional input space into a high dimensional feature space by using a so-called kernel function. In this way, nonlinear classifiers can be created by constructing linear classifiers in the feature space (41). To calibrate the regression model we used 1,000 random sequences for each of ≈10,000 points evenly spaced in the variable space spanned by chain length and base composition. Independent SVMs were trained for μ and σ (see Methods for details).
The accuracy of the SVM regression model was verified by comparing z scores from the SVM approach with z scores obtained by sampling (Fig. 1). We found that the correlation between sampled values and SVM values was as good as the correlation between two independently sampled z scores for the same test sequences at a sample size of 1,000. We therefore can replace the time-consuming sampling procedure by the SVM estimate without a significant loss of accuracy, while saving about a factor of 1,000 in computer time.
Fig. 1.

z scores calculated by SVM regression in comparison with z scores determined from 1,000 random samples for each data point. As test sequences we chose 100 sequences from random locations in the human genome and 100 known ncRNAs from the Rfam database (31). (Upper) Correlation of z scores from two independent samplings (mean squared error: 0.00990). (Lower) Correlation of calculated z scores and sampled z scores (mean squared error: 0.00998)
Classification Based on both Scores. To classify alignments as a functional RNA or other sequence we have to determine the separatrix between functional RNAs and other sequences in the SCI/z-score plane. Again, this is a typical application for SVMs; we therefore trained a binary classification SVM on test sets encompassing all major known classes of ncRNAs.
We generated test alignments by using clustalw of 12 well known ncRNA classes from Rfam (31) as well as random controls for which any native secondary structure is removed by shuffling the alignment positions (see Methods) and computed z score and SCI. Fig. 2 illustrates the results for a test set of tRNAs and 5S rRNAs. Fig. 3, which is published as supporting information on the PNAS web site, shows the results for the other ncRNA classes. We find that the combination of both scores reliably separates the native alignments from the randomized controls in two dimensions.
Fig. 2.

Classification based on z scores and SCI by using a SVM. Alignments of tRNAs and 5S rRNAs with two to four sequences per alignment and mean pairwise identities between 60% and 90% are shown. Green circles represent native alignments, and red crosses represent shuffled random controls. The background color ranging from red to green indicates the RNA class probability for different regions of the z–SCI plane.
To improve the performance of the binary classification SVM we used not only z score and SCI but also the mean pairwise identity and the number of sequences in the alignment as input parameters. In essence, this additional input teaches the SVM to interpret the information contained in the numerical value of the SCI depending on the sequence variation in the alignment. This refinement is necessary because the information content of a multiple alignment strongly depends on these parameters. In the extreme case, an alignment of identical sequences has SCI = 1 but does not contain any information about structural conservation at all. Because we used a randomized control that has the same number of sequences and the same pairwise sequence conservation together with each positive example, the calibration process was not biased by these additional variables.
The class probability P estimated by the SVM provides a convenient significance measure. Table 1 shows the sensitivity and specificity for detecting different ncRNA classes at different probability cutoffs. We used alignments with mean pairwise sequence identities between 60% and 100% and two to four sequences per alignment. At a cutoff of P = 0.9, we can detect on average 75.27% at a specificity of 98.93%.
Table 1. Detection performance for different classes of ncRNAs.
| Cutoff, classification probability
|
|||||||
|---|---|---|---|---|---|---|---|
| 0.5
|
0.9
|
0.99
|
|||||
| ncRNA type | N | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity |
| 5S ribosomal RNA | 297 | 81.48 (242) | 96.63 (10) | 68.69 (204) | 99.33 (2) | 33.00 (98) | 100.00 (0) |
| tRNA | 329 | 94.83 (312) | 93.62 (21) | 90.27 (297) | 97.87 (7) | 75.68 (249) | 99.70 (1) |
| SRP RNA | 464 | 100.00 (464) | 96.55 (16) | 96.55 (448) | 98.92 (5) | 66.16 (307) | 100.00 (0) |
| RNAse P | 291 | 98.97 (288) | 96.22 (11) | 84.19 (245) | 99.31 (2) | 56.70 (165) | 100.00 (0) |
| U2 spliceosomal RNA | 351 | 98.58 (346) | 97.72 (8) | 95.44 (335) | 99.15 (3) | 66.67 (234) | 99.72 (1) |
| U5 spliceosomal RNA | 285 | 91.58 (261) | 98.25 (5) | 81.75 (233) | 100.00 (0) | 70.53 (201) | 100.00 (0) |
| U3 snoRNA | 277 | 83.75 (232) | 98.56 (4) | 62.82 (174) | 99.28 (2) | 44.40 (123) | 99.64 (1) |
| U70 snoRNA | 363 | 61.16 (222) | 96.69 (12) | 35.54 (129) | 98.90 (4) | 17.91 (65) | 99.72 (1) |
| Hammerhead III ribozyme | 271 | 100.00 (271) | 95.20 (13) | 98.15 (266) | 98.89 (3) | 89.67 (243) | 99.26 (2) |
| Group II catalytic intron | 407 | 78.62 (320) | 96.31 (15) | 76.90 (313) | 98.53 (6) | 25.31 (103) | 100.00 (0) |
| tmRNA | 386 | 24.87 (96) | 96.37 (14) | 18.65 (72) | 98.19 (7) | 8.55 (33) | 99.48 (2) |
| MicroRNA mir-10 | 380 | 100.00 (380) | 95.26 (18) | 97.63 (371) | 99.21 (3) | 62.37 (237) | 100.00 (0) |
| Total | 4,101 | 84.17 (3,452) | 96.42 (147) | 75.27 (3,087) | 98.93 (44) | 50.18 (2,058) | 99.80 (8) |
Results for alignments with two to four sequences and mean pairwise identities between 60% and 100% are shown. N is the number of alignments in the test set. For each native alignment, one randomized alignment was produced, and randomized alignments classified as ncRNA were counted as false positives. Sensitivity and specificity are shown in percentage for three cutoffs of the RNA class probability predicted by the SVM. Absolute numbers of true positives and false negatives are shown in parentheses.
The accuracy of the classification depends quite strongly on the type of ncRNA. We can find most RNA classes with high sensitivities in the range of 80% to 100%. Only 2 of the 12 classes in our test set (U70 snoRNA and tmRNA) were difficult to detect. The scatter plots (Fig. 3) show that the U70 is quite stable but not very well conserved, whereas the tmRNA has a conserved secondary structure that is obviously not very stable and moreover contains pseudoknots. Alignments with more sequences are needed to detect these two RNA classes quantitatively.
We emphasize that, although we use here a machine learning approach for classification, we do not train the SVM on specific sequences, sequence patterns, structure motifs, conservation patterns, or base compositions. We use the SVM solely as a guide to interpret the SCI and z score, which represent two diagnostic features that do not contain any information that is specific for a particular class of ncRNAs. In fact, it would be interesting to replace the SVM by a direct statistical model. To demonstrate that our classification procedure is generally applicable and not biased toward ncRNA classes of the training set, we trained the SVM by excluding particular classes of ncRNAs and used those models to classify the excluded ncRNAs and their randomized controls. The sensitivities summarized in Table 1 therefore can also be expected for novel classes of structured ncRNAs.
Comparison to Other Methods. RNaseP and SRP RNAs have repeatedly been used for benchmarking ncRNA detection algorithms (22, 26). We therefore use these data sets here as well. For the comparison to qrna and ddbrna we used pairwise and three-way alignments with mean pairwise identities between 60% and 90%, respectively. In contrast to the previous section, we excluded alignments with identities >90% because both qrna and ddbrna are known to perform poorly on such input data. We used a cutoff of P = 0.9 for rnaz and chose the cutoffs for the other programs in a way that the specificity is at least 90%. Results are summarized in Table 2. We found that rnaz is substantially more sensitive on both pairwise and three-way alignments than qrna and ddbrna and at the same time has a larger specificity.
We also tested our method on larger alignments with 10 sequences as used for benchmarking msari. We generated 150 alignments that had mean pairwise identities between 50% and 70%. Our SVM classification model currently is trained only for up to six sequences so we did not use it for the classification of this test set. It turns out, however, that the simple rule SCI ≥ 0.3 and z ≤ -1.5 perfectly separates the native alignments from the controls with 100% sensitivity and 100% specificity by using either of the two scores without help of a SVM. Although the alignments produced by clustalw are, at this level of sequence similarity, structurally not perfectly correct, our consensus folding algorithm still finds the correct common structure and the SCI is still significant, albeit at lower levels.
At the time this article was written, no executable version of msari was available so we can compare rnaz only with the published results: according to ref. 26, msari achieves at best a sensitivity of 56% at 100% specificity for clustalw alignments of n = 10 RNaseP or SRP RNA sequences.
Implementation and Run Time. The method described above was implemented in rnaz by using the c programming language. The time complexity of our method is 𝒪 (N × n3), where N is the number of sequences and n is the length of the alignment. Table 3 compares the run time for pairwise alignments of different lengths between rnaz and the alternative methods: rnaz is not only more accurate but also significantly faster than the other methods. (A comparison with msari was not possible because no implementation is publicly available. It should have similar run times as rnaz, however, because it also uses the RNA folding routines of the Vienna RNA package as the rate-limiting step.)
Table 3. Computer time in s for 1,000 alignments on an Intel 2.4-GHz Pentium 4.
| Alignment length
|
|||
|---|---|---|---|
| Program | 100 | 200 | 300 |
| qrna | 485 | 4,044 | 14,777 |
| ddbrna | 741 | 921 | 1,522 |
| rnaz | 163 | 375 | 754 |
Screening the Comparative Regulatory Genomics (CORG) Database for Functional RNA Structures. The CORG database is a collection of conserved sequence elements in noncoding, genomic DNA (42). The release 2.0 version contains multiple sequence alignments of conserved elements in the upstream regions (up to 15 kb from the translation start) of orthologous protein-coding genes from human, mouse, rat, Fugu, and zebrafish. We focus here on the 4,263 conserved noncoding blocks (CNBs) that are >50 nt.
We scanned the alignments by using rnaz; after clustering overlapping and redundant CNBs we found 89 distinct regions are predicted as structural RNA with P > 0.5. Of these, 28 score with P > 0.9 (see Table 4). Among the predicted RNAs we can find all known ncRNAs from Rfam (31) and the miRNA registry (43) that are located in the upstream regions of known protein-coding genes. We identified six micro-RNAs with P > 0.99 and the snoRNA U93 with P = 0.72. Furthermore, we also could reliably (P > 0.98) detect known structural cis-acting elements (44); in particular, we encountered four internal ribosome entry sites (45) and one iron response element (46).
Table 4. Top-scoring alignments in the CORG database.
| CORG ID | P | Genomic context | Function |
|---|---|---|---|
| 110355 | 1.000 | 5′ UTR of Di George syndrome critical region gene 8 | IRES |
| 194820 | 1.000 | Micro RNA: mir-196b | |
| 226470 | 1.000 | MicroRNA: mir-10a | |
| 288188 | 1.000 | Micro RNA: mir-10b | |
| 393758 | 1.000 | 5′ UTR of solute carrier family 40 (iron-regulated transporter) | IRE |
| 119596 | 0.999 | Micro RNA: mir-34b | |
| 159932 | 0.999 | Micro RNA: mir-138-2 | |
| 373196 | 0.999 | Not annotated | Unknown |
| 453969 | 0.999 | Coding exon of retinoic acid-induced 17 | Unknown |
| 461749 | 0.999 | Coding exon of CIN85-associated multidomain containing RhoGAP | Unknown |
| 264053 | 0.997 | 5′ UTR of brain chitinase like protein 2 | IRES |
| 376858 | 0.997 | Not annotated | Unknown |
| 405712 | 0.997 | 5′ UTR exon of nuclear factor of activated T cells 5, tonicity-responsive (NFAT5) | Unknown |
| 391315 | 0.996 | Micro RNA: mir196a-2 | |
| 386451 | 0.985 | 5′ UTR of a hypothetical protein | Unknown |
| 260572 | 0.984 | Upstream of a hypothetical protein | Unknown |
| 430443 | 0.983 | Upstream/5′ UTR of Hairy and enhancer of split 1 | Unknown |
| 57635 | 0.980 | 5′ UTR of a hypothetical protein | IRES |
| 238772 | 0.980 | 5′ UTR of a hypothetical protein | IRES |
| 284325 | 0.964 | Intron of skeletal muscle LIM-protein 2 | Unknown |
| 134297 | 0.963 | Not annotated | Unknown |
| 501416 | 0.961 | Coding region of hypothetical protein | Unknown |
| 363131 | 0.950 | Upstream of Eyes absent 1 | Unknown |
| 386639 | 0.950 | 5′ UTR of ribosomal protein L12 | Unknown |
| 143688 | 0.938 | Upstream of zinc finger protein 503 | Unknown |
| 456164 | 0.921 | Intron of the spliced 5′ UTR of checkpoint suppressor 1 | Unknown |
| 154812 | 0.918 | Upstream/5′ UTR of basic helix-loop-helix domain containing, class B 5 | Unknown |
| 406119 | 0.902 | Upstream of zinc finger protein of the cerebellum 3 | Unknown |
IRE, iron response element. IRES, internal ribosome entry site.
Thus only 11 of the 89 rnaz hits are known ncRNAs or cis-acting structures. This leaves us with 78 candidates, 17 of which have rnaz probabilities above P = 0.9. We estimated the specificity in this screen by scoring random controls and found that the P = 0.5 and P = 0.9 cutoffs have associated specificities of 99.2% and 99.9%, respectively. This finding is even higher than in the test examples; we therefore are confident that most of these hits are true positives.
Table 4 lists the top hits and their genomic context. We found several hits in 5′ UTRs of protein-coding genes, as for example in NFAT5, the only known transcription factor involved in osmoregulation in mammalian cells. NFAT5 has a spliced 5′ UTR, and in one exon we found a stable and conserved stem–loop structure (CNB-405712). Interestingly, several splice variants of this mRNA exist, some of which have this exon, whereas others do not. We suspect that CNB-405712 is an important regulatory module of the NFAT5 mRNA.
Significant hits also were found in introns, even though introns are not systematically covered in the current release of CORG. For example, CNB-284325 is a structurally highly conserved element supported by many compensatory mutations in the intron of a muscle-specific LIM domain protein. This structure probably is part of a ncRNA.
Some other hits are not directly related to any known protein-coding genes. CNB-134297 is an exceptionally large (≈1,800 nt) conserved region without any annotation or predicted coding capacity. We scanned alignments >300 in sliding windows of size 300 and slide 50. In this special case, significant RNA structures were predicted in several independent windows. This region is thus a strong candidate for a novel ncRNA.
The CORG database sporadically contains alignments of coding regions, and we also found significant secondary structures in some of them (e.g., CNB-453969: P = 0.999). In some instances we could detect only a signal in the reverse complement strand compared with the mRNA, possibly indicating structured antisense transcripts. For some hits, this prediction was additionally supported by EST data. We routinely scanned the reverse complement for all alignments, because rnaz scores are generally higher if the RNA in question is provided in the correct orientation. The snoRNA U93 found in CNB-470004 is a good example demonstrating the remarkable sensitivity of rnaz. It is predicted as RNA with P = 0.72 in its correct orientation, whereas there is no significant signal in the reverse complement strand (P = 0.06).
A detailed description of all 89 hits can be found at www.tbi.univie.ac.at/papers/SUPPLEMENTS/RNAz, where we provide links to the University of California, Santa Cruz genome browser (47), allowing a detailed study of the genomic context for all hits (annotation, mRNA structure, ESTs, etc.). Unlike other methods, rnaz does not only predict the existence of a functional RNA element, it also predicts an accurate model of the consensus structure. These can also be found at www.tbi.univie.ac.at/papers/SUPPLEMENTS/RNAz together with the annotation of compensatory mutations.
Discussion
We have described here a versatile method for detecting functional RNAs in genomic screens. This approach can reliably detect a surprisingly wide variety of different ncRNAs and cis-acting RNA elements by using only evolutionary conservation and thermodynamic stability as characteristic signal. Although conceptually simple, the SCI proved to be a convenient and effective measure of structural conservation. Our stability measure, on the other hand, shows that, contrary to common belief, thermodynamic stability can be useful for ncRNA detection. As a consensus of several independent sequences in an alignment, stability can be a significant measure. Furthermore, we have demonstrated that a properly normalized stability measure can be directly calculated without the need for time-consuming sampling of shuffled sequences or alignments. Our results show that rnaz is suitable for large-scale genomic annotation whenever alignments can be obtained.
A wealth of genomic data together with new methods for generating high-quality alignments (29) are already available. Aided by visualization tools (47), we aim to draw genomewide maps of significant RNA structures. This approach of “computational RNomics” opens a perspective, which we hope will result in the discovery of additional terrain in the expanding RNA world of cellular mechanisms.
Supplementary Material
Acknowledgments
We thank Christoph Dieterich and Martin Vingron for permission to use release 2.0 of their CORG databases before publication and Paul Gardner and Andrea Tanzer for discussion. This work was supported in part by the Austrian Genome Research Program Bioinformatics Integration Network sponsored by Bundesministerium für Bildung, Wissenschaft und Kultur and Bundesministeriums für Wirtschaft und Arbeit, Austrian Fonds zur Förderung der Wissenschaftlichen Forschung Project P-15893, and the Bioinformatics Initiative of the Deutsche Forschungsgemeinschaft (Grant BIZ-6/1-2).
Abbreviations: ncRNA, noncoding RNA; snoRNA, small nucleolar RNA; SRP, signal-recognition particle; MFE, minimum free energy; SCI, structure conservation index; SVM, support vector machine; CNB, conserved noncoding block; CORG, Comparative Regulatory Genomics.
See Commentary on page 2269.
References
- 1.Eddy, S. R. (2001) Nat. Rev. Genet. 2, 919-929. [DOI] [PubMed] [Google Scholar]
- 2.Storz, G. (2002) Science 296, 1260-1263. [DOI] [PubMed] [Google Scholar]
- 3.Mattick, J. S. (2003) BioEssays 25, 930-939. [DOI] [PubMed] [Google Scholar]
- 4.He, L. & Hannon, G. J. (2004) Nat. Rev. Genet 5, 522-531. [DOI] [PubMed] [Google Scholar]
- 5.Avner, P. & Heard, E. (2001) Nat. Rev. Genet. 2, 59-67. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki, M. & Hayashizaki, Y. (2004) BioEssays 26, 833-843. [DOI] [PubMed] [Google Scholar]
- 7.Cawley, S., Bekiranov, S., Ng, H. H., Kapranov, P., Sekinger, E. A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A. J., et al. (2004) Cell 116, 499-509. [DOI] [PubMed] [Google Scholar]
- 8.Kampa, D., Cheng, J., Kapranov, P., Yamanaka, M., Brubaker, S., Cawley, S., Drenkow, J., Piccolboni, A., Bekiranov, S., Helt, G., et al. (2004) Genome Res. 14, 331-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nudler, E. & Mironov, A. S. (2004) Trends Biochem. Sci. 29, 11-17. [DOI] [PubMed] [Google Scholar]
- 10.McClelland, M., Florea, L., Sanderson, K., Clifton, S. W., Parkhill, J., Churcher, C., Dougan, G., Wilson, R. K. & Miller, W. (2000) Nucleic Acids Res. 28, 4974-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Florea, L., McClelland, M., Riemer, C., Schwartz, S. & Miller, W. (2003) Nucleic Acids Res. 31, 3527-3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423, 241-254. [DOI] [PubMed] [Google Scholar]
- 13.C. elegans Sequencing Consortium (1998) Science 282, 2012-2018. [DOI] [PubMed] [Google Scholar]
- 14.Stein, L. D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M. R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003) PLoS Biol. 1, E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genome Sequencing Consortium (2001) Nature 409, 860-921. [DOI] [PubMed] [Google Scholar]
- 16.International Mouse Genome Sequencing Consortium (2002) Nature 420, 520-562. [DOI] [PubMed] [Google Scholar]
- 17.Rat Genome Sequencing Project Consortium (2004) Nature 428, 493-521.15057822 [Google Scholar]
- 18.Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S. & Haussler, D. (2004) Science 304, 1321-1325. [DOI] [PubMed] [Google Scholar]
- 19.Bejerano, G., Haussler, D. & Blanchette, M. (2004) Bioinformatics 20, Suppl. 1, I40-I48. [DOI] [PubMed] [Google Scholar]
- 20.Thomas, J. W., Touchman, J. W., Blakesley, R. W., Bouffard, G. G., Beckstrom-Sternberg, S. M., Margulies, E. H., Blanchette, M, Siepel, A. C., Thomas, P. J., McDowell, J. C., et al. (2003) Nature 424, 788-793. [DOI] [PubMed] [Google Scholar]
- 21.Margulies, E. H., Blanchette, M., Haussler, D. & Green, E. D. (2003) Genome Res. 13, 2507-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivas, E. & Eddy, S. R. (2001) BMC Bioinformatics 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivas, E., Klein, R. J., Jones, T. A. & Eddy, S. R. (2001) Curr. Biol. 11, 1369-1373. [DOI] [PubMed] [Google Scholar]
- 24.McCutcheon, J. P & Eddy, S. R. (2003) Nucleic Acids Res. 31, 4119-4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.di Bernardo, D., Down, T. & Hubbard, T. (2003) Bioinformatics 19, 1606-1611. [DOI] [PubMed] [Google Scholar]
- 26.Coventry, A, Kleitman, D. J. & Berger, B. (2004) Proc. Natl. Acad. Sci. USA 101, 12102-12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofacker, I. L., Fontana, W., Stadler, P. F., Bonhoeffer, S., Tacker, M. & Schuster, P. (1994) Monatsh. Chemie 125, 167-188. [Google Scholar]
- 28.Hofacker, I. L., Fekete, M. & Stadler, P. F. (2002) J. Mol. Biol. 319, 1059-1066. [DOI] [PubMed] [Google Scholar]
- 29.Blanchette, M., Kent, W. J., Riemer, C., Elnitski, L., Smit, A. F., Roskin, K. M., Baertsch, R., Rosenbloom, K., Clawson, H., Green, E. D., et al. (2004) Genome Res. 14, 708-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Workman, C. & Krogh, A. (1999) Nucleic Acids Res. 27, 4816-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. (2003) Nucleic Acids Res. 31, 439-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenblad, M. A., Gorodkin, J., Knudsen, B., Zwieb, C. & Samuelsson, T. (2003) Nucleic Acids Res. 31, 363-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown, J. W. (1999) Nucleic Acids Res. 27, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Washietl, S. & Hofacker, I. L. (2004) J. Mol. Biol. 342, 19-30. [DOI] [PubMed] [Google Scholar]
- 35.Zuker, M. & Stiegler, P. (1981) Nucleic Acids Res. 9, 133-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter, A. E., Turner, D. H., Kim, J., Lyttle, M. H., Muller, P., Mathews, D. H. & Zuker, M. (1994) Proc. Natl. Acad. Sci. USA 91, 9218-9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathews, D. H., Sabina, J., Zuker, M. & Turner, D. H. (1999) J. Mol. Biol. 288, 911-940. [DOI] [PubMed] [Google Scholar]
- 38.Rivas, E. & Eddy, S. R. (2000) Bioinformatics 16, 583-605. [DOI] [PubMed] [Google Scholar]
- 39.Bonnet, E., Wuyts, J., Rouze, P. & Van De Peer, Y. (2004) Bioinformatics 20, 2911-2917. [DOI] [PubMed] [Google Scholar]
- 40.Le, S. V., Chen, J. H., Currey, K. M. & Maizel, J. V., Jr. (1988) Comput. Appl. Biosci. 4, 153-159. [DOI] [PubMed] [Google Scholar]
- 41.Cristianini, N. & Shawe-Taylor, J. (2000) An Introduction to Support Vector Machines (Cambridge Univ. Press, Cambridge, U.K.).
- 42.Dieterich, C., Wang, H., Rateitschak, K., Luz, H. & Vingron, M. (2003) Nucleic Acids Res. 31, 55-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffiths-Jones, S. (2004) Nucleic Acids Res. 32, D109-D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pesole, G., Liuni, S., Grillo, G., Licciulli, F., Mignone, F., Gissi, C. & Saccone, C. (2002) Nucleic Acids Res. 30, 335-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le, S. Y. & Maizel, J. V., Jr. (1997) Nucleic Acids Res. 25, 362-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hentze, M. W. & Kuhn, L. C. (1996) Proc. Natl. Acad. Sci. USA 93, 8175-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M. & Haussler, D. (2002) Genome Res. 12, 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.