

Abstract
Background
KG-135, a standardized formulation enriched with Rk1, Rg3, and Rg5 ginsenosides, has been shown to inhibit various types of cancer cells; however, the underlying mechanisms are not fully understood. In this study, we explored its effects in A549 human lung cancer cells to investigate the induction of Forkhead Class box O3a (FOXO3a) and autophagy.
Methods
Cell viability was determined by sulforhodamine B staining. Apoptosis and cell cycle distribution were analyzed using flow cytometry. The changes of protein levels were determined using Western blot analysis. Autophagy induction was monitored by the formation of acidic vesicular organelles stained with acridine orange.
Results
KG-135 effectively arrested the cells in G1 phase with limited apoptosis. Accordingly, a decrease of cyclin-dependent kinase-4, cyclin-dependent kinase-6, cyclin D1, and phospho-retinoblastoma protein, and an increase of p27 and p18 proteins were observed. Intriguingly, KG-135 increased the tumor suppressor FOXO3a and induced the accumulation of autophagy hallmark LC3-II and acidic vesicular organelles without an increase of the upstream marker Beclin-1. Unconventionally, the autophagy adaptor protein p62 (sequestosome 1) was increased rather than decreased. Blockade of autophagy by hydroxychloroquine dramatically potentiated KG-135-induced FOXO3a and its downstream (FasL) ligand accompanied by the cleavage of caspase-8. Meanwhile, the decrease of Bcl-2 and survivin, as well as the cleavage of caspase-9, were also drastically enhanced, resulting in massive apoptosis.
Conclusion
Besides arresting the cells in G1 phase, KG-135 increased FOXO3a and induced an unconventional autophagy in A549 cells. Both the KG-135-activated extrinsic FOXO3a/FasL/caspase-8 and intrinsic caspase-9 apoptotic pathways were potentiated by blockade of autophagy. Combination of KG-135 and autophagy inhibitor may be a novel strategy as an integrative treatment for cancers.
Keywords: autophagy, FOXO3a, ginsenoside, KG-135, lung cancer
1. Introduction
Panax ginseng Meyer, an important and popular medicinal plant, has been widely used for several thousand years as a tonic, prophylactic, and restorative agent in Asian countries [1]. Its remedial effects in various disorders has been demonstrated by modern pharmacological studies [2]. Recently, the anticancer functions of P. ginseng have been increasingly recognized [3]. P. ginseng can be classified as white or red ginseng, according to processing conditions. The white ginseng is processed by air drying and the red ginseng is processed by steaming and heating to alter or enhance the pharmacological activities [4]. Several specific ginsenosides are contained in red ginseng but not detected in white ginseng [5]. These red ginseng-specific ginsenosides have been shown to exert more potent cytostatic or cytotoxic activity than other ginsenosides in various cancer cells [6], [7]. Sun ginseng, a specific heat-processed ginseng, was developed in order to increase the content of Rk1, Rg3, and Rg5 ginsenosides, which have not been isolated from white ginseng and thus exhibit more anticancer activities [8]. Recently, increasing the content of ginsenosides Rk1, Rg3, and Rg5 by microwave-assisted processing was shown to enhance the anticancer effects of P. ginseng extract [9], inicating the crucial roles of these three ginsenosides in the anticancer activities of P. ginseng.
The anticancer ginsenosides of sun ginseng are present in a butanol-soluble fraction, known as KG-135 [8], that contains equal amounts of Rk1, Rg3, and Rg5 [5]. This standardized ginsenoside-rich fraction, KG-135, has been shown to inhibit 12-O-tetradecanoylphorbol-13-acetate-induced cyclooxygenase-2 expression in human breast epithelial cells by blocking the c-Jun N-terminal kinase/activator protein-1 signaling pathway [10], suppressing the proliferation of human prostate cancer cells both in vitro and in vivo [8], downregulating G1 cyclin-dependent kinase through the proteasome-mediated pathway [5], and potentiating etoposide-induced apoptosis in human cervical cancer cells [11]. The anticancer effects shown in previous studies of KG-135 are substantial and effective; however, the extent of apoptosis induction by KG-135 in cancer cells seems limited. Some molecular events might serve as prosurvival mechanisms, by which the cancer cells escape from KG-135-induced apoptotic cell death.
Autophagy is an evolutionarily conserves the intracellular degradation process, which sequestrates cytoplasmic components and organelles to form a double-membraned autophagosome for subsequent lysosomal degradation. The degradation products are transported back to the cytoplasm for biosynthesis or energy production [12], [13]. In addition to the turnover of cellular proteins and organelles, autophagy has been implicated in various physiological and pathophysiological functions, including starvation adaptation, cellular differentiation and death, antiaging, elimination of microorganisms, antigen presentation, cancer development, and treatment response, etc., [14], [15], [16]. It is considered an adaptive response to maintain cell survival under different stress conditions by ensuring metabolic supply or removing protein aggregates and abnormal organelles. However, beyond a certain threshold, excessive degradation by autophagy ignites a nonapoptotic (Type II) programmed cell death [15], [17]. Increasing evidence indicates that autophagy constitutes a potential target for cancer therapy [18]. Activation of prosurvival autophagy protects some cancer cells against anticancer treatments by blocking the apoptotic pathway, although some therapies could induce autophagic cell death in cancer cells. Manipulation of autophagy has been suggested as a potential strategy to improve anticancer therapies [18], [19].
In addition to apoptosis, autophagy induction was also observed in cancer cells treated with ginsenoside Rg3 [20] or microwave-irradiated P. ginseng extract containing higher content of Rk1, Rg3, and Rg5 [9]. How autophagy affects the anticancer activity of ginsenoside-rich fractions such as KG-135 has not been reported. It is tempting to investigate the related crucial molecular events.
Lung cancer is the most common cancer and the leading cause of cancer death worldwide [21]. Nonsmall cell lung cancer (NSCLC) constitutes more than 80% of lung cancers. Because NSCLC is relatively resistant to chemotherapy and radiation therapy, the 5-yr survival rate is poor and hardly reaches 20% [22], [23], [24]. New approaches are needed to tackle this disease. As such, natural phytochemicals like ginsenosides or their derivatives have being studied for their potential in NSCLC treatment [25], [26], [27], [28], [29], [30].
In this study, we explore the effects of KG-135 in A549 human NSCLC cells and investigate the role of autophagy in KG-135-mediated anticancer activities. In addition to the regulation of G1/S transition machinery components, our results show KG-135 increases the protein level of tumor suppressor Forkhead Class box O3a (FOXO3a), which positively regulates genes that promote cell cycle arrest and apoptosis [31]. Moreover, we found an unconventional autophagy in KG-135-treated cells as the accumulation of LC3-II was not only without change of Beclin-1, but was also accompanied by an increase of p62 (sequestosome 1). Blockade of autophagy by hydroxychloroquine (HCQ) potentially enhances the KG-135-mediated increase of FOXO3a and its downstream death receptor ligand Fas ligand (FasL), as well as the decrease of survivin and Bcl-2, resulting in massive apoptosis. Autophagy appears to be a survival factor for the cancer cells to escape the apoptotic death induced by KG-135. Combing KG-135 with autophagy inhibitor might be a potential strategy as an integrative medicine for cancer treatment.
2. Materials and methods
2.1. Cell culture
The human NSCLC A549 and normal human fetal lung fibroblast WI-38 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). A549 and WI-38 cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 and Dulbecco's Modified Eagle Medium (Gibco, Grand Island, NY, USA), respectively, supplemented with 10% fetal bovine serum (Caisson, North Logan, UT, USA), 1 × penicillin–streptomycin–glutamine (Corning, Manassas, VA, USA), and 1 × nonessential amino acids (Corning, Manassas, VA, USA). Cells were cultured at 37°C in a water-jacketed 5% CO2 incubator.
2.2. Reagents and chemicals
The quality-controlled standardized ginsenoside formulation KG-135 was prepared as described previously [11] and supplied from the Ginseng Science Inc. (Seoul, Korea). It was dissolved in dimethylsulfoxide (Sigma, St. Louis, MO, USA) and diluted in sterile culture medium immediately prior to use. The final concentrations of dimethylsulfoxide were all below 0.1%. Sulforhodamine B (SRB), trichloroacetic acid, acridine orange, propidium iodide, HCQ, and metformin were purchased from Sigma.
2.3. Assessment of the cell viability
A549 and WI-38 cells were seeded respectively in 96-well plate at a density of 1,600 cells/well for 24 h and then treated with drugs or sterile culture medium for 72 h. The cell viability was measured with SRB binding assay. Briefly, the cells were fixed with 10% trichloroacetic acid and incubated for 1 h at 4°C. The plates were then washed twice with tap water and air dried. The dried plates were stained with 80 μL of 0.4% (w/v) SRB prepared in 1% (v/v) acetic acid for 30 min at room temperature. The plates were rinsed quickly twice with 1% acetic acid to remove unbound SRB, and then air dried until no moisture was visible. The bound dye was solubilized in 20 mmol/L Tris base (200 μL/well) for 5 min on a shaker. Optical densities were read on a microplate reader (ELx800; BioTek, Winooski, VT, USA) at 570 nm. The optical density is directly proportional to the cell number over a wide range.
2.4. Analysis of cell-cycle distribution and sub-G1 apoptotic fraction
One day after being seeded in a six-well plate (5 × 104 cells/mL, 2 mL/well), the A549 cells were treated with agents as indicated in the figure legends for 72 h. At harvest, cells were fixed in ice-cold 70% ethanol and stored at −20°C. Cells were then washed twice with ice-cold phosphate-buffered saline and then incubated with RNase and DNA intercalating dye propidium iodide (50 μg/mL) at room temperature for 20 min. The percentages of sub-G1 apoptotic fraction and cell-cycle distribution were then analyzed using a flow cytometer (Beckman Coulter EPICS XL, Fullerton, CA, USA).
2.5. Western blot
A549 cells were seeded in 10-cm dishes at a density of 4 × 105 cells/dish for 24 h and then treated with agents as described in the figure legends. On the day of harvest, the whole-cell lysates were prepared with 1 × radioimmunoprecipitation lysis buffer (Millipore, Billerica, MA, USA) containing 1 × tyrosine phosphatase inhibitor cocktail (FC0020-0001; BIONOVAS, Toronto, Canada), 1 × protease inhibitor cocktail, full range (FC0070-0001, BIONOVAS), and 1 × serine/threonine phosphatase inhibitor cocktail (FC0030-0001, BIONOVAS). The protein extracts were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (GE Healthcare, Pittsburgh, PA, USA) by electroblotting. The membranes were blocked with 5% bovine serum albumin in Tris-buffered saline and Tween 20 (25mM Tris-HCl, 125mM NaCl, 0.1% Tween 20) for 1 h at room temperature and probed with primary antibody overnight at 4°C and then with horseradish peroxidase–conjugated secondary antibody for 1 h at room temperature. The immune complexes were visualized using an enhanced chemiluminescence detection system (ECL; Perkin Elmer, Waltham, MA, USA) according to the manufacturer's instructions.
2.6. Antibodies
Primary antibodies against phospho-retinoblastoma protein (pRb) (Ser807/811; #9308), p18 (#2896), FOXO3a (#2497), LC3-I/II (#4108), p62/sequestosome 1 (#5114), cleaved caspase-9 (Asp330; #7237), cleaved caspase-9 (Asp315; #9505), and FasL (#4273) were purchased from Cell Signaling (Danvers, MA, USA). Primary antibodies for cyclin-dependent kinase (CDK)4 (ab68266), CDK6 (ab124821), cyclin D1 (ab134175), glyceraldehyde 3-phosphate dehydrogenase (ab8245), and Bcl-2 (ab32124) were purchased from Abcam (Cambridge, MA, USA). Primary antibody for Rb (sc-102) was purchased from Santa Cruz Biotechnology (San Diego, CA, USA). Primary antibodies for p27 (1591-1), Beclin-1 (2026-1), and full-length caspase-8 (1006-1) were purchased from Epitomics (Burlingame, CA, USA).
2.7. Detection of acidic vesicular organelles
Autophagy is featured by the formation of autophagosomes and autolysosomes, which are acidic vesicular organelles (AVOs) [32]. Acridine orange, a weak base dye, moves freely across biological membranes and emits green fluorescence when uncharged. Its protonated form accumulates in AVOs (consisting predominantly of autophagosomes and autolysosomes), where it forms aggregates that emit red fluorescence [32], [33]. To analyze the formation of AVOs, the A549 cells were seeded in a six-well plate and treated as described in the figure legends. After 72 h of treatment, the cells were stained with acridine orange (5 μg/mL) for 20 min at room temperature. After washing out the excess dye, green (510–530 nm) and red (>650 nm) fluorescence emission from cells illuminated with blue (488 nm) excitation light was visually examined under a fluorescence microscope (ECLIPSE TE300; Nikon, Tokyo, Japan) and quantified using flow cytometry (EPICS XL; Beckman Coulter, Fullerton, CA, USA), respectively.
2.8. Photograph of the cells
The phase contrast images of cells were photographed using a digital microscope camera (PAXcam2+,Villa Park, IL, USA) adapted to an inverted microscope (CKX31; Olympus, Tokyo, Japan) at 10× objective lens magnification. The images of acridine orange-stained AVOs were taken using a digital microscope camera (SPOT FLEX; Diagnostic Instruments, Sterling Heights, MI, USA) under a fluorescence microscope (ECLIPSE TE300; Nikon, Tokyo, Japan) at 20× objective lens magnification.
2.9. Statistical analysis
Cell viability data are expressed as mean ± standard error. In Fig. 1, Fig. 2, differences between groups were evaluated using one-way analysis of variance followed by Dunnett's t-test. Significance values are represented by single (p < 0.05), double (p < 0.01), and triple (p < 0.001) asterisks.
Fig. 1.

Effects of KG-135 on the proliferation and cell cycle distribution of A549 human nonsmall cell lung cancer cells. (A) After treatment with KG-135 for 72 h, the cell viability of A549 cells was measured using sulforhodamine B binding assay. (B) The cell cycle distribution was analyzed using flow cytometry. Cell viability data are expressed as mean ± standard error and analyzed as described in Section 2.
Fig. 2.

Effects of KG-135 and autophagy inhibitor hydroxychloroquine (HCQ) on the cell viability of normal human fetal lung fibroblast WI-38 cells. (A) After treatment with KG-135 alone or in the presence of HCQ for 72 h, the cell viability was determined using sulforhodamine B binding assay and (B) examined using phase contrast microscopy, scale bar = 100 μm. Cell viability data are expressed as mean ± standard error and analyzed as described in Section 2.
3. Results
3.1. KG-135 arrests the cell cycle of A549 human NSCLC cells in G1-phase
A549 human NSCLC cells were treated with various concentrations of KG-135 for 72 h and then the cell viability was measured using SRB staining assay. As shown in Fig. 1A, KG-135 dose-dependently inhibited the proliferation of A549 cells, with IC50 at about 86 μg/mL. To determine whether this inhibitory effect of KG-135 was due to apoptosis induction or cell-cycle regulation, analysis of cellular DNA content was performed using flow cytometry after staining the cells with propidium iodide. After 72 h of treatment, the percentage of cells in G1 phase was markedly increased from 66.5% of control to 83.6% of 100 μg/mL KG-135-treated cells (Fig. 1B). Reciprocally, the percentages of cells in S and G2/M phases were decreased from 14% and 19.5% to 6.9% and 9.6%, respectively. Nevertheless, the sub-G1 apoptotic fraction was minimally elevated from 1.8% to 6.3% (Fig. 1B). Apparently, at doses <100 μg/mL, the effect of KG-135 against A549 cells is mainly attributed to G1 phase arrest rather than apoptosis induction.
3.2. KG-135 regulates the protein levels of G1/S transition components and tumor suppressor FOXO3a in A549 human NSCLC cells
We then analyzed the cell cycle regulatory proteins controlling the G1-S phase transition in cells treated with KG-135. The results show that KG-135 markedly decreased the protein levels of CDK4, CDK6, and cyclin D1 (Fig. 3A). As these CDKs and cyclin proteins could inactivate Rb by phosphorylation, the phosphorylation of Rb was dose-dependently inhibited accordingly (Fig. 3A). In addition, the cyclin-dependent kinase inhibitors able to arrest the cell cycle in G1 phase, such as p18 and p27, were upregulated (Fig. 3B). As p27 is a downstream target of tumor suppressor FOXO3a [34], we examined the effect of KG-135 on this tumor suppressor in A549 cells. Intriguingly, we found the protein level of FOXO3a was actually increased after treatment with KG-135 (Fig. 3C).
Fig. 3.

Effects of KG-135 on the cell cycle regulation proteins and tumor suppressor FOXO3a of A549 human nonsmall cell lung cancer cells. (A) After treatment with KG-135 for 72 h, Western blot was used to analyze the protein levels of cyclin-dependent kinase (CDK)4, CDK6, cyclin D1, Rb, and pRb, (B) p18 and p27, and (C) FOXO3a. FOXO3a, Forkhead Class box O3a; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; pRb, phospho-retinoblastoma protein.
3.3. KG-135 induces autophagy in A549 human NSCLC cells without significant increase of Beclin-1
In spite of the substantial effects of KG-135 against the proliferation of A549 cells shown in Fig. 1, Fig. 3, the extent of apoptosis was very limited. As autophagy had been shown to protect some cancer cells against drug treatment-induced apoptotic cell death [19], we analyzed the autophagy markers in KG-135-treated A549 cells. As shown in Fig. 4A, treatment of A549 cells with KG-135 dose-dependently increased the protein of LC3-II, a hallmark of autophagy. Unlike canonical autophagy, the upstream autophagy marker Beclin-1 protein remained almost unchanged in response to KG-135, indicating that such an increase of LC-3-II was Beclin-1 independent. Autophagy induction is often associated with a decrease of the autophagy substrate and adaptor protein, p62 (sequestosome 1) [35]. However, treatment with KG-135 increased p62 rather than decreased it (Fig. 4A). In contrast, as shown in Fig. 4B, metformin induced LC3-II accompanied by an increase of Beclin-1 and a decrease of p62 in A549 cells similar to its effects on conventional autophagy induction in esophageal squamous cell carcinoma cells [36].
Fig. 4.

Effects of KG-135 and metformin on autophagy related proteins in A549 human nonsmall cell lung cancer cells. (A) After treatment with KG-135 or (B) metformin for 72 h, Western blot was used to analyze the protein levels of LC3-I, LC3-II, Beclin-1, and p62 (sequestosome 1). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
We further analyzed the autophagic flux in KG-135-treated A549 cells. According to the guidelines proposed by Mizushima and Yoshimori [37], the LC3-II accumulation by autophagy induction would be enhanced by an autophagy inhibitor like HCQ. As shown in Fig. 5A, the increases of LC3-II induced by 50 μg/mL and 75 μg/mL of KG-135 were both substantially further enhanced by HCQ (25μM), suggesting the induction of autophagic flux.
Fig. 5.

Effects of autophagy inhibitor hydroxychloroquine (HCQ) on KG-135-induced LC3-II accumulation and acidic vesicular organelle (AVO) formation in A549 human nonsmall cell lung cancer cells. (A) After treatment with KG-135 alone or in the presence of HCQ for 72 h, the protein levels of LC3-I and LC3-II were analyzed by Western blot. (B) As described in Section 2, the formation of acridine orange-stained AVOs was examined using fluorescence microscopy, scale bar = 50 μm and (C) quantified using flow cytometry. The AVOs in KG-135-treated cells showed bright red, whereas the cytoplasm and the nucleus showed dominant green fluorescence. The percentage of cells with bright red fluorescence (the upper two FL3-positive quadrants) is indicated in the parentheses of each flow cytometric dot plot. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Autophagy is featured by the formation of autophagosomes and autolysosomes, which are AVOs [32]. As described in Section 2, these AVOs could be stained by acridine orange and fluorescence bright red when excited at 488 nm. The results shown in Fig. 5B show that vital staining of A549 cells with acridine orange revealed the appearance of AVOs after treatment with KG-135 in a dose-dependent manner. The AVOs in KG-135-treated cells showed bright red, whereas the cytoplasm and the nucleus showed dominant green fluorescence. In contrast, the control cells exhibited mainly green fluorescence (Fig. 5B). Because HCQ inhibits autophagy at the late stage, it could also enhance the accumulation of AVOs induced by autophagic flux [38]. In agreement with this, the formation of AVOs induced by KG-135 (50 μg/mL) was also further enhanced when cotreated with 25μM of HCQ (Fig. 5B). The green and red fluorescent intensities in acridine orange-stained A549 cells were quantified by flow cytometric analysis. In Fig. 5C, green (x-axis, 510–530 nm, FL1 channel) and red (y-axis, >650 nm, FL3 channel) fluorescence emission with blue (488 nm) excitation produced from stained A549 cells was measured. The percentage of cells with bright red fluorescence (the upper two FL3-positive quadrants) is indicated in the parentheses of each dot plot. KG-135 dose-dependently increased the percentage from 2.6% to 7.2%, 13.7%, and 30.7% at a dose of 50 μg/mL, 75 μg/mL, and 100 μg/mL, respectively (Fig. 5C). In the presence of HCQ (25μM), the KG-135 (50 μg/mL)-induced positive acridine orange staining was further increased to 48.1%, compared with 20.5% (HCQ) and 7.2% (KG-135) if treated alone (Fig. 5C).
Based on these results, KG-135 induced a Beclin-1-independent noncanonical/unconventional autophagy in A549 human NSCLC cells.
3.4. Blockade of autophagy potentiates KG-135-induced FOXO3a and apoptosis in A549 human NSCLC cells
To investigate the role of autophagy on KG-135 mediated anticancer effects, the survival of KG-135-treated A549 cells was determined in the presence of an autophagy inhibitor HCQ. As shown in Fig. 6A, treatment with HCQ (25μM) alone only slightly reduced the cell viability to 83% of control. In the presence of HCQ (25μM), the cell viability of KG-135 (75 μg/mL)-treated cells was dramatically reduced from 64.3% to 3% of control (Fig. 6A). Combination of these two agents resulted in drastic reduction of cell viability and shrinkage of cell shape (Fig. 6B). To further examine whether this enhancement by HCQ was due to apoptosis induction, the sub-G1 (apoptotic) percentages were analyzed. As shown in Fig. 6C, the sub-G1 fractions induced by HCQ (25μM) and KG-135 (75 μg/mL) alone were only 0.9% and 2.2%, respectively; however, the sub-G1 fraction was dramatically enhanced to 60.1% when cotreated with these two agents (Fig. 6C). Similar enhancement of apoptosis was also observed in A549 cells treated with 50 μg/mL of KG-135 combined with HCQ (25μM; Fig. S1). The enhancement of HCQ on KG-135-mediated anticancer effects was mainly due to apoptosis induction.
Fig. 6.

Effects of autophagy inhibitor hydroxychloroquine (HCQ) on KG-135-mediated growth inhibition and apoptosis induction in A549 human nonsmall cell lung cancer cells. (A) After treatment with KG-135 alone or in the presence of HCQ for 72 h, the cell viability was determined using sulforhodamine B binding assay and (B) examined using phase contrast microscopy, scale bar = 100 μm. (C) The sub-G1 apoptotic fraction was analyzed using flow cytometry.
To evaluate the safety of KG-135 in normal lung cells, we examined the effect of KG-135 on the cell viability of normal human fetal lung fibroblast WI-38 cells. At a dose range of 100 μg/mL, KG-135 has no significant suppressive effect on the viability of WI-38 cells (Fig. 2A). Combining KG-135 (75 μg/mL) with HCQ (25μM) only slightly reduced the cell viability of WI-38 cells to 77% of control (Fig. 2A) and most of the treated cells showed intact morphology (Fig. 2B), in contrast to that of A549 NSCLC cells shown in Fig. 6.
The FOXO3a protein in A549 cells decreases with the culture time (Fig. S2). The enhancement of KG-135-induced FOXO3a by cotreatment with HCQ (25μM) could be observed after 24 h of treatment. Notably, this enhancement became more remarkable after 72 h of treatment, while the FOXO3a in control cells was decreased after culture (Fig. 7A). Accordingly, the induction of the death receptor ligand FasL, a downstream of FOXO3a, and the decrease of full-length caspase-8 were enhanced as well when the cells were cotreated (Fig. 7B). Next, we examined the changes of the protein levels of Bcl-2, survivin, and caspase-9, which play important roles in the intrinsic (mitochondrial) apoptosis pathway. The decrease of Bcl-2 and survivin proteins by KG-135 was markedly enhanced in the presence of HCQ (Fig. 7C). Similar enhancement was observed in the cleavage of caspase-9. Two cleaved forms of caspase-9 (cleavage at Asp315 and Asp330) were obviously more elevated in the cells treated with both KG-135 and HCQ (Fig. 7C), suggesting the contribution of an intrinsic apoptosis pathway in this enhancement of apoptosis induction. Therefore, blockade of autophagy in KG-135-treated A549 cells activates both the mitochondrial intrinsic and death receptor-mediated extrinsic apoptotic pathways.
Fig. 7.

Effects of autophagy inhibitor hydroxychloroquine (HCQ) on KG-135-mediated activation of apoptosis pathways and upregulation of tumor suppressor FOXO3a in A549 human nonsmall cell lung cancer cells. (A) The protein level of FOXO3a was analyzed using Western blot after treatment with KG-135 alone or in the presence of HCQ for 24 h and 72 h. (B) The extrinsic apoptosis-related proteins Fas ligand (FasL) and full-length caspase-8 were analyzed using Western blot after treatment with KG-135 alone or in the presence of HCQ for 48 h. (C) The mitochondrial (intrinsic) apoptosis-related proteins were analyzed using Western blot 48 h (survivin and caspase-9 cleaved at Asp315 and Asp330) and 72 h (Bcl-2) after treatment with KG-135 alone or in the presence of HCQ. FOXO3a, Forkhead Class box O3a; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
4. Discussion
KG-135 is a quality-controlled standardized formulation enriched with the red ginseng-specific antitumorigenic ginsenosides such as Rk1, Rg3, and Rg5 [11]. It had been shown to induce marked G1 cell cycle arrest in prostate and cervical cancer cells. In these previous studies, although a limited amount of early apoptosis was found in KG-135-treated prostate cancer cells (PC3 and DU-145), the late stage of apoptosis (sub-G1 fraction) was minimally elevated even at dose of 150 μg/mL [5], [8]. Similarly, the sub-G1 fraction is also minimally increased in KG-135-treated A549 human NSCLC cells even at dose of 100 μg/mL, in spite of the marked induction of G1 cell-cycle arrest. According to recent findings in the adaptive response of cancer cells to drug treatment [18], it is tempting to speculate that a prosurvival autophagy was induced by KG-135, by which the cancer cells escaped from apoptotic death. In agreement with this speculation, our results demonstrate the autophagy flux induced by KG-135 and show that an autophagy inhibitor like HCQ could dramatically enhance KG-135-induced apoptosis of A549 cells.
Chloroquine (CQ) and its derivative HCQ are the only autophagy inhibitors whose effectiveness and safety in clinical trials have been approved by the Food and Drug Administration [18]. In comparison with CQ, HCQ can be safely dose escalated in cancer patients [18], [39]. Thus, we chose HCQ for autophagy inhibition to potentiate the anticancer efficacy of KG-135 in this study.
The induction of p27 by KG-135 had been shown in Hela human cervical cancer cells by Lee et al. [5]. In A549 NSCLC cells, we also observed the increase of p27 protein level by KG-135 and further unveiled the upregulation of the upstream tumor suppressor FOXO3a, which positively regulates genes promoting cell cycle arrest and apoptosis [31]. When the KG-135-induced FOXO3a was drastically enhanced by HCQ, the downstream death receptor FasL was markedly elevated accompanied by a decrease of full-length caspase-8. Yoo et al. [8], had observed the involvement of death receptor signaling and caspase-8 as well as apoptosis inducing factor in the early apoptosis of KG-135-treated prostate cancer cells, although late apoptosis (sub-G1 fraction) was only minimally elevated. It is interesting to investigate whether these apoptotic pathways in KG-135-treated prostate cancer cells could also be dramatically enhanced by HCQ and result in the appearance of a massive sub-G1 fraction.
Unlike the canonical autophagy [40], KG-135 induced obvious accumulation of autophagy hallmark protein LC3-II without the increase of upstream marker Beclin-1. Moreover, the autophagy substrate and adaptor protein p62 (sequestosome 1) was increased after KG-135 treatment rather than decreased. Because p62 is recognized as an autophagy substrate and is often degraded in autolysosomes, accumulation of p62 could be the result of inhibition of autophagy [35]. Therefore, the results from cells treated with KG-135 alone seemed not be sufficient to determine whether the autophagic flux was promoted or inhibited. To determine the induction of autophagic flux, Mizushima and Yoshimori [37] proposed that the accumulation LC3-II by autophagy induction would be enhanced by an autophagy inhibitor like HCQ and would not be affected if it was caused by autophagy inhibition. According to this guideline, we examined and found the KG-135-induced accumulation of LC3-II was enhanced by HCQ in A549 cells, indicating the induction of autophagic flux by KG-135. Besides LC3-II, HCQ could also enhance the accumulation of AVOs induced by autophagic flux, because it inhibits autophagy at the late stage [38]. In line with this, similar enhancement of KG-135-indcued AVOs by HCQ was observed and further suggested the presence of autophagic flux induction.
Autophagy accompanied by an increase of p62 (sequestosome 1) and without a change of Beclin-1 had also been shown in resveratrol-treated K562 human chronic myelogenous leukemia cells [41], [42]. In that study, suppressing the p62 expression by a specific c-Jun N-terminal kinase inhibitor SP600125 or small interfering RNA drastically reduced the accumulation of LC3-II. The autophagy induced by resveratrol is p62-dependent [41], [42]. However, unlike the case in resveratrol-treated chronic myelogenous leukemia cells, the c-Jun N-terminal kinase inhibitor SP600125 suppressed the increase of p62 but did not reduce the accumulation of LC3-II in KG-135-treated A549 cells (data not shown). As the protein level of p62 can also be changed independent of autophagy [37], additional functions of p62 remain to be determined with regard to its role in KG-135-induced autophagy.
Cheong et al. [20] recently reported that Rg3, one of the active components in KG-135, induced autophagy in HepG2 hepatoma cells evidenced by the increase of LC-3-II. Controversially, the study by Kim et al. [43] shows that Rg3 inhibits the autophagy of HepG2 cells as the Rg3-increased LC3-II could not be further enhanced in the presence of CQ. The explanation for this discrepancy between the results of these two studies remains to be elucidated. However, in A549 cells, we did find that KG-135-induced accumulation of LC3-II was enhanced by HCQ. As different cancer cells were used, one possibility is that the effects of ginsenosides on autophagic flux might be quite different depending on the cellular context. Further investigation is needed to clarify the detailed molecular events.
Taken together, in addition to the regulation of G1/S transition machinery, we found an increase of p27 and its upstream tumor suppressor FOXO3a in KG-135-treated A549 human NSCLC cells. The apoptosis induction of A549 cells by KG-135 is prevented by a prosurvival autophagy, which might be Beclin-1-independent and noncanonical. Blockade of autophagy by HCQ activates not only the caspase-9 but also the FOXO3a/FasL/caspase-8 apoptotic pathways. In another human NSCLC cell line, H460 large-cell lung cancer cells, KG-135 also induces noncanonical/unconventional autophagy, and combination with HCQ also enhances KG-135-induced FOXO3a and apoptosis (Fig. S3). Regarding the future implementation of KG-135 as an integrative anticancer therapeutics, combination of KG-135 and an autophagy inhibitor like HCQ may be a potential novel strategy for cancer treatment.
Conflicts of interests
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Acknowledgments
This work was supported by the joint grant of Wan Fang Hospital, Taipei Medical University and Everglory Bio-Tech Ltd., Taipei (Grant W196) and National Health Research Institutes (Grant CA-104-PP-35), and Health and Welfare surcharge of tobacco products, Taiwan (MOHW104-TDU-B-212-124-001). We thank the Ginseng Science Inc., Korea for providing the original KG-135 powder.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jgr.2016.04.003.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Fig. S1.
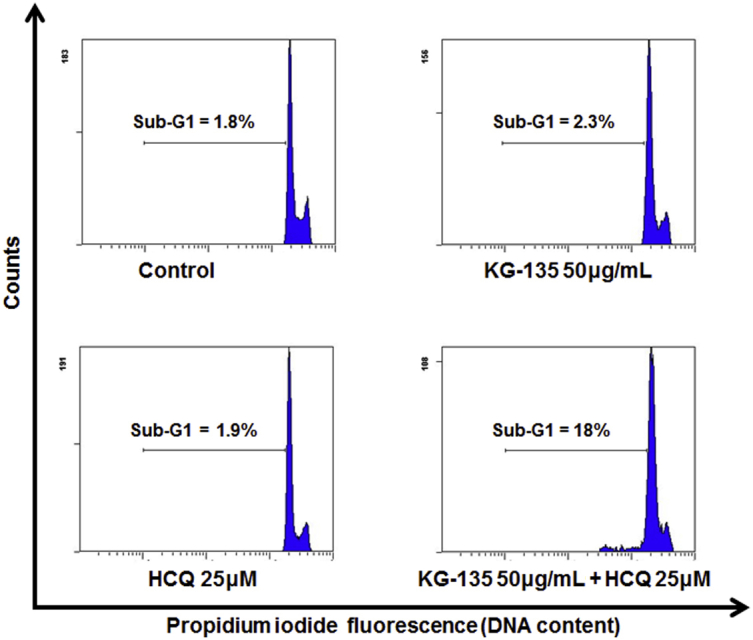
Effect of autophagy inhibitor hydroxychloroquine (HCQ) 25μM combined with 50 μg/mL of KG-135 on apoptosis induction in A549 human nonsmall cell lung cancer cells. After treatment with KG-135 (50 μg/mL) alone or in the presence of HCQ (25μM) for 72 h, the sub-G1 apoptotic fraction was analyzed with flow cytometry.
Fig. S2.

The FOXO3a protein in A549 cells decreases with the culture time. The protein level of FOXO3a was analyzed using Western blot after treatment with KG-135 for 4 h, 24 h, and 72 h. FOXO3a, Forkhead Class box O3a; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Fig. S3.
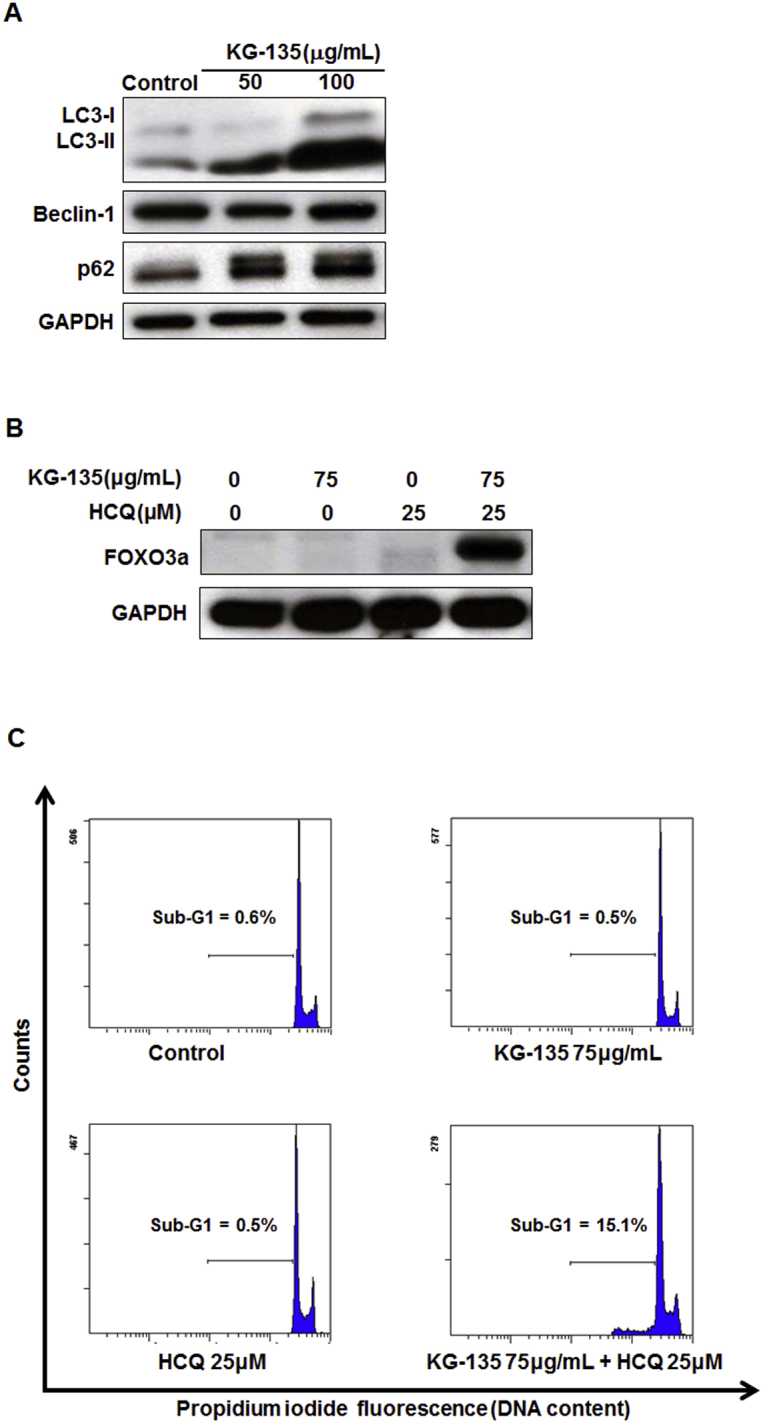
Effects of KG-135 in H460 human large-cell lung cancer cells. (A) After treatment with KG-135 for 72 h, Western blot was used to analyze the protein levels of LC3-I, LC3-II, Beclin-1, and p62. (B) The protein level of FOXO3a and (C) sub-G1 apoptotic fraction were analyzed after treatment with KG-135 alone or in the presence of hydroxychloroquine (HCQ) for 72 h. H460 cells were maintained and treated with drugs in the same condition as that for A549 cells. FOXO3a, Forkhead Class box O3a; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
References
- 1.Xiang Y.Z., Shang H.C., Gao X.M., Zhang B.L. A comparison of the ancient use of ginseng in traditional Chinese medicine with modern pharmacological experiments and clinical trials. Phytother Res. 2008;22:851–858. doi: 10.1002/ptr.2384. [DOI] [PubMed] [Google Scholar]
- 2.Choi K.T. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng Meyer. Acta Pharmacol Sin. 2008;29:1109–1118. doi: 10.1111/j.1745-7254.2008.00869.x. [DOI] [PubMed] [Google Scholar]
- 3.Wong A.S., Che C.M., Leung K.W. Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview. Nat Prod Rep. 2015;32:256–272. doi: 10.1039/c4np00080c. [DOI] [PubMed] [Google Scholar]
- 4.Choi H.J., Kim E.J., Shin Y.W., Park J.H., Kim D.H., Kim N.J. Protective effect of heat-processed ginseng (sun ginseng) in the adenine-induced renal failure rats. J Ginseng Res. 2012;36:270–276. doi: 10.5142/jgr.2012.36.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee W.H., Choi J.S., Kim H.Y., Park J.H., Lee S.K., Surh Y.J. Heat-processed neoginseng, KG-135, down-regulates G1 cyclin-dependent kinase through the proteasome-mediated pathway in HeLa cells. Oncol Rep. 2009;21:467–474. [PubMed] [Google Scholar]
- 6.Wang C.Z., Aung H.H., Ni M., Wu J.A., Tong R., Wicks S., He T.C., Yuan C.S. Red American ginseng: ginsenoside constituents and antiproliferative activities of heat-processed Panax quinquefolius roots. Planta Med. 2007;73:669–674. doi: 10.1055/s-2007-981524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yun T.K., Lee Y.S., Lee Y.H., Kim S.I., Yun H.Y. Anticarcinogenic effect of Panax ginseng C.A. Meyer and identification of active compounds. J Korean Med Sci. 2001;16(Suppl):S6–S18. doi: 10.3346/jkms.2001.16.S.S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo J.H., Kwon H.C., Kim Y.J., Park J.H., Yang H.O. KG-135, enriched with selected ginsenosides, inhibits the proliferation of human prostate cancer cells in culture and inhibits xenograft growth in athymic mice. Cancer Lett. 2010;289:99–110. doi: 10.1016/j.canlet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Park J.Y., Choi P., Kim H.K., Kang K.S., Ham J. Increase in apoptotic effect of Panax ginseng by microwave processing in human prostate cancer cells: in vitro and in vivo studies. J Ginseng Res. 2016;40:62–67. doi: 10.1016/j.jgr.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park S.A., Kim E.H., Na H.K., Surh Y.J. KG-135 inhibits COX-2 expression by blocking the activation of JNK and AP-1 in phorbol ester-stimulated human breast epithelial cells. Ann N Y Acad Sci. 2007;1095:545–553. doi: 10.1196/annals.1397.059. [DOI] [PubMed] [Google Scholar]
- 11.Lee W.H., Choi J.S., Kim H.Y., Park J.H., Park B.D., Cho S.J., Lee S.K., Surh Y.J. Potentiation of etoposide-induced apoptosis in HeLa cells by co-treatment with KG-135, a quality-controlled standardized ginsenoside formulation. Cancer Lett. 2010;294:74–81. doi: 10.1016/j.canlet.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 13.Eskelinen E.L., Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 15.de Bruin E.C., Medema J.P. Apoptosis and nonapoptotic deaths in cancer development and treatment response. Cancer Treat Rev. 2008;34:737–749. doi: 10.1016/j.ctrv.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl. 2):1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 17.Kaminskyy V.O., Piskunova T., Zborovskaya I.B., Tchevkina E.M., Zhivotovsky B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy. 2012;8:1032–1044. doi: 10.4161/auto.20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui X., Chen R., Wang Z., Huang Z., Kong N., Zhang M., Han W., Lou F., Yang J., Zhang Q. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:1–12. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kondo Y., Kanzawa T., Sawaya R., Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 20.Cheong J.H., Kim H., Hong M.J., Yang M.H., Kim J.W., Yoo H., Yang H., Park J.H., Sung S.H., Kim H.P. Stereoisomer-specific anticancer activities of ginsenoside Rg3 and Rh2 in HepG2 cells: disparity in cytotoxicity and autophagy-inducing effects due to 20(S)-epimers. Biol Pharm Bull. 2015;38:102–108. doi: 10.1248/bpb.b14-00603. [DOI] [PubMed] [Google Scholar]
- 21.Ferlay J., Shin H.R., Bray F., Forman D., Mathers C., Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 22.Bartucci M., Svensson S., Romania P., Dattilo R., Patrizii M., Signore M., Navarra S., Lotti F., Biffoni M., Pilozzi E. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012;19:768–778. doi: 10.1038/cdd.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jemal A., Siegel R., Xu J., Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 24.Collins L.G., Haines C., Perkel R., Enck R.E. Lung cancer: diagnosis and management. Am Fam Physician. 2007;75:56–63. [PubMed] [Google Scholar]
- 25.Wang L., Li X., Song Y.M., Wang B., Zhang F.R., Yang R., Wang H.Q., Zhang G.J. Ginsenoside Rg3 sensitizes human nonsmall cell lung cancer cells to gamma-radiation by targeting the nuclear factor-kappaB pathway. Mol Med Rep. 2015;12:609–614. doi: 10.3892/mmr.2015.3397. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y.J., Choi W.I., Jeon B.N., Choi K.C., Kim K., Kim T.J., Ham J., Jang H.J., Kang K.S., Ko H. Stereospecific effects of ginsenoside 20-Rg3 inhibits TGF-beta1-induced epithelial-mesenchymal transition and suppresses lung cancer migration, invasion and anoikis resistance. Toxicology. 2014;322:23–33. doi: 10.1016/j.tox.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Bi X., Xia X., Mou T., Jiang B., Fan D., Wang P., Liu Y., Hou Y., Zhao Y. Anti-tumor activity of three ginsenoside derivatives in lung cancer is associated with Wnt/beta-catenin signaling inhibition. Eur J Pharmacol. 2014;742:145–152. doi: 10.1016/j.ejphar.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L.H., Jia Y.L., Lin X.X., Zhang H.Q., Dong X.W., Zhao J.M., Shen J., Shen H.J., Li F.F., Yan X.F. AD-1, a novel ginsenoside derivative, shows anti-lung cancer activity via activation of p38 MAPK pathway and generation of reactive oxygen species. Biochim Biophys Acta. 2013;1830:4148–4159. doi: 10.1016/j.bbagen.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 29.An I.S., An S., Kwon K.J., Kim Y.J., Bae S. Ginsenoside Rh2 mediates changes in the microRNA expression profile of human non-small cell lung cancer A549 cells. Oncol Rep. 2013;29:523–528. doi: 10.3892/or.2012.2136. [DOI] [PubMed] [Google Scholar]
- 30.Cheng C.C., Yang S.M., Huang C.Y., Chen J.C., Chang W.M., Hsu S.L. Molecular mechanisms of ginsenoside Rh2-mediated G1 growth arrest and apoptosis in human lung adenocarcinoma A549 cells. Cancer Chemother Pharmacol. 2005;55:531–540. doi: 10.1007/s00280-004-0919-6. [DOI] [PubMed] [Google Scholar]
- 31.Ruvolo P.P. The Herculean task of killing cancer cells: suppression of FOXO3A in acute leukemia involves a hydra of multiple survival kinases. Cell Cycle. 2012;11:2589. doi: 10.4161/cc.21233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Navas R., Munder M., Mollinedo F. Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy. 2012;8:1557–1576. doi: 10.4161/auto.21315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Traganos F., Darzynkiewicz Z. Lysosomal proton pump activity: supravital cell staining with acridine orange differentiates leukocyte subpopulations. Methods Cell Biol. 1994;41:185–194. doi: 10.1016/s0091-679x(08)61717-3. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y., Fei M., Wang Y., Lu M., Cheng C., Shen A. Expression of Foxo3a in non-Hodgkin's lymphomas is correlated with cell cycle inhibitor p27. Eur J Haematol. 2008;81:83–93. doi: 10.1111/j.1600-0609.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- 35.Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Y., Ke C., Tang Q., Dong H., Zheng X., Lin W., Ke J., Huang J., Yeung S.C., Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizushima N., Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki K., Tsuno N.H., Sunami E., Tsurita G., Kawai K., Okaji Y., Nishikawa T., Shuno Y., Hongo K., Hiyoshi M. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370. doi: 10.1186/1471-2407-10-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunja N., Roberts D., McCoubrie D., Lamberth P., Jan A., Simes D.C., Hackett P., Buckley N.A. Survival after massive hydroxychloroquine overdose. Anaesth Intensive Care. 2009;37:130–133. doi: 10.1177/0310057X0903700112. [DOI] [PubMed] [Google Scholar]
- 40.Codogno P., Mehrpour M., Proikas-Cezanne T. Canonical and noncanonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 41.Puissant A., Robert G., Fenouille N., Luciano F., Cassuto J.P., Raynaud S., Auberger P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70:1042–1052. doi: 10.1158/0008-5472.CAN-09-3537. [DOI] [PubMed] [Google Scholar]
- 42.Puissant A., Auberger P. AMPK- and p62/SQSTM1-dependent autophagy mediate resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy. 2010;6:655–657. doi: 10.4161/auto.6.5.12126. [DOI] [PubMed] [Google Scholar]
- 43.Kim D.G., Jung K.H., Lee D.G., Yoon J.H., Choi K.S., Kwon S.W., Shen H.M., Morgan M.J., Hong S.S., Kim Y.S. 20(S)-Ginsenoside Rg3 is a novel inhibitor of autophagy and sensitizes hepatocellular carcinoma to doxorubicin. Oncotarget. 2014;5:4438–4451. doi: 10.18632/oncotarget.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]