

Abstract
Systemic acidosis has detrimental effects on the skeleton, and local acidosis coincides with bone destruction in inflammatory and metastatic diseases. Acidification dramatically enhances osteoclastic resorption, although the underlying mechanism has remained elusive. We investigated the effect of acidosis on the osteoclastogenic transcription factor NFATc1, which upon dephosphorylation translocates from the cytoplasm to nuclei. Lowering extracellular pH dramatically increased accumulation of NFATc1 in nuclei of rat and rabbit osteoclasts to levels comparable with those induced by the proresorptive cytokine receptor activator of NF-κB ligand (RANKL). Activation of NFATc1 by RANKL was mediated by means of prolonged stimulation of the Ca2+/calmodulin-dependent protein phosphatase, calcineurin. In contrast, NFATc1 activation by acidosis involved stimulation of calcineurin and suppression of NFATc1 inactivation. Acidosis, like RANKL, induced transient elevation of cytosolic free Ca2+ concentration ([Ca2+]i), which persisted in Ca2+-free media and was abolished by inhibition of phospholipase C or depletion of intracellular Ca2+ stores. Real-time-PCR of osteoclast-like cells generated from RAW 264.7 cells revealed high levels of expression of ovarian cancer G protein-coupled receptor 1, which links extracellular acidification to elevation of [Ca2+]i. In addition, the calcineurin inhibitor cyclosporin A suppressed the stimulatory effect of acidification on resorption, implicating NFAT in mediating the actions of acidosis on osteoclast activity. In summary, acidification and RANKL induce signals in osteoclasts that converge on the Ca2+/calcineurin/NFAT pathway. Acidosis acts directly on osteoclasts to activate NFATc1 and stimulate resorption.
Keywords: bone resorption, cyclosporin A, ovarian cancer G protein-coupled receptor 1, phospholipase C, protons
The detrimental effects of acidosis on bone have been recognized for at least 85 years (1). Resorption of mineral counteracts systemic acidosis but frequently results in osteoporosis (2, 3). In addition, local acidosis in inflammation and malignancy stimulates bone resorption, thereby contributing to the pathogenesis of rheumatoid arthritis, periodontitis, and bone metastases (2). Acidification potently stimulates resorptive activity of osteoclasts in vitro and in vivo (4–7) but has little effect on osteoclastogenesis (8). However, the mechanism by which acidification activates osteoclasts has remained elusive.
The TNF-related cytokine, receptor activator of NF-κB ligand (RANKL), stimulates the formation and resorptive activity of osteoclasts. RANKL, expressed by osteoblasts, stromal cells, and activated lymphocytes, signals through its receptor RANK, present on osteoclasts and their precursors (9). Osteoprotegerin (OPG) is a soluble decoy receptor, which binds RANKL blocking its interaction with RANK. Acidosis up-regulates expression of RANKL in bone, which may then stimulate osteoclasts (10). Signaling through RANK involves multiple pathways leading to activation of mitogen-activated protein kinases, c-Jun N-terminal kinase, extracellular signal-regulated kinase and p38, and transcription factors NF-κB and AP-1 (9). We have recently shown that RANKL activates phospholipase C (PLC), leading to release of Ca2+ from intracellular stores and a transient rise in cytosolic free Ca2+ concentration ([Ca2+]i) (11).
NFATc1 was recently found to be the transcription factor that is most strongly induced during RANKL-stimulated osteoclast differentiation, and it was found to be essential for osteoclastogenesis (12, 13). Inactive nuclear factor of activated T cells (NFAT) is maintained in the cytoplasm in a hyperphosphorylated state. Elevation of [Ca2+]i stimulates the phosphatase calcineurin, which dephosphorylates multiple serine residues, exposing a nuclear localization signal that permits translocation of NFAT to nuclei (14). Kinases, such as c-Jun N-terminal kinases, p38, casein kinase 1 (CK1) and 2 (CK2), and glycogen synthase kinase 3β (GSK3β), phosphorylate NFAT, promoting its inactivation (15, 16).
In this study, we examined whether acidosis and RANKL activate NFATc1 in mature osteoclasts. We used single-cell techniques to study authentic osteoclasts or generated osteoclast-like cells in vitro from RAW 264.7 cells. We found that both acidosis and RANKL cause nuclear accumulation of NFATc1 in authentic osteoclasts. Calcineurin mediates NFATc1 activation in both cases; however, distinct mechanisms maintain its nuclear residence. We show that osteoclasts express ovarian cancer G protein-coupled receptor (GPCR) 1 (OGR1), a proton-sensing GPCR that couples extracellular acidification to elevation of [Ca2+]i. Acidosis acts directly on osteoclasts to induce elevation of [Ca2+]i, leading to activation of NFATc1 and stimulation of resorption.
Methods
Cell Cultures and RT-PCR. Osteoclasts were isolated from the long bones of neonatal Wistar rats or New Zealand White rabbits (11). Cells were maintained in medium 199 buffered with HCO3- (26 mM) and Hepes (25 mM) (Invitrogen) supplemented with 15% heat-inactivated FBS and 1% antibiotics (10,000 units/ml penicillin/10,000 μg/ml streptomycin/25 μg/ml amphotericin B) at 37°C in 5% CO2 (pH 7.3). These procedures were approved by the Council on Animal Care of the University of Western Ontario. Osteoclastic resorptive activity was assessed by using a pit-formation assay (17). Details of this procedure are given in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
The RAW 264.7 mouse monocytic cell line was obtained from the American Type Culture Collection and cultured in DMEM buffered with HCO3- (18 mM) and supplemented with 10% FBS and 1% antibiotics at 37°C in 5% CO2. To generate osteoclast-like cells, RAW 264.7 cells were plated at a density of 0.5 × 104 cells per cm2 and grown in the presence of RANKL (100 ng/ml) for 4–5 days (medium was changed on day 3). Osteoclast-like cells were multinucleated and stained positively for tartrate-resistant acid phosphatase. UMR-106 osteoblast-like cells and CFK2 prechondrocytic cells were cultured as described (18, 19). Mouse OGR1 (Gpr68; GenBank accession no. NM_175493) transcripts were detected by conventional RT-PCR and real-time RT-PCR, as described in Supporting Materials and Methods.
Protocol for Extracellular Acidification. Osteoclasts were preincubated for 1 h in supplemented medium 199 under alkaline conditions (0% CO2). Two protocols were then used to change extracellular pH. First, the pH of supplemented 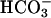 -free medium 199 buffered with 25 mM Hepes was adjusted by titration with NaOH or HCl, to mimic metabolic acidosis. Second, where indicated, CO2 levels were varied from 0–10%, with 10% CO2 mimicking respiratory acidosis. pH was measured by using a glass combination electrode (PHM82; Radiometer, Copenhagen), and for media incubated at different CO2 levels, the pH of selected samples was determined by blood-gas analysis (ABL700; Radiometer). For studies involving OPG or kinase inhibitors, test agents were added for the final 20 or 30 min, respectively, of the 1-h preincubation period.
-free medium 199 buffered with 25 mM Hepes was adjusted by titration with NaOH or HCl, to mimic metabolic acidosis. Second, where indicated, CO2 levels were varied from 0–10%, with 10% CO2 mimicking respiratory acidosis. pH was measured by using a glass combination electrode (PHM82; Radiometer, Copenhagen), and for media incubated at different CO2 levels, the pH of selected samples was determined by blood-gas analysis (ABL700; Radiometer). For studies involving OPG or kinase inhibitors, test agents were added for the final 20 or 30 min, respectively, of the 1-h preincubation period.
Test Compounds. Soluble RANKL (murine recombinant 158–316) and OPG (human recombinant 21–194 fused at the N terminus to the Fc domain of human IgG1) were provided by Amgen Biologicals. Salmon calcitonin was obtained from Bachem. U73122 and U73343 were obtained from Calbiochem and prepared as described (11). Cyclosporin A, SB202190, PD169316, and SB202474 were obtained from Calbiochem; thapsigargin and emodin were obtained from Sigma; and 4,5,6,7-tetrabromobenzotriazole (TBB) was provided by L. A. Pinna (University of Padua, Padua, Italy). Stock solutions of inhibitors were prepared in DMSO.
Immunofluorescence. As described in Supporting Materials and Methods, osteoclasts on glass coverslips were fixed with paraformaldehyde, permeabilized with Triton X-100, and blocked with normal goat serum. For NFATc1 localization, we used an mAb (7A6, catalog no. sc-7294, Santa Cruz Biotechnology) established to be specific for NFATc1 (20). Staining was completed with biotinylated goat-antimouse IgG (Vector Laboratories, Burlingame, CA) and fluorescein-conjugated streptavidin (Vector Laboratories). For some experiments, a rabbit polyclonal Ab broadly reactive with different NFAT isoforms (K-18R, catalog no. sc-1149, Santa Cruz Biotechnology) was used, followed by Texas red-conjugated goat anti-rabbit secondary Ab (Molecular Probes). Staining for the p65 subunit of NF-κB was performed as described (11). We assessed localization of fluorescent label in all osteoclasts on each coverslip (usually 40–70 cells per coverslip) by using an LSM 510 confocal microscope (Zeiss). Osteoclasts were rated positive for nuclear localization of NFAT or NF-κB if fluorescence intensity of nuclei exceeded that of the cytoplasm (Fig. 7, which is published as supporting information on the PNAS web site). In some experiments, nuclei were counterstained by using TOTO-3 (Molecular Probes). Localization of OGR1 was assessed in fixed samples of rat osteoclasts, RAW 264.7 cells, and osteoclast-like cells by using a rabbit polyclonal Ab (catalog no. LS-R3870, LifeSpan BioSciences, Seattle, WA), followed by biotinylated goat anti-rabbit IgG (Vector Laboratories) and fluorescein-conjugated streptavidin.
Fluorescence Measurement of [Ca2+]i. [Ca2+]i of isolated rat osteoclasts, undifferentiated RAW 264.7 cells, and osteoclast-like cells was monitored by using microspectrofluorimetry (11). Cells loaded with fura-2 were superfused at room temperature with buffer containing 130 mM NaCl, 5 mM KCl, 10 mM glucose, 1 mM MgCl2, 1 mM CaCl2, and 20 mM Hepes (280–290 mosml/liter), adjusted with NaOH to pH 7.3–7.6. Changes in pH were induced by bath addition of HCl or NaOH, followed by gentle mixing. As a control, the same amount of vehicle was added by using the same protocol. In some studies, cells were superfused with nominally Ca2+-free buffer supplemented with 0.5 mM EGTA.
Results
Effect of Extracellular pH on NFAT Accumulation in Osteoclast Nuclei. We used immunofluorescence to monitor the spatial distribution of NFATc1, which accumulates in the nuclei upon activation. In most rat and rabbit osteoclasts maintained at pH 7.6, NFATc1 was located predominantly in the cytoplasm for the duration of the experiment (Fig. 1A). Extracellular acidification to pH 7.0 (for 30–75 min) induced redistribution of NFATc1 to the nuclei, most often to all of the nuclei within individual osteoclasts (Fig. 1B). The same results were observed when NFAT localization was assessed in rat osteoclasts by using an Ab broadly reactive with different NFAT isoforms (data not shown).
Fig. 1.

Effect of extracellular pH on nuclear accumulation of NFATc1. Rat osteoclasts were exposed to different pH levels for 45 min and then fixed. NFATc1 localization was assessed by immunofluorescence (green) and nuclei were stained by using TOTO-3 (red). (A) In most osteoclasts maintained at pH 7.6, NFATc1 was localized predominantly in the cytoplasm. (B) Extracellular acidification to pH 7.0 induced nuclear accumulation of NFATc1. Yellow in Right (Superimposed) indicates nuclear localization of NFATc1. Calibration bars indicate 20 μm.
To investigate the pH dependence of NFAT nuclear accumulation, we induced acidification by imitating respiratory or metabolic acidosis. At alkaline pH (7.6–7.9), only 30 ± 4% of osteoclasts exhibited NFATc1 nuclear accumulation (Fig. 2A). Lowering pH increased the percentage of osteoclasts exhibiting NFATc1 nuclear accumulation, reaching a maximum of 71 ± 3% at pH 7.0. NFATc1 activation was most sensitive to pH between 7.4 and 7.2, similar to the pH dependence of resorptive activity of osteoclasts (7). In the range of pH 7.6–7.0, no effect on NFATc1 localization was observed in osteoblast-like UMR-106 cells or chondrocytic precursor CFK2 cells 15–60 min after acidification was induced (data not shown), indicating cell-type specificity.
Fig. 2.

Effects of pH and RANKL on nuclear accumulation of NFATc1. NFATc1 nuclear accumulation was assessed by using immunofluorescence and expressed as a percentage of the total number of rat osteoclasts. Data are means ± SEM. Differences were assessed by one-way ANOVA, followed by a Bonferroni test. (A) pH dependence of NFATc1 nuclear accumulation. pH of the medium was adjusted to the indicated level by using metabolic (•) or respiratory (○) protocols. Osteoclasts were incubated for 60 min and then fixed. NFATc1 nuclear accumulation at the three most acidic points was significantly greater than that at the three most alkaline points (P < 0.05, from three to nine independent experiments). Sigmoid curve was fit by nonlinear regression. (B) Effect of RANKL on nuclear accumulation of NFATc1. Osteoclasts were incubated in media of indicated pH with or without RANKL (1 μg/ml) for 60 min then fixed. *, Significant difference for the effect of RANKL (P < 0.001, from three to 11 independent experiments). (C) OPG prevented the effect of RANKL but not the effect acidification on NFATc1 nuclear accumulation in osteoclasts. Osteoclasts were incubated in medium (i) containing OPG (10 μg/ml) or vehicle, (ii) at the indicated pH, and (iii) with or without RANKL (1 μg/ml) for 45 min and fixed. *, Significant difference for the effect of OPG (P < 0.05, from three independent experiments). (D) The calcineurin inhibitor cyclosporin A prevented nuclear accumulation of NFATc1 induced by acidosis and RANKL. Cyclosporin A (CsA, 1 μM) or vehicle was added for the final 30 min of preincubation at pH 7.6. Osteoclasts were then incubated in medium (i) containing CsA or vehicle, (ii) at the indicated pH, and (iii) with or without RANKL (1 μg/ml) for 60 min and fixed. *, Significant difference for the effect of CsA (P < 0.01, from three independent experiments).
Effect of RANKL on NFATc1 Accumulation in Osteoclast Nuclei. The proresorptive cytokine RANKL (0.1–1 μg/ml, pH 7.6) induced redistribution of NFATc1 to osteoclast nuclei (Fig. 8, which is published as supporting information on the PNAS web site). The 2-fold increase in nuclear accumulation induced by RANKL at pH 7.6 was comparable with the effect of acidification to pH 7.0. Concurrent acidification to pH 7.0 and treatment with RANKL did not enhance NFATc1 activation beyond that induced by each stimulus alone (Fig. 2B).
Because acidosis can up-regulate expression of RANKL in bone (10), we considered whether acidification affects osteoclasts indirectly through stimulation of RANKL expression by other cell types present in the sample. The decoy receptor OPG prevented NFATc1 activation by RANKL but had no effect on NFATc1 activation by acid (Fig. 2C). Moreover, RANKL induced nuclear translocation of NF-κB in rat osteoclasts at pH 7.4. In contrast, acidosis did not stimulate nuclear accumulation of NF-κB in parallel samples (Fig. 9, which is published as supporting information on the PNAS web site). Together, these findings indicate a direct effect of acidosis on mature osteoclasts.
Role of Calcineurin in NFATc1 Activation Induced by Acidosis and RANKL. When applied before acidification or RANKL, the calcineurin inhibitor cyclosporin A abolished subsequent NFATc1 activation (Fig. 2D). Because NFAT undergoes rapid shuttling between cytoplasm and nuclei (21), we next investigated the role of calcineurin in maintaining NFATc1 in the nuclei (Fig. 3). Osteoclasts were first stimulated by acidification to pH 7.0 or RANKL. Within 15 min, both stimuli induced significant NFATc1 activation, which was maximal for acid-stimulated cells at 45 min (68 ± 3%, Fig. 3A, green circles) and for RANKL-treated cells at 30–75 min (65 ± 2%, Fig. 3B, red circles). In both cases, NFATc1 activation was transient, declining toward basal levels by 90 min. Addition of cyclosporin A to samples already activated by acidification did not affect the rate of NFATc1 inactivation (Fig. 3A, yellow triangles), indicating that calcineurin activity is not required to maintain nuclear localization of NFATc1 in this case. However, when acid-activated samples were transferred to alkaline medium (pH 7.6), NFATc1 disappeared promptly from the nuclei (Fig. 3A, blue squares), suggesting that acidosis suppressed NFATc1 inactivation. A strikingly different response was observed when cyclosporin A was added to samples stimulated by RANKL. In this case, cyclosporin A promptly decreased nuclear NFATc1 (Fig. 3B, yellow triangles), indicating that calcineurin activity is required to maintain NFATc1 activation after treatment with RANKL. In contrast, removal of RANKL did not affect the kinetics of NFATc1 inactivation (Fig. 3B, blue squares). Together, these data indicate that calcineurin mediates initial NFATc1 activation by acidification and RANKL; however, distinct mechanisms are involved in maintaining nuclear residence of NFATc1.
Fig. 3.

Kinetics of NFATc1 activation and inactivation. NFATc1 nuclear accumulation was assessed by using immunofluorescence and expressed as a percentage of the total number of rat osteoclasts. Data are means ± SEM (from three to 14 independent experiments), and differences were assessed by one-way ANOVA, followed by a Bonferroni test. (A) At time 0, osteoclasts were placed in medium at pH 7.6 (Control, white circles) or 7.0 (green circles). At 45 min (arrowhead), some samples were moved to medium at pH 7.6 (blue squares) or containing cyclosporin A (CsA, 2 μM) at pH 7.0 (yellow triangles). *, Significant difference for pH 7.6 (blue) or CsA (yellow) compared with pH 7.0 (green) for the same time points (P < 0.05). All control samples (white), except at 0 and 90 min, were significantly different from pH 7.0 (green) (P < 0.05). (B) At time 0, osteoclasts were placed in medium at pH 7.6 with RANKL (1 μg/ml, red circles) or vehicle (Control, white circles, repeated from A). At 45 min (arrowhead), some samples were moved to medium at pH 7.6 without RANKL (Vehicle, blue squares) or containing cyclosporin A (CsA, 2 μM) in the continued presence of RANKL (yellow triangles). *, Significant difference for Vehicle (blue) or CsA (yellow) compared with RANKL (red) for the same time points (P < 0.05). All control samples (white), except at 0 and 90 min, were significantly different from RANKL (red) (P < 0.05).
Role of Kinases in Inactivation of NFATc1. We used pharmacological inhibitors to examine the roles of p38, GSK3β, and CK2 in the inactivation of NFATc1 in osteoclasts stimulated by acidification or RANKL (Table 1). Osteoclasts were treated with inhibitors or vehicle, then exposed to acidosis or RANKL or left unstimulated (Control). Nuclear accumulation was assessed at 15 min, when activation of NFATc1 was submaximal. In control osteoclasts, only inhibition of CK2 significantly increased NFATc1 nuclear accumulation. In RANKL-stimulated cells, inhibition of p38 or GSK3β increased nuclear accumulation of NFATc1, with additive effects. However, when osteoclasts were stimulated by acidosis, no significant effects of kinase inhibition were observed. Vehicles and the inactive p38 inhibitor analog SB202474 (10 μM) had no significant effects. These data establish distinct roles for kinases in regulating NFATc1 inactivation in osteoclasts stimulated by acidosis and RANKL, and they suggest that acidosis acts in part by suppressing kinase activity.
Table 1. Effect of inhibitors of protein kinases on nuclear accumulation of NFAT.
| Change in NFAT nuclear accumulation, %
|
||||
|---|---|---|---|---|
| Inhibited kinase | Control | Acidosis | RANKL | n |
| CK2 | +22 ± 6* | -2 ± 4 | 0 ± 3 | 6 |
| p38 | +9 ± 6 | 0 ± 4 | +19 ± 4* | 6 |
| GSK3β | +15 ± 12 | +2 ± 2 | +18 ± 5* | 5 |
| CK2 plus GSK3β | +38 ± 10* | +13 ± 6 | +28 ± 4* | 3 |
| p38 plus GSK3β | +5 ± 23 | +5 ± 4 | +43 ± 11*† | 4 |
CK2 was inhibited by using emodin (60 μM) or TBB (60 μM) (22); p38 kinase was inhibited by using SB202190 (10 μM) or PD169316 (10 μM); and GSK3β was inhibited by using LiCl (10 mM) (23). GSK3β and CK2 were inhibited with LiCl and either emodin or TBB. GSK3β and p38 were inhibited with LiCl and either SB202190 or PD169316. Osteoclasts in medium containing one or two kinase inhibitors or vehicle were incubated for 15 min at pH 7.6 (Control), at pH 7.0 (Acidosis), or with RANKL (1 μg/ml) at pH 7.6 (RANKL). Samples were then fixed, and NFAT localization was assessed by using immunofluorescence. For each condition, the difference in NFAT nuclear accumulation between inhibitor- and vehicle-treated osteoclasts was expressed as a percentage of vehicle-treated samples. Data are means ± SEM; n values indicate the number of independent experiments. Differences were assessed by Student's t test.
Significant difference compared with vehicle-treated samples (P < 0.001)
Significant difference compared with the effect of single inhibitor (P < 0.001)
Effect of Acidosis on [Ca2+]i. We have shown previously that RANKL transiently elevates [Ca2+]i in 60% of mature osteoclasts (11). We examined whether acidosis also leads to changes in [Ca2+]i. Osteoclasts had stable basal [Ca2+]i of 73 ± 7 nM. Most osteoclasts (16 of 18) responded to acidification with elevation of [Ca2+]i, which peaked at 280 ± 23 nM above basal levels and then declined slowly, with a plateau evident in 67% of cases (Fig. 4A). Upon washout of acidic bathing solution, Ca2+ returned to basal levels. Alkalinization had no effect on [Ca2+]i (Fig. 4B), and no responses were observed when osteoclasts were stimulated with vehicle (n = 15).
Fig. 4.

Acidification elicits transient rise of [Ca2+]i in osteoclasts. Single rat osteoclasts were loaded with fura-2, and [Ca2+]i was monitored by microspectrofluorimetry. Bars below the Ca2+ traces indicate extracellular pH. (A) Upon acidification, [Ca2+]i peaked and then declined to a plateau, returning to basal levels after alkalinization. Response is representative of 16 of 18 tested osteoclasts. (B) Alkalinization had no effect on [Ca2+]i (representative of five of six trials on four osteoclasts). Time scale is the same for A and B. (C) Acidification induced elevation of [Ca2+] in Ca2+i-free buffer containing 0.5 mM EGTA (0 Ca2+). Response is representative of 13 of 14 osteoclasts tested. (D) Inhibition of PLC or depletion of Ca2+ stores abolished acid-induced elevation of [Ca2+]i. Osteoclasts were pretreated with vehicle (Control), the PLC inhibitor U73122 (1 μM, 10 min), its inactive analog U73343 (1 μM, 10 min), or the endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (5 μM, 30 min). Amplitudes of acid-induced Ca2+ transients under each condition are shown expressed as a percentage of matched controls. Data are means ± SEM assessed by one-way ANOVA and a Bonferroni test for the effects of U73122 and U73343 (*, P < 0.05, n = 5 for vehicle, n = 7 for U73122, and n = 6 for U73343) or by Student's t test for the effect of thapsigargin (*, P < 0.0005, n = 6 for vehicle and n = 6 for thapsigargin).
We next investigated the source of Ca2+ contributing to acid-induced elevation of [Ca2+]i. When osteoclasts were bathed in Ca2+-free extracellular solution, acidification still induced elevation of [Ca2+]i (rising by 190 ± 26 nM), consistent with release of Ca2+ from intracellular stores (Fig. 4C). Ca2+ release from stores often involves PLC-mediated production of inositol 1,4,5-trisphosphate. We have shown that the PLC inhibitor U73122 blocks Ca2+ elevations induced by ATP acting through P2Y GPCRs in osteoclasts (24). The acid-induced rise of [Ca2+]i was blocked by U73122 but not by vehicle or the control compound U73343. Moreover, depletion of intracellular Ca2+ stores using thapsigargin, an inhibitor of endoplasmic reticulum Ca2+-ATPases, prevented acid-induced Ca2+ elevations (Fig. 4D and Fig. 10, which is published as supporting information on the PNAS web site). Thus, acidosis leads to activation of PLC and release of Ca2+ from intracellular stores, consistent with involvement of a pH-sensing GPCR.
Expression of OGR1. OGR1 was recently shown to be activated by protons (25) in the same pH range that we observed regulates [Ca2+]i and NFATc1 in osteoclasts. In other cell types, OGR1 couples to Gq, leading to production of inositol phosphates and elevation of [Ca2+]i. We first examined OGR1 expression in osteoclast-like cells differentiated in vitro from RAW 264.7 cells by using RANKL. In undifferentiated RAW 264.7 cells, acidification from 7.4 to 7.0 induced only a small rise of Ca2+ (Fig. 5A Left). In contrast, osteoclast-like cells responded to acidosis with robust elevation of [Ca2+]i, peaking 500 ± 60 nM above basal levels (Fig. 5A Center). Conventional RT-PCR showed expression of OGR1 in osteoclast-like cells (Fig. 11 A and B, which is published as supporting information on the PNAS web site). Real-time RT-PCR analysis revealed strong induction of OGR1 expression upon differentiation of RAW 264.7 cells with RANKL (Fig. 5A Right), consistent with the increased amplitude of the acid-induced Ca2+ response. Levels of OGR1 transcripts in osteoclast-like cells were at least 10-fold greater than in lung (data not shown). Using immunofluorescence, we found that both osteoclast-like cells and authentic rat osteoclasts stained positively for OGR1. Some label was evident at the plasma membrane, consistent with the presence of functional receptors on these cells. The staining of undifferentiated RAW 264.7 cells was less intense than differentiated osteoclast-like cells, consistent with the differences in transcript levels (Fig. 11 C–F).
Fig. 5.

Expression and function of OGR1 in osteoclast-like cells and osteoclasts. (A) RAW 264.7 cells were grown for 5 or 6 days without (RAW) or with RANKL to induce differentiation of osteoclast-like cells (OCL). (Left and Center) Acidification from 7.4 to 7.0 was induced where indicated by bar below the Ca2+ traces. (Left) Response is representative of nine of nine samples of tested undifferentiated RAW 264.7 cells. (Center) Response is representative of seven of eight tested single osteoclast-like cells. (Right) Quantitative real-time RT-PCR was performed by using a primer/TaqMan probe set specific for OGR1 on RNA isolated from undifferentiated RAW 264.7 cells and differentiated osteoclast-like cells. OGR1 mRNA levels were normalized to endogenous ribosomal RNA and expressed relative to levels in liver. Data are means ± SEM, and difference was assessed by using Student's t test. *, P < 0.005, from three independent experiments. (B) OGR1 antagonist Zn2+ inhibits acid-induced elevation of [Ca2+]i in authentic rat osteoclasts. (Left) Representative response in control rat osteoclast. (Center) Response is diminished after treatment with Zn2+ (100 μM, 10 min in the bath). (Right) After treatment with Zn2+, amplitudes of acid-induced Ca2+ transients are significantly decreased. *, P < 0.001. Data are means ± SEM. Difference was assessed by using Student's t test; n = 16 for control (C) and n = 9 for Zn2+-treated (Zn) osteoclasts.
OGR1-mediated production of inositol phosphates is blocked by Zn2+ (25). Treatment of rat osteoclasts with Zn2+ (100 μM) significantly blunted acid-induced elevations of [Ca2+]i (Fig. 5B). Thus, acidification may act through the proton-sensing receptor OGR1 to stimulate PLC, which in turn leads to release of Ca2+ from intracellular stores, activation of calcineurin and translocation of NFAT.
Role of Calcineurin/NFAT Pathway in Mediating the Effects of Acidosis on Resorption. To quantify resorptive activity, we plated rabbit osteoclasts on resorbable dentin slices for 24 h. Consistent with refs. 5 and 7, acidification increased resorptive activity (Fig. 6). More pits were observed under acidic conditions, although average pit area was not significantly altered (Fig. 12, which is published as supporting information on the PNAS web site). Thus, acidosis appears to increase the frequency of initiation of resorptive events. Under the conditions tested, pH did not affect the number of osteoclasts per slice (mean numbers of osteoclasts per slice were 193 ± 36 at pH 7.6 and 168 ± 30 at pH 7.0). Cyclosporin A inhibited the effect of acidification on resorption, decreasing the total area resorbed as effectively as calcitonin, a peptide hormone established to suppress osteoclastic resorption (Fig. 6B). Interestingly, whereas calcitonin did not affect the number of pits per slice, it significantly reduced pit size. In contrast, cyclosporin A reduced the number of pits, without significantly affecting pit area (Fig. 12). Thus, cyclosporin A specifically antagonizes the action of acidosis to increase the number of pits per slice, implicating NFAT in mediating the effects of acidosis on initiation of resorption.
Fig. 6.

Role of NFAT in mediating the effects of acidosis on osteoclastic resorption. Rabbit osteoclasts were plated on dentin slices and treated with calcitonin (10 μM) or cyclosporin A (CsA, 1 μM) for 30 min at pH 7.6. In some samples, the pH was then altered to 7.0 by increasing the partial pressure of CO2 (respiratory acidosis). Experiments were stopped at 24 h, and the number of osteoclasts was assessed. The mean number of osteoclasts per slice was 170 ± 9, and no significant differences were observed among conditions. (A) Phase-contrast micrograph of resorption pits. (B) Quantification of the total area resorbed per slice. Data are means ± SEM, for three independent experiments, each performed in triplicate. Differences were assessed by using Student's t test. *, Significant difference for the effect of acidosis; #, significant difference for the effects of calcitonin or CsA, compared with untreated samples at the same pH.
Discussion
In this article, we report that acidosis and RANKL induce nuclear accumulation of the transcription factor NFATc1 in mature osteoclasts. In both cases, activation of NFATc1 was triggered by calcineurin, and inhibition of calcineurin suppressed the stimulatory effects of acidosis on resorptive activity. pH changes over the range used in our studies occur in severe systemic acidosis, and locally at sites of inflammation and neoplasia (2, 26), pathological conditions associated with increased osteoclastic resorption.
This article links pH changes to activation of NFAT. In other cell types, NFAT shuttles rapidly between the cytoplasm and nuclei and its maintenance in the nucleus requires sustained activation of calcineurin (21, 27). Consistent with this dogma, we found that prolonged activation of calcineurin was required to maintain NFATc1 activation in osteoclasts stimulated with RANKL. In contrast, a more complex mechanism mediates nuclear accumulation of NFATc1 in acid-treated cells, as indicated by three lines of evidence. First, cyclosporin A did not affect NFATc1 distribution in cells already activated by acidosis, suggesting that prolonged activation of calcineurin is not required to maintain NFATc1 in its activated state, consistent with acidification suppressing NFATc1 inactivation. Second, alkalinization led to prompt removal of NFATc1 from osteoclast nuclei, suggesting that acidosis directly affects NFATc1 inactivation. Consistent with this possibility, persistent extracellular acidification induces only a transient decrease in cytosolic pH of osteoclasts (28), with time course for recovery of cytosolic pH similar to that observed for NFATc1 inactivation in the present study. Third, upon inhibition of kinases, NFATc1 activation was increased in both control and RANKL-stimulated osteoclasts; whereas acidosis-treated osteoclasts were unaffected. These data indicate that, in control and RANKL-stimulated osteoclasts, NFATc1 is actively phosphorylated, promoting its removal from the nuclei. In contrast, phosphorylation does not contribute to NFATc1 inactivation in cells activated by acidosis. Acidosis could diminish NFATc1 inactivation by inhibiting kinase activity or suppressing nuclear export. Consistent with the former possibility, purified CK2 and GSK have an alkaline pH optimum of 7.8–8.0 (29, 30). Together, we have found that acidosis activates NFATc1 in osteoclasts through a mechanism initiated by activation of calcineurin and sustained by suppression of NFATc1 inactivation.
Acidification activates bone resorption through direct (7) and indirect mechanisms, including induction of expression of RANKL in bone (10). However, several of our findings are consistent with a direct effect on mature osteoclasts; these include 1) specific activation of NF-κB by RANKL but not acidification, 2) failure of OPG to prevent acid-induced activation of NFATc1, and 3) distinct effects of acid and RANKL on NFATc1 inactivation. We did not observe acidosis-induced activation of NFATc1 in an osteoblastic and a chondrocytic cell line (that did not express OGR1, data not shown), ruling out a nonspecific effect of acidification on NFATc1 activation. Lack of OGR1 expression in the UMR-106 cell line is not representative of osteoblasts in general (25).
NFAT activation is triggered by elevation of cytosolic Ca2+, which acts through calmodulin to stimulate calcineurin activity (14). We found that acidosis leads to release of Ca2+ from intracellular stores in a PLC-dependent manner, consistent with GPCR-mediated signaling. Although a number of proton-sensing receptors have been identified, most of them are ion channels (31). OGR1 is the only receptor that has been shown to act as a proton sensor and to stimulate formation of inositol phosphates and release of Ca2+ from intracellular stores (25), responses induced by acidosis in osteoclasts. Moreover, expression of OGR1 is strongly induced during osteoclast differentiation. Lower expression of OGR1 in precursors may explain why acidosis does not stimulate osteoclast differentiation. The OGR1 antagonist Zn2+ (25), which blocked acid-induced Ca2+ elevations in rat osteoclasts, has previously been shown to inhibit resorption by isolated rat osteoclasts (32). Together, OGR1 is the most likely candidate for mediating acidosis-induced elevation of [Ca2+]i, activation of NFATc1 and stimulation of resorption.
NFATc1 was recently established as a critical transcription factor in osteoclast differentiation (13), with differentiation accompanied by sustained activation of NFATc1 (12). However, we demonstrate that NFATc1 is dynamically regulated in mature osteoclasts by pH as well as RANKL. Our findings are consistent with acidification acting through NFAT to increase the frequency of initiation of resorptive events. Thus, in addition to its critical role in osteoclast differentiation, NFAT appears to regulate the genetic program required for initiating resorptive activity. Indeed, promoters for genes encoding tartrate-resistant acid phosphatase, matrix metalloproteinase-9, cathepsin K and carbonic anhydrase II contain multiple binding sites for NFAT and its transcriptional partner AP-1 (12, 33, 34). Interestingly, acidosis is known to induce expression of carbonic anhydrase II in osteoclasts (35). It has been reported that osteoclastic resorption is stimulated synergistically by low pH and RANKL (17), suggesting the involvement of additional pathways. For example, RANKL may act through NF-κB to enhance osteoclast survival or resorptive activity (36), or acidification may prolong the activation of NFATc1 induced by RANKL.
Together, our data indicate that direct acid-induced activation of calcium/calcineurin/NFAT signaling in osteoclasts stimulates resorption. The degree to which this pathway contributes to the skeletal effects of acidosis in vivo is yet to be determined. Nevertheless, our findings point to the calcium/calcineurin/NFAT pathway as a potential therapeutic target for preventing osteoclast activation associated with local or systemic acidification.
Supplementary Material
Acknowledgments
We thank Dr. F. Valiyeva for assistance with quantification of pit formation, Dr. P. Solano for help with preliminary experiments, Dr. S. M. Bernier (University of Western Ontario) for providing the CFK2 cell line, and Drs. D. W. Litchfield and F. Beier for helpful discussions. We also thank Amgen Biologicals for providing soluble RANKL and OPG and Dr. L.A. Pinna (University of Padua) for the gift of TBB. This work was supported by operating grants from the Canadian Institutes of Health Research and the Canadian Arthritis Network.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RANKL, receptor activator of NF-κB ligand; [Ca2+]i, cytosolic free Ca2+ concentration; CK1/2, casein kinase 1/2; GPCR, G protein-coupled receptor; GSK3β, glycogen synthase kinase 3β; OGR1, ovarian cancer GPCR 1; OPG, osteoprotegerin; PLC, phospholipase C; NFAT, nuclear factor of activated T cells; TBB, 4,5,6,7-tetrabromobenzotriazole.
References
- 1.Goto, K. (1918) J. Biol. Chem. 36, 355-376. [Google Scholar]
- 2.Arnett, T. (2003) Proc. Nutr. Soc. 62, 511-520. [DOI] [PubMed] [Google Scholar]
- 3.Bushinsky, D. A. (2001) Eur. J. Nutr. 40, 238-244. [DOI] [PubMed] [Google Scholar]
- 4.Kraut, J. A., Mishler, D. R. & Kurokawa, K. (1984) Kidney Int. 25, 608-612. [DOI] [PubMed] [Google Scholar]
- 5.Arnett, T. R. & Dempster, D. W. (1986) Endocrinology 119, 119-124. [DOI] [PubMed] [Google Scholar]
- 6.Goldhaber, P. & Rabadjija, L. (1987) Am. J. Physiol. 253, E90-E98. [DOI] [PubMed] [Google Scholar]
- 7.Arnett, T. R. & Spowage, M. (1996) Bone 18, 277-279. [DOI] [PubMed] [Google Scholar]
- 8.Meghji, S., Morrison, M. S., Henderson, B. & Arnett, T. R. (2001) Am. J. Physiol. 280, E112-E119. [DOI] [PubMed] [Google Scholar]
- 9.Boyle, W. J., Simonet, W. S. & Lacey, D. L. (2003) Nature 423, 337-342. [DOI] [PubMed] [Google Scholar]
- 10.Frick, K. K. & Bushinsky, D. A. (2003) J. Bone Miner. Res. 18, 1317-1325. [DOI] [PubMed] [Google Scholar]
- 11.Komarova, S. V., Pilkington, M. F., Weidema, A. F., Dixon, S. J. & Sims, S. M. (2003) J. Biol. Chem. 278, 8286-8293. [DOI] [PubMed] [Google Scholar]
- 12.Takayanagi, H., Kim, S., Koga, T., Nishina, H., Isshiki, M., Yoshida, H., Saiura, A., Isobe, M., Yokochi, T., Inoue, J., et al. (2002) Dev. Cell 3, 889-901. [DOI] [PubMed] [Google Scholar]
- 13.Ishida, N., Hayashi, K., Hoshijima, M., Ogawa, T., Koga, S., Miyatake, Y., Kumegawa, M., Kimura, T. & Takeya, T. (2002) J. Biol. Chem. 277, 41147-41156. [DOI] [PubMed] [Google Scholar]
- 14.Rao, A., Luo, C. & Hogan, P. G. (1997) Annu. Rev. Immunol. 15, 707-747. [DOI] [PubMed] [Google Scholar]
- 15.Porter, C. M., Havens, M. A. & Clipstone, N. A. (2000) J. Biol. Chem. 275, 3543-3551. [DOI] [PubMed] [Google Scholar]
- 16.Murphy, L. L. & Hughes, C. C. (2002) J. Immunol. 169, 3717-3725. [DOI] [PubMed] [Google Scholar]
- 17.Hoebertz, A. & Arnett, T. R. (2003) Methods Mol. Med. 80, 53-64. [DOI] [PubMed] [Google Scholar]
- 18.Santhanagopal, A. & Dixon, S. J. (1999) Am. J. Physiol. 277, E423-E432. [DOI] [PubMed] [Google Scholar]
- 19.Bernier, S. M. & Goltzman, D. (1993) J. Bone Miner. Res. 8, 475-484. [DOI] [PubMed] [Google Scholar]
- 20.Boss, V., Abbott, K. L., Wang, X. F., Pavlath, G. K. & Murphy, T. J. (1998) J. Biol. Chem. 273, 19664-19671. [DOI] [PubMed] [Google Scholar]
- 21.Zhu, J. & McKeon, F. (2000) Cell Mol. Life Sci. 57, 411-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litchfield, D. W. (2003) Biochem. J. 369, 1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein, P. S. & Melton, D. A. (1996) Proc. Natl. Acad. Sci. USA 93, 8455-8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weidema, A. F., Dixon, S. J. & Sims, S. M. (2001) Am. J. Physiol. 280, C1531-C1539. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig, M. G., Vanek, M., Guerini, D., Gasser, J. A., Jones, C. E., Junker, U., Hofstetter, H., Wolf, R. M. & Seuwen, K. (2003) Nature 425, 93-98. [DOI] [PubMed] [Google Scholar]
- 26.Martin, G. R. & Jain, R. K. (1994) Cancer Res. 54, 5670-5674. [PubMed] [Google Scholar]
- 27.Crabtree, G. R. & Olson, E. N. (2002) Cell 109, Suppl., S67-S79. [DOI] [PubMed] [Google Scholar]
- 28.Nordstrom, T., Shrode, L. D., Rotstein, O. D., Romanek, R., Goto, T., Heersche, J. N., Manolson, M. F., Brisseau, G. F. & Grinstein, S. (1997) J. Biol. Chem. 272, 6354-6360. [DOI] [PubMed] [Google Scholar]
- 29.Li, H. & Roux, S. J. (1992) Plant Physiol. 99, 686-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Payne, M. E., Schworer, C. M. & Soderling, T. R. (1983) J. Biol. Chem. 258, 2376-2382. [PubMed] [Google Scholar]
- 31.Waldmann, R. (2001) Adv. Exp. Med. Biol. 502, 293-304. [DOI] [PubMed] [Google Scholar]
- 32.Moonga, B. S. & Dempster, D. W. (1995) J. Bone Miner. Res. 10, 453-457. [DOI] [PubMed] [Google Scholar]
- 33.David, J. P., Rincon, M., Neff, L., Horne, W. C. & Baron, R. (2001) J. Cell. Physiol. 188, 89-97. [DOI] [PubMed] [Google Scholar]
- 34.Anusaksathien, O., Laplace, C., Li, X., Ren, Y., Peng, L., Goldring, S. R. & Galson, D. L. (2001) J. Biol. Chem. 276, 22663-22674. [DOI] [PubMed] [Google Scholar]
- 35.Biskobing, D. M. & Fan, D. (2000) Calcif. Tissue Int. 67, 178-183. [DOI] [PubMed] [Google Scholar]
- 36.Lee, Z. H. & Kim, H. H. (2003) Biochem. Biophys. Res. Commun. 305, 211-214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.