

Abstract
Dehydrocostus lactone (DHE), a natural sesquiterpene lactone, has been used for treatment of various diseases with its anti-inflammatory activity. Recently, it has caused extensive interest in researchers due to it has anti-cancer abilities in some types of carcinomas. However, the anti-cancer effect and mechanism of DHE in glioma remains unclear. The present study conducted to determine the biological effects of DHE on the glioblastoma cells, as well as the mechanisms underlying these effects. After treatment with DHE, the glioblastoma (U118, U251 or U87) cells were significantly inhibited in their viability, proliferation and migration. At the meantime, DHE also induced mitochondria-mediated apoptosis by promoting the release of cytochrome c into cytosol, which activating caspase signaling pathway. Furthermore, our results fully demonstrate that DHE significantly suppressed COX-2 expression by inhibiting the phosphorylation of IKKβ via targeting the ATP-binding site, thereby abrogating NF-κB binding and p300 recruitment to COX-2 promoter. Moreover, the current study firstly demonstrated that DHE can cross blood-brain barrier (BBB). In addition, treatment with DHE markedly inhibited neoplastic weight and volume without the notable adverse effects in the xenograft nude mice model, and these effects may be mediated through inhibition of the IKKβ/NF-κB/COX-2 signaling pathway. These findings provide the pharmacological evidence for development of DHE as a potential agent against glioma.
Keywords: Dehydrocostus lactone, glioblastoma multiforme, blood brain barrier, COX-2, NF-κB, IKKβ
Introduction
Glioblastoma multiforme (GBM) is the most common form of malignant brain tumor in adults, has a poor prognosis. Due to its extremely high proliferation and invasiveness etc, patients merely have a median survival of approximately 14 months, although with currently therapeutic intervention, such as surgery, chemotherapy and radiotherapy [1,2]. Therefore, it is necessary to find some novel therapy strategies to prolong survival and improve patients’ life quality whom suffered from glioma.
Recently, increasing evidence suggests that inflammatory cells and molecules in the tumor microenvironment, influenced almost every aspects of cancer progress [3,4]. Epidemiologic studies have highlighted associations between the regular use of nonsteroidal anti-inflammatory drugs (NSAID) and reduced glioma risks in humans [5,6]. Cyclooxygenase-2 (COX-2), as the rate-limiting enzymes for the synthesis of prostaglandins from arachidonic acid, involved in inflammatory progression, in the meanwhile, is causally linked to the carcinogenesis of many human cancers [7-9]. Previous studies have indicated that COX-2 protein was highly expressed in human glioma specimens and the level of expression was associated with the WHO grade and prognosis [10,11]. Therefore, inhibition of COX-2 expression might be an effective alternative approach to suppress glioma development.
It is well-known that activation of nuclear factor-κb (NF-κB) contributes to an inflammatory response and plays a critical role in cancer, including proliferation, invasion, metastasis and resistance to apoptosis [12]. Like other malignancies, GBM demonstrates high constitutive NF-κB activity [13-15]. In the canonical pathway, NF-κB is regulated by two kinases, IKKα and IKKβ. The latter is particularly important as it phosphorylates IκBs, which is binding to NF-κB dimers in unstimulated cells [16]. Then phosphorylated IκBs are rapidly ubiquitinated and degraded by the proteasome [17,18]. Consequently, the liberated NF-κB proteins which are mostly comprised of p50/p65 heterodimers translocate from the cytoplasm to the nucleus where they bind to specific promoters and regulate target genes expression [17,19]. Importantly, COX-2 is one of its crucially downstream target genes. Therefore, it is essential to identify a small molecule inhibitor selectively suppressing NF-κB/COX-2 signaling pathway activation in glioma.
Traditional Chinese medicines (TCM), an important novel source with a wide range of pharmaceutical potential, and more than 3,000 plants species have long history in the treatment of cancer [20]. Dehydrocostus lactone (DHE) (Figure 1A), a natural sesquiterpene lactone, is derived from many species of medicinal plants, such as Inulahelenium L. and Saussurea lappa [21]. Previous studies have shown that DHE has anti-inflammatory [22], anti-ulcer [23], immunomodulatory [24] and anti-tumor properties [25,26]. In recent years, it has caused extensive interest in researchers due to its potential anti-cancer activities for various types of cancer, such as, leukemia [27], lung cancer [28], breast cancer [29,30], liver cancer [31,32], ovarian cancer [33], prostatic cancer [34,35], bladder cancer [36] and colorectal cancer [37] etc. However, as a herbal monomer agent, the anti-cancer effect and mechanism of DHE in glioma have yet to be elucidated, and the targeting proteins of DHE are not fully clarified.
Figure 1.
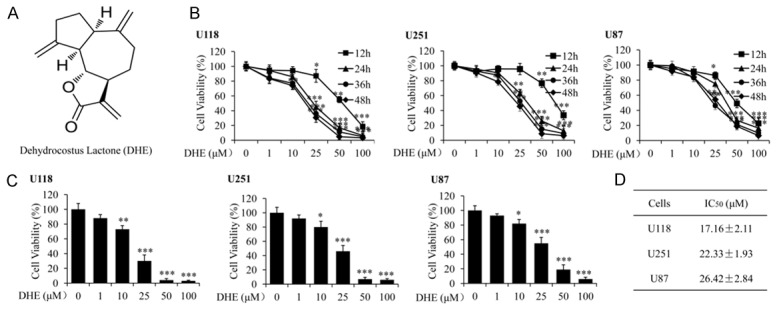
DHE inhibited growth of glioblastoma cells in vitro. A: Chemical structure of dehydrocostus lactone (DHE). B: Effect of DHE on growth inhibition of U118, U251 and U87 glioblastoma cell lines. Cells were treated with various concentrations of DHE (0, 1, 10, 25, 50 and 100 μM) and incubated for 12, 24, 36 or 48 h, and then cell viability was determined by MTT assay. Data were represented as percentage of vehicle-treated control. C: U118, U251 and U87 cells were exposed to DHE at the indicated doses for 48 h, and cell viability was measured by MTT assay. D: The IC50 values were calculated by interpolation from dose-response curves. The data are presented as the mean ± standard deviation of three independent experiments. (*P<0.05, **P<0.01 and ***P<0.001 as compared with the control group).
In the current study, we investigated whether DHE inhibited viability, proliferation, migration and apoptotic resistance of glioblastoma cells, and revealed that DHE could inactivate NF-κB/COX-2 signaling pathway by directly targeting IKKβ in vitro and in vivo. In addition, we demonstrated that DHE can cross blood-brain barrier. Hence, our results suggested that DHE would serve as a potential candidate drug on targeting IKKβ to suppress COX-2 expression in GBM treatment.
Materials and methods
Chemicals and reagents
Dehydrocostus lactone (DHE) was isolated from Inulahelenium L. by Dr. Xiaochi Ma (Dalian Medical University, Liaoning, China), with purity of 98.7%. In present study, DHE was dissolved in dimethyl sulfoxide (DMSO) as a 100 mM stock solution and stored at -20°C. DHE was diluted to obtain the desired concentration in cell culture medium, where the final concentration of DMSO was less than 0.1%. Control cultures received the carrier solvent (0.1% DMSO).
Antibodies and other materials
Antibodies specific to cleaved caspase-3, cleaved caspase-9, COX-2, p-IKKα/β, IKKα, IKKβ, p-IκBα, IκBα, p-p65, p65, β-actin and all the secondary antibodies were purchased from Cell Signaling Technology (Cell Signaling Technology, Inc, USA). Antibodies specific to cytochrome c, p300 and p50 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies specific to Bcl-2, BAX and Lamin B1 were purchased from Proteintech Group (Proteintech, Inc, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS) and Trypsin were purchased from Gibco. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) unless otherwise specified.
Cell culture
Human glioblastoma cell lines U-118 MG (U118, p53-/-), U-251 MG (U251, p53-/-) and U-87 MG (U87, p53+/+) were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), and grown at 37°C in a humidified atmosphere with 5% CO2.
Cell viability assay
U118 (6×103 cells/well), U251 (8×103 cells/well) and U87 (7×103 cells/well) cells were counted and allowed to adhere to obtain 70% confluent monolayer. Following this, the cells were treated with different concentrations of DHE as indicated. After incubation for 12, 24, 36 or 48 h, 10 μl of MTT (5 mg/ml) was added to each well and the cells were incubated for another 4 h. Subsequently, the medium was replaced with 100 μl of DMSO to dissolve formazan crystals. The absorbance was measured at 570 nm using a microplate reader (Bio-Rad, CA, USA). Each experiment was repeated at least three times. The IC50 values were calculated by interpolation from dose-response curves.
Colony formation assay
U87 and U251 cells were seeded at a density of 4×103 cells per well in six-well plates. After adherence, the cells were treated with different concentrations of DHE for 3 h. Subsequently, the supernatant was replaced with fresh medium, and the cells were cultured until colonies were large enough to be visualized. After that, the colonies were fixed with 4% paraformaldehyde and then stained with 0.1% crystal violet solution, dried and then imaged.
Wound healing assay
U118 and U251 cells were seeded in 6-well culture plates and allowed to form a fully confluent monolayer. After 6 h of serum starvation, the layer of cells was scraped with a 20-200 µl sterile pipette tip to create some wounds with the same width. Subsequently, the cells were treated with serum-free medium containing various concentrations of DHE for 48 h. Images of the cells were captured at 0 and 48 h using a Leica DM 14000B microscope fitted with digital camera and the migration rate was counted from five randomly selected fields.
Confocal immunofluorescence analysis
For immunofluorescence analysis, U87 cells were seeded on coverslips, and treated with different concentrations of DHE for 48 h. After that, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% TritonX-100, and then blocked in 5% BSA. Subsequently, the cells were incubated with diluted primary antibodies against cytochrome c, p300, p65 or p50 overnight at 4°C. Following this, the cells were incubated with fluorescein isothiocyanate or rhodamine isothiocyanate-conjugated secondary antibodies. Finally, DAPI was added to each sample for nuclear counterstaining and fluorescent images were examined using a Leica DM 14000B confocal microscope.
Western blot analysis
Proteins from cell lysates or streptavidin-agarose pulldown assay were subjected to sodium dodecyl sulfate-polyacrylamide minigels (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked in 5% non-fat powdered milk buffer before being incubated with specific primary antibodies and secondary antibodies. Protein bands were visualized by enhanced chemiluminescence (ECL) and integrated optical density of bands was quantitated by the ImageQuant software (GE Healthcare). The concentration of proteins was determined by BCA method. Similar experiments were performed at least three times.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from U87 and U251 cells by using Trizol reagent (TaKaRa Bio Inc, Dalian, China) according to manufacturer’s instructions. Following this, reverse transcription was performed using the Prime Script TM RT-PCR Kit (TaKaRa Bio Inc, Dalian, China), and then the cDNA was used for PCR with the following primers (synthesized by TaKaRa Bio Inc, Dalian, China): for COX-2 (sense: 5’-TCACAGGCTTCCATTGACCAG-3’, antisense: 5’-CCGAGGCTTTTCTACCAGA-3’); for GAPDH (sense: 5’-AATCCCATCACCATCTTCC-3’; antisense: 5’-CATCACGCCACAGTTTCC-3’). The PCR products were separated on 1.5% agarose gel electrophoresis and visualized under ultraviolet light and the band density was measured through quantitative analysis. Similar experiments were performed at least three times.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation assay were performed with procedures modified from the protocol by previously described [38]. Briefly, the DHE-treated cells were harvested after cross-linked with 1% formaldehyde, and sonicated break chromatin DNA to an average length of ~500 bp. The lysate was incubated with antibodies against p300, p65, p50 or IgG (as negative control) overnight at 4°C, and immunoprecipitated by protein A/G agarose beads. Cross-linking of protein-DNA complexes was reversed at 65°C, followed by treatment with proteinase K solution. Finally, the DNA was extracted with phenol/chloroform and precipitated with ethanol. The purified DNA was subjected to PCR amplification using specific COX-2 promoter primers (sense: 5’-ACGTGACTTCCTCGACCCTC-3’, antisense: 5’-AAGACTGAAAACCAAGCCCA-3’). The PCR products were analyzed by 1.5% agarose gel electrophoresis.
Streptavidin-agarose pulldown assay
Nuclear extract proteins (400 μg) were incubated in a 400 μl mixture of containing biotinylated DNA probe (4 μg), streptavidin-conjugated agarose beads (40 μl) and supplemented with PBSi (PBS buffer with 1 mM EDTA, 1 mM DTT and protease inhibitor cocktail complete) buffer at room temperature for 5 h in a rotating shaker. Thereafter, the beads were pelleted by centrifugation, and dissociated in 50 μl of 2× Laemmli sample buffer by boiled at 100°C for 10 min. The supernatant was analyzed by western blot.
Molecular modeling
The molecular docking studies were performed to explore the potential binding mode between DHE and IKKβ protein complex. DHE was optimized using the semi-empirical PM3 method with the Polak-Ribie’re conjugate gradient algorithm with an RMS gradient of 0.01 kcal mol-1 Å-1 as convergence criterion. The optimized structure of DHE was docked into the active site of IKKβ with ligand K-252A (PDB Code: 4KIK). The crystallographic ligand was extracted from the active site, and the residues within a 6.5 Å radius around IKKβ molecule were defined as the active pocket. The SurflexDock program was used for the docking calculations with default parameters. MOLCAD surfaces were generated for visualizing the binding mode of the dock protein-ligand complexes.
Animals study
All animals maintenance and procedures were performed in according with protocols approved by the Animal Care and Ethics Committee of Dalian Medical University. The female athymic nude mice (4-6 weeks old) and Sprague Dawley rats (8-10 weeks old) were obtained from SPF Laboratory Animal Center at Dalian Medical University. The animals were housed under controlled conditions.
U87 (1×107) cells were resuspended in 100 μl PBS solution and subcutaneously injected into the left axillary fossa of each nude mice. After two weeks, the mice were randomly divided into three groups (n = 6 per group), and daily intraperitoneal administered with PBS (control), DHE (10 mg/kg) or DHE (20 mg/kg) for 14 days. The body weight of each mice and the volume of the implanted tumor were recorded every 2 days. The tumor volume was calculated according to the formula: V = 1/2 (length × width2). All mice were euthanized on day 30. The implanted tumors were excised and weighed, and then fixed in 10% formalin before paraffin embedding. The expression of COX-2, p-p65 and p-IKKβ in implanted tumors was detected by immunohistochemical staining. The images were captured under a Leica DM 4000B fluorescence microscope fitted with a digital camera.
Statistical analysis
All of the data were presented as the mean ± SD for at least three independent experiments. Statistical analysis was performed with SPSS 17.0 software. One-way analysis of variance (ANOVA) or Student’s t-tests was used to evaluate the statistical significance between controls with treated groups. Results were considered statistically significant at the level of P<0.05. One asterisk, two asterisks, and three asterisks represent P<0.05, P<0.01 and P<0.001, respectively.
Results
DHE inhibited growth of glioblastoma cells in vitro
To evaluate the anti-glioblastoma effect of DHE, three glioblastoma cell lines (U118, U251 and U87) were treated with different concentrations (0, 1, 10, 25, 50 and 100 μM) of DHE for 12 h, 24 h, 36 h or 48 h, respectively. And then a MTT assay was performed to determine cell viability. We found that DHE could inhibit these cells growth in a dose-dependent and time-dependent manner (Figure 1B). At 48 h after treatment, the IC50 values of DHE against U118, U251 and U87 cells were 17.16±2.11, 22.33±1.93 and 26.42±2.84 μM, respectively (Figure 1C and 1D). Taken together, our findings indicate that DHE might be a potent inhibitor in glioblastoma cells proliferation.
DHE suppressed clonogenic and migratory ability of glioblastoma cells
Enabling replicative immortality is one core hallmark of cancers, which is manifested by unlimited replicative potential and high clonogenic ability [39]. Thus we employed colony formation assay to evaluate the effect of DHE on the clonogenic capacity of U87 and U251 cells. As shown in Figure 2A and 2B, treatment of these cells with DHE at the different concentrations (10 and 20 μM) result in a dose-dependently reduced the number of colonies, compared with untreated control cells.
Figure 2.
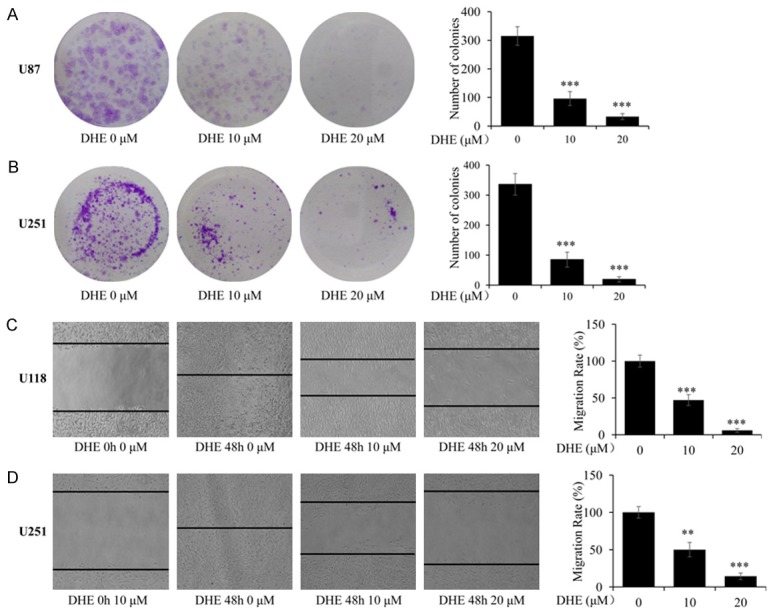
DHE suppressed clonogenic and migratory ability of glioblastoma cells. (A, B) U87 and U251 cells were treated with different concentrations of DHE (0, 10 and 20 μM) for 3 h, and then cultured for an additional 15 days. Representative images of the U87 (A) and U251 (B) cells were captured using a microscope fitted with digital camera (magnification, ×100). And the colonies number of the U87 and U251 cells was calculated. (C, D) Cells were treated with DHE (0, 10 and 20 μM) for 48 h after scratch. Representative images of the U118 (C) and U251 (D) cells were captured at 0 h and 48 h using a microscope fitted with digital camera (magnification, ×100). And the migration rate of the U118 and U251 cells was calculated. The data are presented as the mean ± SD of three independent experiments. (*P<0.05, **P<0.01 and ***P<0.001 as compared with the control group).
Likewise, migration is the other important hallmark of cancers [39]. To identify the effect of DHE on the migration ability of glioblastoma cell lines, wound healing assay was performed under serum-free conditions. The results indicated that the migration rate of cells significantly decreased with increasing concentrations of DHE in U118 and U251 cells (Figure 2C, 2D) compared to the control group at 48 h (P<0.01). These results demonstrated that DHE exhibits strong properties in suppressing colony formation and migration for glioblastoma cells.
DHE induced apoptosis through mitochondrial pathway in U87 glioblastoma cells
The ability of anti-apoptosis is another important hallmark of cancers [39]. Many of the signals that elicit apoptosis converge on the mitochondria, which respond to proapoptotic signals by releasing cytochrome c into cytosol, a potent catalyst of apoptosis [40]. Therefore, confocal immunofluorescence microscopy was performed to detect whether DHE resulted in cytochrome c release from mitochondria to the cytosol in U87 cells. As shown in Figure 3A, in the control group, cytochrome c was mainly localized in the mitochondria as evidenced by yellow-orange staining due to merging of Mito-Tracker red and cytochrome c-associated green fluorescence. In the treated group, increased concentrations of DHE resulted in elevated levels of cytosolic cytochrome c as evidenced by a diffused green fluorescence. Afterward, western blot was used to determine the extent of cytochrome c release from the mitochondria into cytosol during treatment. As shown in Figure 3B, DHE statistically increased the levels of cytosolic cytochrome c in a concentration-dependent manner. To gain better insight into DHE-induced apoptosis in U87 cells, we measured the expression of major mitochondrial apoptosis signaling associated protein, using western blot. The results demonstrated that DHE remarkably reduction of Bcl-2 levels and anti-apoptotic Bcl-2/BAX ratio. Furthermore, DHE resulted in cleavage of procaspase-9 and procaspase-3, generating the activated fragment in a dose-dependent manner (Figure 3C). Taken together, these results showed that DHE indeed induce apoptosis via the intrinsic mitochondrial pathways in glioblastoma cells.
Figure 3.
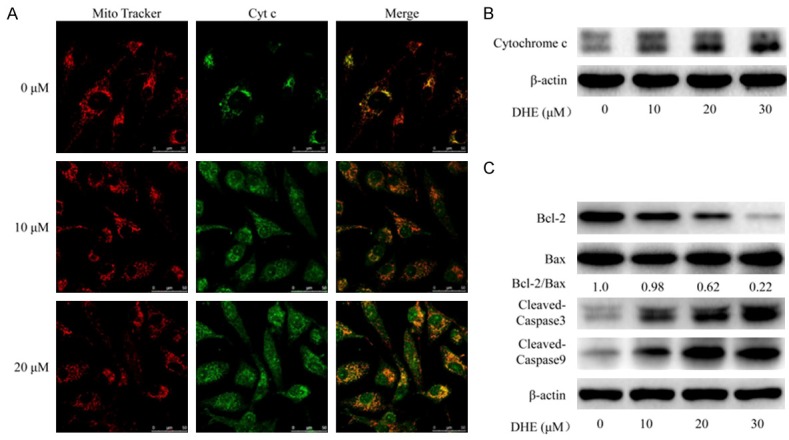
DHE induced apoptosis through mitochondrial pathway in U87 cells. U87 cells treated with DHE at the indicated concentrations for 48 h. A: Laser scanning confocal microscope immunofluorescence analysis of cytochrome c (green) and mitochondria (red) co-localization in U87 cells. B: Cytoplasmic protein were extracted from the U87 cells and subjected to western blot analysis for cytochrome c and β-actin. C: And the levels of Bcl-2, BAX and cleaved caspase-3/9 proteins in total cell lysates from U87 cells were evaluated by western blot. The β-actin served as the protein loading control. Densitometric ratios of Bcl-2 and BAX proteins were quantified by Image J software.
DHE modulated the expression of COX-2 in glioblastoma cells
The overexpression of COX-2 in human glioma specimens has been identified by previous study [11], and it led to induce proliferation, migration, and development of apoptotic resistance in cancer cells [41]. Firstly, the expression of COX-2 was studied in three human glioblastoma cell lines (U118, U251 and U87) by western blot. Representative immunoblot was shown in Figure S1. Analysis of the band densities revealed that COX-2 protein was strong expressed in U87 and U251 cells, whereas the expression of COX-2 was relatively weak in U118 cells. Thus, we employed U87 and U251 cell lines to determine whether the tumor growth inhibitory effect of DHE was correlated with COX-2 expression. And the effect of DHE treatment on COX-2 protein and mRNA levels was evaluated by western blot analysis and RT-PCR, respectively. As shown in Figure 4, DHE dose-dependently inhibited COX-2 protein expression in U87 (Figure 4A) and U251 (Figure 4B) cell lines. Consistent with the western blot analysis results, DHE significantly and concentration-dependently reduced COX-2 mRNA expression in U87 (Figure 4C) and U251 (Figure 4D) cell lines. Collectively, these results suggested that DHE effectively inhibited COX-2 expression at both protein and mRNA levels in glioblastoma cells by a dose-dependently manner. Consequently, we considered that DHE should effect on COX-2 mRNA transcriptional regulation in glioblastoma cells.
Figure 4.
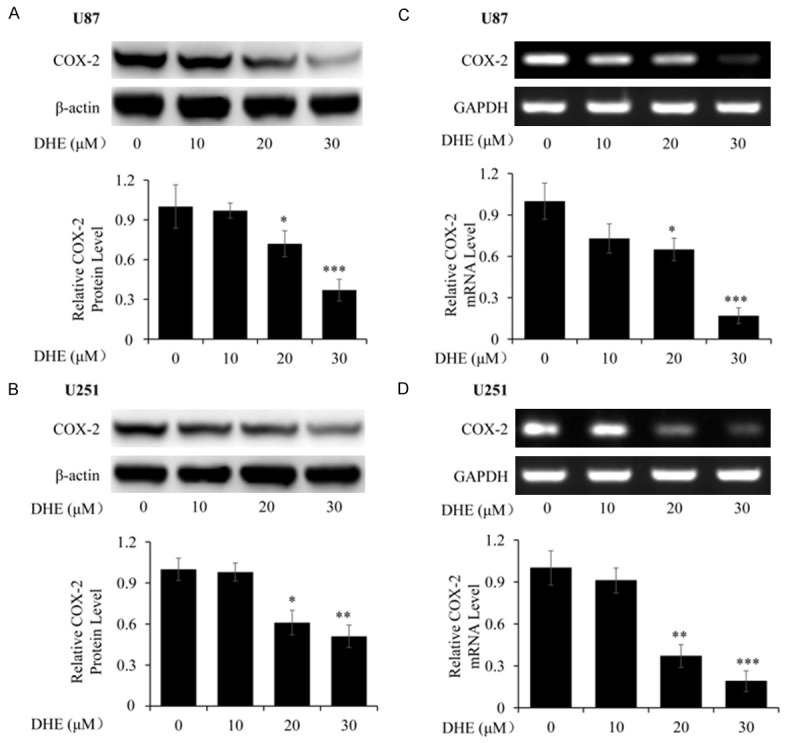
Effects of DHE on COX-2 protein and mRNA expression in U87 and U251 cells. The U87 and U251 cells were treated with various concentrations of DHE (0, 10, 20 and 30 μM) for 48 h. And the expression of COX-2 protein and mRNA in the cells were evaluated by western blot (A, B) and RT-PCR (C, D), respectively. The β-actin and GAPDH served as the protein and mRNA loading control, respectively. Quantitative analyses of the band density were normalized to each loading control. The data are presented as the mean ± SD of five independent experiments. (*P<0.05, **P<0.01 and ***P<0.001 as compared with the control group).
DHE inhibited transcriptional activation of COX-2 by effect on p300 recruitments and p50/p65 NF-κB nuclear translocation
The transcription activation of genes is regulated by the binding activities of transactivators and coactivators on gene promoter structure. Sequence analysis of the 5’-flanking region of the human COX-2 gene has identified several potential transcriptional regulatory elements, of which two nuclear factor kappa B (NF-κB) binding sites are essential for transcriptional activation of COX-2 [42,43]. Previous study established that the transcriptional coactivator p300 recruitments to promoter-bound transactivators and plays a central role in transcriptional activation of COX-2 by regulating p50/p65 NF-κB [44]. To determine whether p300 and p50/p65 NF-κB are involved in the transcriptional regulation of DHE on COX-2 by binding directly to the promoter in U87 glioblastoma cells, nuclear extracts were analyzed by ChIP using specific antibodies directed against p300, p50 and p65. As shown in Figure 5A, DHE inhibited p300 recruitments and p50/p65 NF-κB binding directly to the COX-2 promoter in a dose-dependent manner, in contrast, the DNA binding was undetectable when using a normal IgG negative control.
Figure 5.
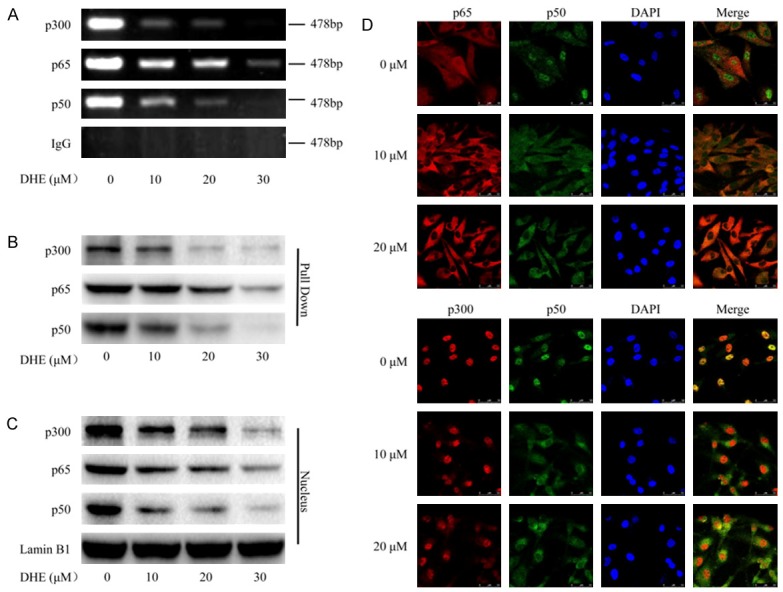
DHE inhibited transcriptional activation of COX-2 by effect on p300 recruitments and p50/p65 NF-κB nuclear translocation. The U87 cells were treated with DHE at the indicated concentrations for 48 h. A: The nuclear extracts were harvested for ChIP assay by using specific antibodies directed against p300, p65 and p50 to immunoprecipitate formaldehyde-fixed chromatin, followed by regular PCR with COX-2 primers. Normal IgG served as a negative control. B: The binding activities of p300, p65 and p50 to COX-2 promoter were analyzed by streptavidin-agrose pulldown assay. C: The total nuclear extracts were subjected to western blot analysis for p300, p65 and p50. The Lamin B1 used as a loading control. D: Laser scanning confocal microscope immunofluorescence assay was performed to detect the subcellular localization of p65, p50 and p300 and the co-localization of p50 with p65 or p300 by using specific antibodies against p65 (red), p50 (green) and p300 (red). And the typical morphology of cells was presented in the above.
To further evaluate the effect of DHE on the binding activities of p300 and p50/p65 NF-κB on COX-2 promoter-transactivator complex, we employed the streptavidin-agarose pull-down assay. Nuclear extracts from U87 cells treated with or without DHE were incubated with the 5’-biotinylated COX-2 promoter probe and streptavidin-agarose beads. After centrifugation, transcription activators p50/p65 NF-κB and coactivators p300 present in the complex were analyzed by western blot. Consistent with the ChIP assays, p300 and p50/p65 NF-κB binding to the COX-2 promoter region were decreased by treated with DHE (Figure 5B). Following this, we detected quantitative information of p300 and p50/p65 NF-κB in total nuclear extracts by western blot. As expected, both p300 and p50/p65 NF-κB protein levels in nucleus decreased significantly after DHE treatment (Figure 5C). To further confirm that DHE inhibited nuclear translocation and interaction between p300 and p50/p65 NF-κB protein, we performed immunoflurescence staining to investigate the distribution of p300 and p50/p65 NF-κB in U87 cells. As shown in Figure 5D, both coactivators p300 and p50/p65 NF-κB obviously decreased in nuclear localization and increased in cytoplasmic localization following DHE treatment. Taken together, these results suggested that DHE inhibited transcriptional activation of COX-2 by effect on p300 recruitments and p50/p65 NF-κB nuclear translocation.
DHE inhibited p50/p65 NF-κB activity by targeting IKKβ kinase in glioblastoma cells
The canonical activation pathway of NF-κB major relies on IKKβ kinase activity which leads to phosphorylation of IκBα. And then, phosphorylated IκBα are degraded by the proteasome, thereby allowing the liberated and phosphorylated p50/p65 NF-κB dimers to translocate from cytoplasm to nucleus where they serve as transcriptional regulators [17-19]. Hence, to fully understand the related molecular mechanism of DHE treatment on glioblastoma cells, we next employed U87 and U251 cells to investigate the effects of DHE on IKKβ/ IκBα/NF-κB signaling pathway. As shown in Figure 6A and 6B, the levels of p-IKKα/β, p-IκBα and p-p65 protein were markedly down-regulated by treated with DHE in a dose-dependent manner, however, the whole levels of IKKα, IKKβ, IκBα and NF-κB p65 did not significantly change during this study. These results indicated that DHE might suppress NF-κB activation through inhibiting IKKβ kinase activity.
Figure 6.
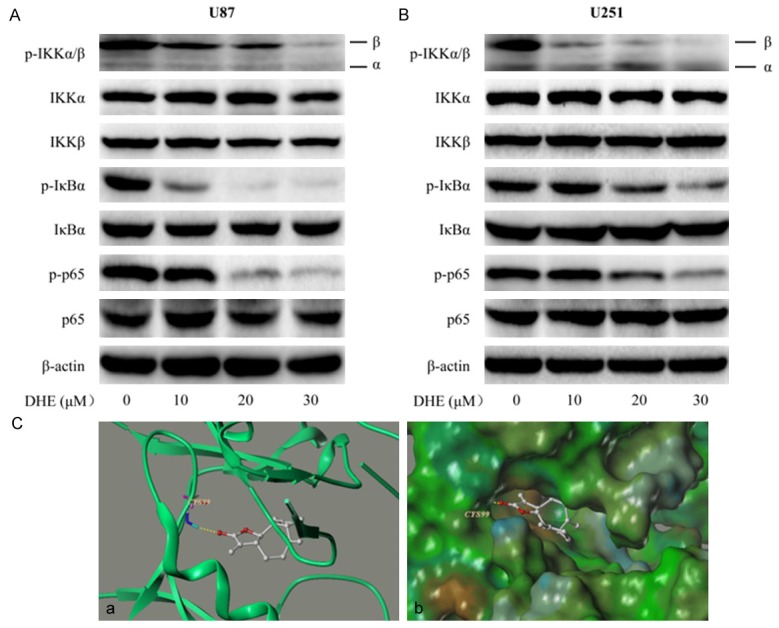
DHE inhibited p50/p65 NF-κB activity by targeting IKKβ kinase in glioblastoma cells. U87 (A) and U251 (B) cells were treated with different concentrations of DHE (0, 10, 20 and 30 μM) for 48 h. The expression of p-IKKα/β, IKKα, IKKβ, p-IκBα, IκBα, p-p65 and p65 proteins in the glioblastoma cells were evaluated by western blot. The β-actin served as the loading control. Representative immunoblot of three independent experiments is shown. (C) The best ranked pose of DHE in the ATP binding site of IKKβ generated with docking. (a) Interactions of DHE and IKKβ are delineated by ribbon structure, hydrogen bonds are displayed as yellow dashed lines, and the participating amino acid residues are marked. (b) MOLCAD representation the molecular lipophilic potential surface upon the bioactive pose of DHE in the ATP binding site of IKKβ. The blue denotes the hydrophilic, brown for the lipophilic and green corresponds to the neutral moiety.
Therefore, we hypothesized that IKKβ may be served as a target of DHE in the treatment of glioma. To test this hypothesis, computer molecular modeling assay was performed to simulate the interactions between DHE and IKKβ. Molecular docking studies predicted that DHE could bind to ATP binding site of IKKβ. Specifically, as shown in Figure 6Ca, DHE formed one hydrogen bond with the ATP binding pocket of the IKKβ kinase domain. The CO motif at the lactonic ring of DHE forms a hydrogen bond with the backbone NH of Cys99. The result of MOLCAD surface modeling indicated that the lacton ring of DHE extends into the deep hydrophobic cavity of the ATP-binding pocket (Figure 6Cb). All of these results supported that IKKβ was a target site of DHE in the NF-κB signaling pathway to suppressed COX-2 expression.
DHE could cross blood-brain barrier and suppress the growth of glioblastoma xenograft in vivo
The mass spectrometry method was used to determine whether the DHE penetrate into mouse brain. Figure S2 shows the representative chromatograms of the cerebrospinal fluid (CSF) samples after intravenous administration of DHE at the dose of 100 mg/kg for 1 h. The mass spectrometry analysis of the CSF samples showed a single peak at a retention time of 1.81 min. The result suggested that DHE can rapidly cross blood-brain barrier in mouse model.
Thus, we established a xenograft nude mice model to further determine the anti-cancer effect of DHE on glioblastoma in vivo. The U87 cells were subcutaneously inoculated into the left axillary fossa of each mouse. After two weeks, the mice were intraperitoneal administered with different doses of DHE (10 or 20 mg/kg/d), or vehicle (PBS) for 14 days. As shown in Figure 7A, the body weights of mice were almost not changed between each group in the tested durations, suggesting that DHE administrations had no obvious side effect on mice. As expected, compared with the control group, both the tumor volume (Figure 7B) and the tumor weights (Figure 7C) were dramatically reduced in the treated group. Therefore, these results may indicate that DHE exhibits strong properties in suppressing the growth of glioblastoma in vivo. Furthermore, to determine the potential mechanism, we detected the expression of COX-2, p-p65 and p-IKKβ in transplanted tumors using immunohistochemistry analysis. When probed with COX-2, p-p65 or p-IKKβ antibodies, there was a predominantly positive staining in the xenografts from control group, and staining intensities gradually grew weaker in the DHE treated groups with increasing dose when it being sparsely observed in the high dose group (Figure 7D). Hence, the above results indicated that DHE could cross blood-brain barrier and suppress the activation of the IKKβ/NF-κB/COX-2 pathway in U87 cells transplanted tumors, which might be partially responsible for the inhibition of the xenograft growth.
Figure 7.
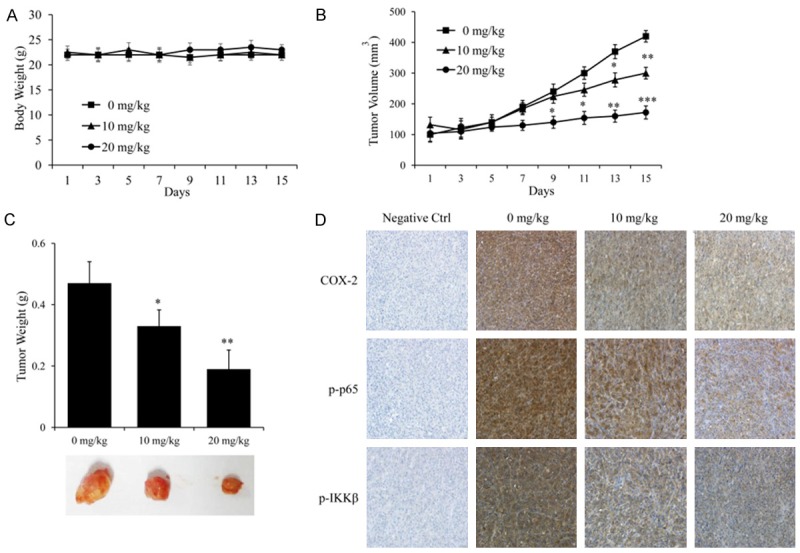
In vivo antitumor efficacy of DHE in U87 tumor xenograft model. Body weights (A) and tumor volumes (B) were recorded every 2 days during the experiment. All mice were euthanized on day 30. (C) The implanted tumors were excised and weighed, and representative tumor photographs of each group were shown. (D) The expression of COX-2, p-p65 and p-IKKβ in implanted tumors was detected by immunohistochemical staining. Representative images of each group were shown. The results are represented as means ± SD (n = 6). (*P<0.05, **P<0.01 and ***P<0.001 as compared with the control group).
Discussion
Dehydrocostus lactone (DHE), a natural sesquiterpene lactone, has been reported as major and biologically active compound of the roots of Saussurea lappa, a well-known Chinese traditional herbal medicine. Currently, clinically available DHE-containing drugs, such as Compound Ancklandia and Berberine Tablets, have been used for treatment of digestive tract diseases with its anti-inflammatory, anti-microbial activities. Recently, a few reports have showed that DHE has anti-cancer abilities in some types of carcinomas [21]. However, the anti-cancer effect and mechanism of DHE in glioma remains unknown. In the current study, we found that DHE has the potential to powerfully suppress the biological characteristics of glioma in many aspects. First of all, treatment of human glioblastoma cell lines with DHE result in a dose-dependently inhibition of viability, proliferation and migration. Meanwhile, DHE also induced mitochondria-mediated apoptosis via down-regulating anti-apoptotic Bcl-2/BAX ratio and promoting mitochondrial release of cytochrome c into cytosol which activating caspase signaling pathway. Moreover, the present study firstly investigated the brain penetration of DHE by using mass spectrometry method, and the result indicated that DHE can rapidly penetrate into the brain. Furthermore, in the xenograft nude mice model, treatment with DHE (10 and 20 mg/kg/d) significantly inhibited neoplastic growth without the notable adverse effects.
A lot of evidence has established that inflammation regulates different stages of tumor development, such as initiation, promotion, invasion and metastasis [3]. Cyclooxygenase (COX) is the rate-limiting enzyme catalyzing the conversion of arachidonic acid to prostaglandins, which is an important inflammatory mediator. Two isoforms of COX have been identified: COX-1 and COX-2. In the central nervous system, COX-1 is constitutively expressed in neurons, astrocytes and microglial cells, and involved in the maintenance of tissue homeostasis, whereas COX-2 is scarcely produced in normal brain tissues until being induced by some stimulations, such as inflammation and cancer [11,45-48]. Previous studies have indicated that COX-2 overexpression in glioma and plays a critical role in the progression of cancer by increasing proliferation, invasiveness and apoptotic resistance [10,11,49-51]. In our study, we found that DHE contributed to transcriptional regulation of COX-2 gene, as a consequence, significantly inhibited COX-2 expression at both mRNA and protein levels in a dose-dependently manner, along with inhibiting glioblastoma viability, proliferation, migration and apoptotic resistance.
The promoter region of the COX-2 gene contains the binding site for NF-κB [52-54]. NF-κB is a crucial transcription factor modulated many of the central hallmarks of cancer including: cellular proliferation, migration and resistance to apoptosis [39]. Moreover, the critical role of NF-kB in promoting inflammation further contributes to the malignant phenotype in glioma [55,56]. Therefore, agents capable of suppressing NF-κB activation might be have the potential to inhibit glioma development. As previous described, in the canonical NF-κB pathway, the activation of p50/p65 NF-κB mainly mediated by IKKβ which induced phosphorylation of IκBα. The phosphorylation and ubiquitination events alter the configuration of IκBα, which is consequently degraded by proteasomes. Subsequently, p50/p65 NF-κB is released, phosphorylated and translocated to the nucleus, and binding to the κB sites of the target genes, and then recruiting coactivators p300 to induce the transcription activation of multiple genes [57,58]. In current study, we determine the inhibitory effects of DHE on IKKβ activity, IκBα phosphorylation and degradation, coactivators p300 recruitments and p50/p65 NF-κB nuclear translocation, and their DNA binding activity on COX-2 promoter. As expected, our results showed that DHE significantly inhibited p50/p65 NF-κB activation, nuclear translocation, and DNA-binding capacity by inhibition of IKKβ activity, and IκBα phosphorylation and degradation. Furthermore, the importance of the IKKβ in regulating NF-κB activation, coupled with the drug gable nature of kinase activity, has made IKKβ become a primary target for pharmacotherapy. While many IKKβ inhibitors have been used in the treatment of peripheral cancers, only a few have been studied in glioma [56]. Therefore, we hypothesized that DHE might bind to IKKβ and subsequently inhibited its kinase activity. To test this hypothesis, computer molecular docking assay was conducted to simulate the interactions between DHE and IKKβ. Molecular docking studies predicted that DHE could bind to the ATP binding site of IKKβ.
In summary, our result indicates that DHE can rapidly cross blood-brain barrier, and our data reveals that DHE has the potential to powerfully suppress the biological characteristics of glioma in many aspects. Furthermore, it is possible that the anti-cancer properties of the DHE in glioma may be mediated, at least in part, through inhibition of the NF-κB/COX-2 signaling pathway by targeting IKKβ in vitro and in vivo. These findings provide the preclinical evidence for development of DHE as a potential agent against glioma.
Acknowledgements
We thank the NSFC (81622047, 81473334 and 81172180), Dalian Outstanding Youth Science and Technology Talent (2015J12JH201), Distinguished professor of Liaoning Province, Project sponsored by Liaoning BaiQianWan Talents Program and Innovation Team of Dalian Medical University for financial support.
Disclosure of conflict of interest
None.
Author’s contribution
XM, ZY and YX initiated the work, designed the experiments. JW, ZY, CW and XT performed the experiments. XH, YW, CS, LF, BZ and QY analyzed the data. JW, ZY, CW and XH prepared the figures. JW, ZY, YX and XM wrote the paper. All authors reviewed the manuscript.
Supporting Information
References
- 1.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 2.Mangiola A, Anile C, Pompucci A, Capone G, Rigante L, De Bonis P. Glioblastoma therapy: going beyond Hercules Columns. Expert Rev Neurother. 2010;10:507–514. doi: 10.1586/ern.09.158. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald N. Chronic inflammatory states: their relationship to cancer prognosis and symptoms. J R Coll Physicians Edinb. 2011;41:246–253. doi: 10.4997/JRCPE.2011.315. [DOI] [PubMed] [Google Scholar]
- 5.Scheurer ME, El-Zein R, Thompson PA, Aldape KD, Levin VA, Gilbert MR, Weinberg JS, Bondy ML. Long-term anti-inflammatory and antihistamine medication use and adult glioma risk. Cancer Epidemiol Biomarkers Prev. 2008;17:1277–1281. doi: 10.1158/1055-9965.EPI-07-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivak-Sears NR, Schwartzbaum JA, Miike R, Moghadassi M, Wrensch M. Case-control study of use of nonsteroidal antiinflammatory drugs and glioblastoma multiforme. Am J Epidemiol. 2004;159:1131–1139. doi: 10.1093/aje/kwh153. [DOI] [PubMed] [Google Scholar]
- 7.Marks F, Muller-Decker K, Furstenberger G. A causal relationship between unscheduled eicosanoid signaling and tumor development: cancer chemoprevention by inhibitors of arachidonic acid metabolism. Toxicology. 2000;153:11–26. doi: 10.1016/s0300-483x(00)00301-2. [DOI] [PubMed] [Google Scholar]
- 8.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381–2386. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 9.Nakanishi M, Rosenberg DW. Roles of cPLA2alpha and arachidonic acid in cancer. Biochim Biophys Acta. 2006;1761:1335–1343. doi: 10.1016/j.bbalip.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myung J, Cho BK, Kim YS, Park SH. Snail and Cox-2 expressions are associated with WHO tumor grade and survival rate of patients with gliomas. Neuropathology. 2010;30:224–231. doi: 10.1111/j.1440-1789.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- 11.Joki T, Heese O, Nikas DC, Bello L, Zhang J, Kraeft SK, Seyfried NT, Abe T, Chen LB, Carroll RS, Black PM. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000;60:4926–4931. [PubMed] [Google Scholar]
- 12.Luqman S, Pezzuto JM. NFkappaB: a promising target for natural products in cancer chemoprevention. Phytother Res. 2010;24:949–963. doi: 10.1002/ptr.3171. [DOI] [PubMed] [Google Scholar]
- 13.Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007;85:39–47. doi: 10.1007/s11060-007-9390-7. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–951. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- 15.Nagai S, Washiyama K, Kurimoto M, Takaku A, Endo S, Kumanishi T. Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant astrocytoma. J Neurosurg. 2002;96:909–917. doi: 10.3171/jns.2002.96.5.0909. [DOI] [PubMed] [Google Scholar]
- 16.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 18.Zandi E, Karin M. Bridging the gap: composition, regulation, and physiological function of the IkappaB kinase complex. Mol Cell Biol. 1999;19:4547–4551. doi: 10.1128/mcb.19.7.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 20.Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 21.Lin X, Peng Z, Su C. Potential anti-cancer activities and mechanisms of costunolide and dehydrocostuslactone. Int J Mol Sci. 2015;16:10888–10906. doi: 10.3390/ijms160510888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho JY, Baik KU, Jung JH, Park MH. In vitro anti-inflammatory effects of cynaropicrin, a sesquiterpene lactone, from Saussurea lappa. Eur J Pharmacol. 2000;398:399–407. doi: 10.1016/s0014-2999(00)00337-x. [DOI] [PubMed] [Google Scholar]
- 23.Yoshikawa M, Hatakeyama S, Inoue Y, Yamahara J. Saussureamines A, B, C, D, and E, new anti-ulcer principles from Chinese Saussureae Radix. Chem Pharm Bull (Tokyo) 1993;41:214–216. doi: 10.1248/cpb.41.214. [DOI] [PubMed] [Google Scholar]
- 24.Pandey MM, Rastogi S, Rawat AK. Saussurea costus: botanical, chemical and pharmacological review of an ayurvedic medicinal plant. J Ethnopharmacol. 2007;110:379–390. doi: 10.1016/j.jep.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 25.Ko SG, Kim HP, Jin DH, Bae HS, Kim SH, Park CH, Lee JW. Saussurea lappa induces G2-growth arrest and apoptosis in AGS gastric cancer cells. Cancer Lett. 2005;220:11–19. doi: 10.1016/j.canlet.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Ko SG, Koh SH, Jun CY, Nam CG, Bae HS, Shin MK. Induction of apoptosis by saussurea lappa and pharbitis nil on AGS gastric cancer cells. Biol Pharm Bull. 2004;27:1604–1610. doi: 10.1248/bpb.27.1604. [DOI] [PubMed] [Google Scholar]
- 27.Butturini E, Cavalieri E, de Prati AC, Darra E, Rigo A, Shoji K, Murayama N, Yamazaki H, Watanabe Y, Suzuki H, Mariotto S. Two naturally occurring terpenes, dehydrocostuslactone and costunolide, decrease intracellular GSH content and inhibit STAT3 activation. PLoS One. 2011;6:e20174. doi: 10.1371/journal.pone.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung JY, Hsu YL, Ni WC, Tsai YM, Yang CJ, Kuo PL, Huang MS. Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lung cancer cells. Lung Cancer. 2010;68:355–365. doi: 10.1016/j.lungcan.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 29.Choi EJ, Kim GH. Evaluation of anticancer activity of dehydrocostuslactone in vitro. Mol Med Rep. 2010;3:185–188. doi: 10.3892/mmr_00000238. [DOI] [PubMed] [Google Scholar]
- 30.Pitchai D, Roy A, Banu S. In vitro and in silico evaluation of NF-kappaB targeted costunolide action on estrogen receptor-negative breast cancer cells--a comparison with normal breast cells. Phytother Res. 2014;28:1499–1505. doi: 10.1002/ptr.5155. [DOI] [PubMed] [Google Scholar]
- 31.Liu CY, Chang HS, Chen IS, Chen CJ, Hsu ML, Fu SL, Chen YJ. Costunolide causes mitotic arrest and enhances radiosensitivity in human hepatocellular carcinoma cells. Radiat Oncol. 2011;6:56. doi: 10.1186/1748-717X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu YL, Wu LY, Kuo PL. Dehydrocostuslactone, a medicinal plant-derived sesquiterpene lactone, induces apoptosis coupled to endoplasmic reticulum stress in liver cancer cells. J Pharmacol Exp Ther. 2009;329:808–819. doi: 10.1124/jpet.108.148395. [DOI] [PubMed] [Google Scholar]
- 33.Sun CM, Syu WJ, Don MJ, Lu JJ, Lee GH. Cytotoxic sesquiterpene lactones from the root of Saussurea lappa. J Nat Prod. 2003;66:1175–1180. doi: 10.1021/np030147e. [DOI] [PubMed] [Google Scholar]
- 34.Kim EJ, Lim SS, Park SY, Shin HK, Kim JS, Park JH. Apoptosis of DU145 human prostate cancer cells induced by dehydrocostus lactone isolated from the root of Saussurea lappa. Food Chem Toxicol. 2008;46:3651–3658. doi: 10.1016/j.fct.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 35.Kim EJ, Hong JE, Lim SS, Kwon GT, Kim J, Kim JS, Lee KW, Park JH. The hexane extract of Saussurea lappa and its active principle, dehydrocostus lactone, inhibit prostate cancer cell migration. J Med Food. 2012;15:24–32. doi: 10.1089/jmf.2011.1735. [DOI] [PubMed] [Google Scholar]
- 36.Rasul A, Bao R, Malhi M, Zhao B, Tsuji I, Li J, Li X. Induction of apoptosis by costunolide in bladder cancer cells is mediated through ROS generation and mitochondrial dysfunction. Molecules. 2013;18:1418–1433. doi: 10.3390/molecules18021418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun X, Kang H, Yao Y, Chen H, Sun L, An W, Jiang E, Wang S, Hu X. Dehydrocostus lactone suppressed the proliferation, migration, and invasion of colorectal carcinoma through the downregulation of eIF4E expression. Anticancer Drugs. 2015;26:641–648. doi: 10.1097/CAD.0000000000000229. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Y, Wang J, Qin Y, Xuan Y, Jia Y, Hu W, Yu W, Dai M, Li Z, Yi C, Zhao S, Li M, Du S, Cheng W, Xiao X, Chen Y, Wu T, Meng S, Yuan Y, Liu Q, Huang W, Guo W, Wang S, Deng W. Ku80 cooperates with CBP to promote COX-2 expression and tumor growth. Oncotarget. 2015;6:8046–8061. doi: 10.18632/oncotarget.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 41.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 42.Bogar LJ, Bartula LL, Parkman HP, Myers SI. Enhanced bradykinin-stimulated prostaglandin release in the acutely inflamed guinea pig gallbladder is due to new synthesis of cyclooxygenase 1 and prostacyclin synthase. J Surg Res. 1999;84:71–76. doi: 10.1006/jsre.1999.5612. [DOI] [PubMed] [Google Scholar]
- 43.Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002;68-69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 44.Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nose F, Ichikawa T, Fujiwara M, Okayasu I. Up-regulation of cyclooxygenase-2 expression in lymphocytic thyroiditis and thyroid tumors: significant correlation with inducible nitric oxide synthase. Am J Clin Pathol. 2002;117:546–551. doi: 10.1309/9CCJ-XQ8P-PMFM-M65K. [DOI] [PubMed] [Google Scholar]
- 46.Klimp AH, Hollema H, Kempinga C, van der Zee AG, de Vries EG, Daemen T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumorassociated macrophages. Cancer Res. 2001;61:7305–7309. [PubMed] [Google Scholar]
- 47.Rajnakova A, Moochhala S, Goh PM, Ngoi S. Expression of nitric oxide synthase, cyclooxygenase, and p53 in different stages of human gastric cancer. Cancer Lett. 2001;172:177–185. doi: 10.1016/s0304-3835(01)00645-0. [DOI] [PubMed] [Google Scholar]
- 48.Son HJ, Kim YH, Park DI, Kim JJ, Rhee PL, Paik SW, Choi KW, Song SY, Rhee JC. Interaction between cyclooxygenase-2 and inducible nitric oxide synthase in gastric cancer. J Clin Gastroenterol. 2001;33:383–388. doi: 10.1097/00004836-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 50.Cianchi F, Cortesini C, Fantappie O, Messerini L, Sardi I, Lasagna N, Perna F, Fabbroni V, Di Felice A, Perigli G, Mazzanti R, Masini E. Cyclooxygenase-2 activation mediates the proangiogenic effect of nitric oxide in colorectal cancer. Clin Cancer Res. 2004;10:2694–2704. doi: 10.1158/1078-0432.ccr-03-0192. [DOI] [PubMed] [Google Scholar]
- 51.Jones DJ, Lamb JH, Verschoyle RD, Howells LM, Butterworth M, Lim CK, Ferry D, Farmer PB, Gescher AJ. Characterisation of metabolites of the putative cancer chemopreventive agent quercetin and their effect on cyclo-oxygenase activity. Br J Cancer. 2004;91:1213–1219. doi: 10.1038/sj.bjc.6602091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T. Structure of the human cyclo-oxygenase-2 gene. Biochem J. 1994;302:723–727. doi: 10.1042/bj3020723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang YJ, Mbonye UR, DeLong CJ, Wada M, Smith WL. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog Lipid Res. 2007;46:108–125. doi: 10.1016/j.plipres.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harper KA, Tyson-Capper AJ. Complexity of COX-2 gene regulation. Biochem Soc Trans. 2008;36:543–545. doi: 10.1042/BST0360543. [DOI] [PubMed] [Google Scholar]
- 55.Sen E. Targeting inflammation-induced transcription factor activation: an open frontier for glioma therapy. Drug Discov Today. 2011;16:1044–1051. doi: 10.1016/j.drudis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Cahill KE, Morshed RA, Yamini B. Nuclear factor-kappaB in glioblastoma: insights into regulators and targeted therapy. Neuro Oncol. 2016;18:329–339. doi: 10.1093/neuonc/nov265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Natoli G. NF-kappaB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol Rev. 2012;246:183–192. doi: 10.1111/j.1600-065X.2012.01103.x. [DOI] [PubMed] [Google Scholar]
- 58.Gilmore TD, Garbati MR. Inhibition of NFkappaB signaling as a strategy in disease therapy. Curr Top Microbiol Immunol. 2011;349:245–263. doi: 10.1007/82_2010_105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.