

Abstract
The recently developed ultrasensitive bio-barcode assay was used to measure the concentration of amyloid-β-derived diffusible ligands (ADDLs), a potential soluble pathogenic Alzheimer's disease (AD) marker, in the cerebrospinal fluid (CSF) of 30 individuals. ADDL concentrations for the subjects diagnosed with AD were consistently higher than the levels in the CSF taken from nondemented age-matched controls. Studies of ADDLs or for any other potential pathogenic AD markers in CSF have not been possible because of their low concentration in CSF (<1 pM). This study is a step toward a diagnostic tool, based on soluble pathogenic markers for the debilitating disease.
Keywords: bio-barcode
Alzheimer's disease (AD) is the most common neurodegenerative dementia with an average death prognosis of 9 years (1). There is no definitive diagnosis of the disease other than postmortem identification of senile plaques and neurofibrillary tangles in the brain (2, 3). Premortem diagnosis, based on a patient's clinical history; in vivo brain imaging (4–7); and neuropsychological, cognitive, and neurological tests, is only ≈85% accurate (8). There are two general approaches for detecting soluble markers for AD. One approach is based on measuring the total tau protein or amyloid-β protein concentration in cerebrospinal fluid (CSF) or plasma (9–12). This approach is hampered by significant overlap of such marker levels in healthy and unhealthy subjects and has led to inconclusive results (13–16). The other approach targets only the suspected pathogenic markers, such as cleaved tau protein, phosphorylated tau protein (17), or amyloid-β-derived diffusible ligands (ADDLs). Although this approach to detecting pathogenic markers might lead to more definitive results, the concentrations of such markers in CSF are so low in the early stages of the disease that they cannot be identified accurately with conventional ELISA or blotting assays. Here, we demonstrate that the ultrasensitive nanoparticle-based bio-barcode assay can be used to determine the approximate ADDL concentration in CSF taken from 30 subjects, 15 of whom were diagnosed with the disease through postmortem analysis of the brain and 15 of whom were found not to have the disease.
Since its invention, the bio-barcode assay has become a powerful analytical tool for the detection of both protein and nucleic acid targets (18, 19) (Scheme 1). In a previous report, we were able to detect prostate-specific antigen with a sensitivity six orders of magnitude greater than the conventional ELISAs for the same target (18). This extraordinary sensitivity opens up the possibility of considering for disease diagnosis biomarkers that could not be used with conventional technology. ADDLs and many of the other potential pathogenic markers for AD provide an excellent test case to evaluate the capabilities of the barcode assay and, in the process, could provide important leads to useful diagnostic systems for studying and ultimately treating the disease (1, 20–22).
Scheme 1.

The bio-barcode amplification assay. The assay uses MMPs functionalized with mAbs that recognize and bind ADDLs. The ADDLs are then sandwiched with an NP probe, modified with double-stranded DNA and an anti-ADDL pAb. After repeated washing while using a magnet to immobilize the MMPs, a dehybridization step releases hundreds of barcode DNA strands for each antigen-binding event.
mAbs and polyclonal Abs (pAbs) specific to ADDLs, which are synaptotoxic oligomers of Aβ, were developed (23, 24) and used to demonstrate that ADDLs are elevated significantly in the soluble brain extracts of transgenic mice and AD patients (25, 26). The elevated levels of ADDLs in the brain suggest that diffusible oligomers would also appear in the CSF, but presumably at much lower levels. Although high concentrations of ADDLs have been detected in buffer solutions by using a localized surface plasmon resonance nanosensor (27), proof of their presence in CSF does not exist, and therefore, one cannot consider the potential of correlating the concentration of ADDL or related markers with the state of the disease.
The key to the bio-barcode assay is the homogeneous isolation of specific antigens by means of a sandwich process involving oligonucleotide-modified Au nanoparticles (NPs; with biobarcodes) and magnetic microparticles (MMPs), both functionalized with Abs specific to the antigen of interest. The increased sensitivity advantage derives mainly from the very effective sequestration of antigen and the amplification process that occurs as a result of the large number of barcode DNA strands released for each antigen recognition and binding event (Scheme 1). By using this approach and adequate Abs, one can routinely detect as low as 30 copies of a target protein, often in a complex sample such as plasma.
Materials and Methods
DNA Synthesis. The DNA strands were synthesized and purified in-house according to standard published procedures (28) by using an automated synthesizer (Expedite) and HPLC (1100 HPLC series, Hewlett–Packard), respectively. All of the reagents required for the phosphoramidite synthesis, including 3′- and 5′-thiol modifiers, were purchased from Glen Research (Sterling, VA). Thiol modification was carried out manually by following the procedures given in ref. 28. Absorption and extinction spectra were recorded by using an 8452a diode array spectrophotometer (Hewlett–Packard). The concentrations of stock DNA solutions were calculated based on the extinction coefficient of each strand. All buffers and aqueous washes were based on Nanopure water (18 mΩ; Barnstead), and reagents were used as received unless indicated otherwise. The following DNA strands were synthesized for the ADDL assay: complementary probe NP, 5′-TACGAGTTGAGACCGTTAAGACGAGGCAATCATGCAATCCTGAATGCGA10-(CH2)6-SH-3′; barcode, 5′-CGCATTCAGGATTGCATGATTGCCTCGTCTTAACGGTCTCAACTCGTA-3′; capture, 5′-HS-(CH2)6-A10-TACGAGTTGAGACCGTTAAGACGA-3′; and NP label, 5′-GGCAATCATGCAATCCTGAATGCGA10-(CH2)6-SH-3′.
CSF Preparation. Premortem samples (provided by John Lee, Loyola University Medical School, Maywood, IL) were obtained via lumbar puncture and kept frozen until used. Postmortem samples were obtained from the Religious Order Study repository maintained by Rush University Medical School (Chicago). The latter samples were obtained postmortem from 10 mild- to severe-AD-diagnosed subjects and 10 age-matched noncognitively impaired control cases. For the latter 20 subjects, results of the National Institute on Aging–Reagan Institute and Mini Mental State Examination (MMSE) neuropsychological tests are also available on request.
Antigen Isolation and Ab Expression for ADDLs. Aβ1–42 peptide (California Peptide Research, Napa, CA) was used to prepare synthetic ADDLs according to published protocols (23). An aliquot of Aβ1–42 was dissolved in anhydrous DMSO to a concentration of 22.5 mg/ml (5 mM), pipette-mixed, and further diluted into ice-cold F12 medium (phenol-red-free) (1:50 dilution; BioSource International, Camarillo, CA). The mixture was quickly vortexed, incubated at 6–8°C for 24 h, and centrifuged at 14,000 × g for 10 minutes, and the oligomers were collected from the supernatant. The concentration of synthetic ADDLs was determined by using a microBCA assay (Pierce, Rockford, IL). Abs targeting ADDLs in the bio-barcode assay (M90 pAb and 20C2 mAb) were generated and characterized as described (23).
NP Synthesis and Modification. Citrate-stabilized Au NP probes (13 nm diameter) were prepared by following the standard methods given in ref. 28, and 30-nm-diameter Au NP (0.5 nM) were purchased and used as received from BB International (Cardiff, U.K.). The 30-nm particles (1 ml) were initially functionalized with 1 μg of pAb antigen-specific Ab (M90) in a basic aqueous solution (pH 9). The particles were modified subsequently with thiolated DNA (final concentration, 2 μM) by slow salt aging (40 h) to a final concentration of PBS (0.1 M NaCl in 0.01 M of phosphate buffer, pH 7; denoted as PBS unless indicated otherwise). Unbound thiolated DNA was removed by repetitive centrifugation (15,700 × g for 30 min) of the particles, followed by rinsing and resuspension in PBS. The thiolated DNA is complementary to the bio-barcode DNA strand that serves as the amplification target. The barcode DNA was added (final concentration, 2 μM) to the NP solution and allowed to hybridize at room temperature for 4 h. The NPs were centrifuged at 15,700 × g, and the supernatant containing excess barcode DNA was removed. The particles were resuspended in 0.1 M PBS, and the procedure was repeated three times.
The NPs used in the scanometric assay were prepared according to the methods published in ref. 28. The concentration of the NPs was calculated based on extinction spectra by using known values (28) of the extinction coefficients for gold NPs:
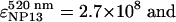 |
 |
The diameters of the synthesized NPs were determined by transmission electron microscopy by using an 8100 instrument (Hitachi, Tokyo).
Functionalization of MMPs. The amino-functionalized MMPs (100 μl, 50 mg/ml aqueous solution, 1 μm diameter polyamine particles with iron oxide cores, Polysciences) were modified with 100 μg of antibodies according to the manufacturer's protocol. The Abs were mAbs specific to ADDL (20C2).
Functionalization of Glass Slides. Functionalized glass slides were modified with half-complementary thiolated capture DNA strands (100 μM) using a microarrayer (Affymetrix, Santa Clara, CA) according to a standard published procedure (18). The DNA strands were covalently immobilized on the chip, the unbound strands were washed away with H2O, and the residual binding sites were passivated by immersion in 40 mM mercaptosuccinic acid for 30 min, followed by repetitive washing with H2O.
Bio-Barcode Assay. In a typical assay, 10 μl of CSF or 10-μl aliquots of ADDL at known concentrations ranging from 100 aM to 100 fM were added to 50 μl of MMP solution (5 mg/ml) and allowed to react under vigorous stirring at 37°C for 1 h. After magnetically immobilizing the MMPs, the unbound antigens were removed by repeated washing with PBS. The MMPs and antigen–target complexes were magnetically separated, and 50 μl of 0.1 nM NP probe (Ab-functionalized and DNA-modified) was added and stirred vigorously at 37°C for 30 min to bind the target antigen–MMP complex. The sandwich complexes were then magnetically separated and washed four times with 100 μl of PBS solution. In the final step, 50 μl of H2O was added and the solutions were stirred vigorously at 60°C for 30 min to allow for full dehybridization of the target DNA. The complexes were again separated magnetically, and the supernatant containing the barcode DNA was collected for quantification by the scanometric detection method (29).
Scanometric Detection. Light scattering by the silver-amplified gold particles was quantified with a commercial image analyzer (Verigene ID, Nanosphere, Northbrook, IL) and the scattering image of the whole slide was collected as a .tif file (16-bit resolution, maximum 216 = 65,536 arbitrary units) with a typical acquisition time of 20 ms. The grayscale intensity was quantified by using the commercially available software imagequant.
Results
In a typical assay, tested samples consisted of either known concentrations of synthetic ADDLs in PBS buffer or CSF samples from subjects obtained via lumbar puncture or extraction from the posterior ventricle. A known volume of each sample was mixed with NPs modified with double-stranded oligonucleotides and MMPs, both functionalized with antigen-specific Abs (see Scheme 1). Both MMPs and NPs are present in excess so that the target antigens are captured effectively, thereby overcoming slow binding kinetics. Binding of the Ab-modified particles to the target antigen results in a sandwich complex involving the NPs and magnetic particles. Magnetic separation of the sandwiched complexes results in efficient separation from the rest of the sample. The double-stranded DNA strands on the NPs are then dehybridized, releasing hundreds of barcodes per target protein into the supernatant. The released barcode DNA strands are easily detected by using the scanometric method (29), and the results are recorded (Scheme 2) with a Verigene ID system (Nanosphere, Northbrook, IL).
Scheme 2.

Schematic representation of scanometric detection. The method is based on capturing the barcode DNA on a microarray with spots of oligonucleotides that are complementary to half of the barcode DNA sequence. NPs with oligonucleotides that are complementary to the other half of the barcode DNA are hybridized to the captured barcode strands. The signal is enhanced by using silver amplification, and the results are recorded with the Verigene ID system, which measures scattered light intensity from each spot (see Materials and Methods). Depending on the silver amplification time and the experimental conditions, the response can vary for each slide. For this reason, the grayscale intensity of the developed spots is then measured and averaged for each ADDL concentration (Inset).
Before analyzing subject samples, a calibration curve for synthetic ADDL concentration in PBS buffer was established (Fig. 1). The assay is semiquantitative and exhibits an analytical target concentration range of three orders of magnitude, with a lower limit of detection at ≈100 aM. At high target concentrations (>500 fM), the assay response becomes nonlinear, and eventually, the signal plateaus. The calibration curve shown in Fig. 1 finally exhibits a plateau at high concentrations (>500 fM) because of the stoichiometry changes from 1:1:1 to 1:>1:>1 (MMP/ADDL/NP), whereas at high ADDL concentrations the MMPs bind more than one NP carrying barcode DNA. The resulting calibration curve establishes the analytical range available to the bio-barcode assay for this soluble marker under the stated conditions, that spans from low attomolar to high femtomolar.
Fig. 1.

A normalized grayscale intensity calibration curve for synthetic ADDL concentration based on a serial dilution study. Each point is an average of at least four responses.
CSF samples from thirty subjects were then evaluated with the assay (Fig. 2). We obtained 10 samples by means of premortem lumbar puncture and 20 samples by means of postmortem extraction from the posterior ventricle of the brain. All cases underwent a standard and uniform neuropathological evaluation for the diagnosis of AD using well established diagnostic criteria, including the Braak and Braak score (30, 31) and the CERAD (Consortium to Establish a Registry for Alzheimer's Disease) criteria (32). Brain extract from a definitively diagnosed AD patient, in which ADDL concentration is known to be high (>1 pM), served as a positive control yielding a saturated signal, and brain extract obtained from a healthy subject without clinical and pathological signs of AD was used as a negative control. The 30 assayed samples were obtained from subjects whose AD histopathological symptoms ranged from null to severe. The ADDL concentration in the CSF for each patient is estimated by comparison of grayscale intensity in the scanometric assay with the calibration curve in Fig. 1.
Fig. 2.

Scatter plot from the scanometric detection of barcode DNA released from the bio-barcode assay for 30 subjects. The response for the negative human control subject (brain extract) was similar to that observed for the chip control (dashed line in Fig. 1). The positive control exhibited a response that was off scale and could not be quantified. The data points are averages of three separate experiments normalized for each assay based on the highest response in a series of runs. The mean values for ADDL concentrations (solid lines) are estimated for each group based on the calibration curve in Fig. 1.
Discussion
The assays for control subjects showed a consistently lower level of ADDLs than the assays for subjects diagnosed with the disease (Fig. 2). The ADDL concentration medians of the two groups are ≈200 aM and 1.7 fM, respectively (statistical value from unpaired t test, P < 0.0001). The highest ADDL concentration in the control group is <0.5 fM, and most of the ADDL concentrations in the AD group are >0.5 fM. Only two individuals diagnosed with AD exhibited overlap with the control population of healthy subjects. One subject was an aged male that scored very high on the Mini Mental State Examination (MMSE), which is inconsistent with the later pathological stages of the disease, whereas the other subject had pathological signs of infarctions in addition to AD. These two AD subjects that are outliers in our assay could be false positives of current diagnostic methods that consider both clinical and pathological data. Interestingly, one nondemented subject (the lowest of control group) had a moderate number of plaques but no ADDLs in CSF. These data are consistent with the idea that soluble oligomers, rather than plaques, may be the Aβ species most relevant to dementia. The presence of ADDLs in CSF may provide a soluble marker to establish AD, and ADDL concentration, in conjunction with the present neuropsychological tests, may act as an accurate marker to delineate the different stages or forms of dementia.
In conclusion, this study is significant for a number of reasons. First, it shows that the ultra-high-sensitivity bio-barcode assay can be used to measure the concentration of the pathogenic ADDL in CSF at clinically relevant concentrations. This advance opens up the opportunity to study a wide range of potential pathogenic markers in CSF, which could lead to very informative diagnostic tools for the disease. Second, it suggests that the soluble pools of ADDLs that exist in the human brain extend to the CSF, and that elevated levels of ADDLs correlate with the presence of the disease. Third, the approach of using pathogenic markers in CSF combined with the barcode assay points toward a potential reliable detection method for diagnosing AD that is faster, higher throughput, and less expensive than current imaging techniques. Before this assay can be used clinically, it needs to be statistically validated with an even larger patient pool.
Acknowledgments
D.G.G. thanks Dr. Rongchao Jin for helpful discussions on DNA-NP probe preparation. We thank the hundreds of nuns, priests, and brothers who participated in the Religious Orders Study for their altruism and support. C.A.M. and W.L.K. were supported by the National Science Foundation through Nanoscale Science and Engineering Center Grant EEC-0118025 and National Institutes of Health Grants AG14449, AG11385, and AG10161. C.A.M. was also supported by a National Institutes of Health Director's Pioneer Award and a Defense University Research Initiative on Nanotechnology from the Department of Defense.
Author contributions: D.G.G., L.C., W.L.K., and C.A.M. designed research; D.G.G., L.C., J.-M.N., and C.S.T. performed research; D.G.G., L.C., W.L.K., and C.A.M. analyzed data; E.J.M., C.A.M., and W.L.K. contributed new reagents/analytic tools; and C.A.M. wrote the paper.
Abbreviations: AD, Alzheimer's disease; ADDL, amyloid-β-derived diffusible ligand; pAb, polyclonal Ab; CSF, cerebrospinal fluid; NP, nanoparticle; MMP, magnetic microparticle.
See Commentary on page 2263.
M.F.H. is a member of the scientific advisory board of Lexrite Labs.
References
- 1.Citron, M. (2002) Nat. Neurosci. 5, 1055-1057. [DOI] [PubMed] [Google Scholar]
- 2.Prussiner, S., Rosenberg, R., Di Mauro, S. & Barche, R., eds. (1997) The Molecular and Genetic Basis of Neurological Disease (Butterworth–Heinemann, Boston), 2nd Ed.
- 3.Phelps, C., Khachaturian, Z. & Trojanowski, J. Q. (1997) Neurobiol. Aging 18, S1-S2.9330978 [Google Scholar]
- 4.Bookheimer, S. Y., Strojwas, M. H., Cohen, M. S., Saunders, A. M., Pericak-Vance, M. A., Mazziotta, J. C. & Small, G. W. (2000) N. Engl. J. Med. 343, 450-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heckl, S., Pipkorn, R., Nägele, T., Vogel, U., Küker, W. & Voight, K. (2004) Histol. Histopathol. 19, 651-668. [DOI] [PubMed] [Google Scholar]
- 6.Calderón, G. P. L., Parra, R. M. A., Llibre, R. J. J. & Gutiérrez, J. V. (2004) Rev. Neurol. 38, 422-427. [PubMed] [Google Scholar]
- 7.Oksengaard, A. R., Haakonsen, M., Dullerud, R., Engedal, K. & Laake, K. (2003) Int. J. Geriatr. Psychiatry 4, 308-312. [DOI] [PubMed] [Google Scholar]
- 8.Knopman, D. S., DeKosky, S. T., Cummings, J. L., Chui, H., Corey-Bloom, J., Relkin, N., Small, G. W., Miller, B. & Stevens, J. C. (2001) Neurology 56, 1143-1153. [DOI] [PubMed] [Google Scholar]
- 9.Clark, C. M. (2003) Arch. Neurol. 60, 1696-1702. [DOI] [PubMed] [Google Scholar]
- 10.Andreasen, N., Minthon, L., Davidsson, P., Vanmechelen, E., Vanderstichele, H., Winblad, B. & Blennow, K. (2001) Arch. Neurol. 58, 373-279. [DOI] [PubMed] [Google Scholar]
- 11.Blennow, K. (2004) J. Intern. Med. 256, 224-234. [DOI] [PubMed] [Google Scholar]
- 12.Blennow, K. & Hampel, H. (2003) Lancet Neurol. 2, 605-613. [DOI] [PubMed] [Google Scholar]
- 13.Teunissen, C. E., de Vente, J., Steinbusch, H. W. & De Bruijn, C. (2002) Neurobiol. Aging 23, 485-508. [DOI] [PubMed] [Google Scholar]
- 14.Sunderland, T., Linker, G., Mirza, N., Putnam, K. T., Friedman, D. L., Kimmel, L. H., Bergeson, J., Manetti, G. J., Zimmermann, M., Tang, B., et al. (2003) J. Am. Med. Assoc. 289, 2094-2103. [DOI] [PubMed] [Google Scholar]
- 15.Hulstaert, F., Blennow, K., Ivanoiu, A., Schoonderwaldt, H. C., Riemenschneider, M., De Deyn, P. P., Bancher, C., Cras, P., Wiltfang, J., Mehta, P. D., et al. (1999) Neurology 52, 1555-1662. [DOI] [PubMed] [Google Scholar]
- 16.Galasko, D., Clark, C., Chang, L., Miller, B., Green, R. C., Motter, R. & Seubert, P. (1997) Neurology 48, 632-635. [DOI] [PubMed] [Google Scholar]
- 17.Maddalena, A., Papassotiropoulos, A., Müller-Tillmanns, B., Jung, H. H., Hegi, T., Nitsch, R. M. & Hock, C. (2003) Arch. Neurol. 60, 1202-1206. [DOI] [PubMed] [Google Scholar]
- 18.Nam, J.-M., Thaxton, C. S. & Mirkin, C. A. (2003) Science 301, 1884-1886. [DOI] [PubMed] [Google Scholar]
- 19.Nam, J.-M., Stoeva, S. I. & Mirkin, C. A. (2004) J. Am. Chem. Soc. 126, 5932-5933. [DOI] [PubMed] [Google Scholar]
- 20.Hardy, J. & Selkoe, D. J. (2002) Science 297, 353-356. [DOI] [PubMed] [Google Scholar]
- 21.Mattson, M. P. (2004) Nature 430, 631-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H. & LaFerla, F. M. (2004) Neuron 43, 321-332. [DOI] [PubMed] [Google Scholar]
- 23.Lambert, M. P., Viola, K. L., Chromy, B. A., Chang, L., Morgan, T. E., Yu, J., Venton, D. L., Krafft, G. A., Finch, C. E., Klein, W. L. (2001) J. Neurochem. 3, 595-605. [DOI] [PubMed] [Google Scholar]
- 24.Chromy, B. A., Nowak, R. J., Lambert, M. P., Viola, K. L., Chang, L., Velasco, P. T., Jones, B. W., Fernandez, S. J., Lacor, P. N., Horowitz, P., et al. (2003) Biochemistry 42, 12749-12760. [DOI] [PubMed] [Google Scholar]
- 25.Chang, L., Bakhos, L., Wang, Z., Venton, D. L. & Klein, W. L. (2003) J. Mol. Neurosci. 20, 305-313. [DOI] [PubMed] [Google Scholar]
- 26.Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., Krafft, G. A. & Klein, W. L. (2003) Proc. Natl. Acad. Sci. USA 100, 10417-10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haes, A. J., Hall, L., Chang, W. P., Klein, W. L. & Van Duyne, R. P. (2004) Nano Lett. 4, 1029-1034. [Google Scholar]
- 28.Jin, R. C., Wu, G. S., Li, Z., Mirkin, C. A. & Schatz, G. C. (2003) J. Am. Chem. Soc. 125, 1643-1654. [DOI] [PubMed] [Google Scholar]
- 29.Taton, T. A., Mirkin, C. A. & Letsinger, R. L. (2000) Science 289, 1757-1760. [DOI] [PubMed] [Google Scholar]
- 30.Braak, H. & Braak, E. (1991) Acta Neuropathol. 82, 239-259. [DOI] [PubMed] [Google Scholar]
- 31.Braak, H. & Braak, E. (1998) J. Neural Transm. Suppl. 53, 127-140. [DOI] [PubMed] [Google Scholar]
- 32.Mirra, S. S. Neurobiol. Aging (1997) 4, S91-S94. [DOI] [PubMed] [Google Scholar]