

Abstract
Lipocalin-2 (also known as NGAL) levels are elevated in obesity and diabetes yet relatively little is known regarding effects on the heart. We induced pressure overload (PO) in mice and found that lipocalin-2 knockout (LKO) mice exhibited less PO-induced autophagy and NLRP3 inflammasome activation than Wt. PO-induced mitochondrial damage was reduced and autophagic flux greater in LKO mice, which correlated with less cardiac dysfunction. All of these observations were negated upon adenoviral-mediated restoration of normal lipocalin-2 levels in LKO. Studies in primary cardiac fibroblasts indicated that lipocalin-2 enhanced priming and activation of NLRP3-inflammasome, detected by increased IL-1β, IL-18 and Caspase-1 activation. This was attenuated in cells isolated from NLRP3-deficient mice or upon pharmacological inhibition of NLRP3. Furthermore, lipocalin-2 induced release of HMGB1 from cells and NLRP3-inflammasome activation was attenuated by TLR4 inhibition. We also found evidence of increased inflammasome activation and reduced autophagy in cardiac biopsy samples from heart failure patients. Overall, this study provides new mechanistic insight on the detrimental role of lipocalin-2 in the development of cardiac dysfunction.
Keywords: Lipocalin-2, pressure overload, NLRP3 inflammasome, HMGB1, toll-like receptor (TLR)-4
Introduction
Heart failure (HF) incidence is increased in patients with obesity and diabetes and inflammation is one underlying mechanism [1]. Lipocalin-2 (LCN2) is a proinflammatory adipokine which is elevated in obesity and diabetes [2] and clinical studies have also established strong positive correlations between circulating LCN2 and various types of HF [3]. Thus, LCN2 has been proposed as an important contributor to the pathophysiology of HF and potentially a useful biomarker for HF. Neutrophils are a major source of LCN2 which is also known as neutrophil gelatinase-associated lipocalin (NGAL) [4]. Mice lacking LCN2 were first shown to exhibit an increased susceptibility to bacterial infections due to lack of antibacterial innate immune response [5] and subsequently shown to be protected from obesity- and aging-associated insulin resistance, endothelial dysfunction and hypertension [6-9]. However, the precise mechanisms via which LCN2 can regulate the progression of HF remain to be resolved.
In contrast to the paucity of information on cardiac effects of LCN2, the pathophysiological role of LCN2/NGAL in kidney dysfunction is much better understood and measurement of LCN2/NGAL has recently become established as a common clinical diagnostic test for acute kidney damage [10]. Inflammation, including NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome activation, and autophagy have recently been established as important mechanisms regulating cardiac dysfunction [11,12] although their regulation by LCN2 is unclear. We have recently shown that LCN2 attenuates cardiomyocyte autophagy [13].
We hypothesized that LKO mice would have less pressure overload-induced cardiac dysfunction than wild type (WT) and that reduced autophagy or elevated NLRP3 inflammasome activation may be potential mechanisms of action. To examine this, we induced cardiac pressure overload (PO) in WT and LCN2 knockout mice (LKO) ± restoration of normal circulating LCN2 levels using adenovirus (L2AV). We first examined functional outcomes using echocardiography, then analyzed mechanisms via which LCN2 contributed to cardiac dysfunction by focusing on fibrosis, autophagy and NLRP3 inflammasome activation in these animal models as well as in primary cells isolated from heart of WT and NLRP3-deficient mice.
Material and methods
Animals, induction of pressure overload and analysis of cardiac function
Male WT and LKO mice aged eight weeks with C57BL/6J background were studied using protocols approved by the Animal Care Committee at York University and the Committee on the Use of Live Animals for Teaching and Research of the University of Hong Kong and all methods were performed in accordance with these guidelines and regulations. Pressure overload was induced by transverse aortic banding as described previously [14]. Briefly, surgery was performed on the transverse aortae of mice under general anesthesia (ip; xylazine 0.15 mg/g; ketamine 0.03 mg/g) with a titanium microligation clip using banding calipers calibrated to a 27-g needle. Sham surgery was performed as outlined above without the placement of a ligation clip. The recombinant adenovirus for LCN2 (108 plaque-forming units) was injected via tail vein of mice one day prior to surgery to achieve normal circulating levels [7] and echocardiography was performed using the Vevo2100 system (Visual Sonics) [15]. Sera were collected every week from tail vein and hearts were harvested at the ends points (four weeks).
Cardiac biopsy from human subjects with or without heart failure (HF)
Patients with end-stage dilated cardiomyopathy (DCM) who met institutional criteria for LVAD device implantation as a bridge to transplant or as destination therapy at the New York Presbyterian-Columbia Campus were included in this study and processed as previously described [14,16]. Cardiac biopsy tissue used in this study (n=5) were all male, aged 41.0±21.4 years and HF etiology ischemic or diabetic cardiomyopathy. All tissue obtained at LVAD implantation represented a decompensated heart failure state. Normal patients were subjects with no known cardiopulmonary disease whose organs were listed but were unable to be placed at the time of organ recovery for heart transplantation and who consented to tissue for research purposes by the New York Organ Donor Network were included in this study. This study met all institutional guidelines of the Institutional Review Board of Columbia University and New York State organ donation guidelines regarding the use of clinical data, ethical treatment of patients adhering to the Declaration of Helsinki principles, and procurement of tissue for research. All subjects were recruited at the New York Presbyterian Hospital-Columbia University campus between 2008-2014, and informed consent was waived for use of discarded, de-identified tissue.
Analysis of cardiac function using echocardiography
Echocardiography was performed using the Vevo2100 system (Visual Sonics, MS550D transducer) as previously described [15]. Cardiac systolic functions of ejection fraction, fractional shortening and strain rate calculation were based on M-mode images of the parasternal short-axis view at papillary level. All parameters were averaged over at least 3 cardiac cycles for analysis.
Histological analysis of cardiac structure
Animals were sacrificed and hearts were isolated and perfused with ice-cold cardioplegic solution (30 mM KCl in PBS) and stored in 10% neutral-buffered formalin 24 hours for further processing. Paraffin sections (5 μm) were prepared for trichrome staining and immunostaining using antibodies against HMGB1, desmin, vimentin or α-SMA as described previously [17].
Immunofluorescent analysis in cultured cells
Rat neonatal fibroblast cells were cultured in 12-well plates, after treatment with or without murine recombinant LCN2 (1 μg/ml) protein for 48 hours. Cells were fixed in 4% paraformaldehyde (PFA) for 15 minutes, quenched with 1% glycine for 10 minutes, and permeabilized with 0.1% Triton X-100 for one minute. After blocking with 3% BSA, cells were incubated with murine LCN2 (1:200) and HMGB1 primary antibody (1:100) overnight at 4°C, and incubated with Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies) at 1:1000 for one hour at room temperature. Images were taken using a 60 objective with confocal microscope (Olympus, BX51).
Transmission electron microscopy (TEM)
TEM was performed as described previously [15]. In brief, LV myocardium was cut to 1 mm3 and fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight at 4°C. After brief washing, the specimens were post-fixed in 1% osmium tetroxide, dehydrated, embedded in epoxy resin, and polymerized overnight at 60°C. Ultra-thin sections (100 nm) were cut and post-stained with Reynold’s lead citrate and uranyl acetate to increase contrast. Sections were viewed and photographed using a P FEI CM100 TEM and Kodak Megaplus camera.
Western blot analysis
Heart tissue was snap frozen and then lysed in modified RIPA buffer by the Tissue Lyser II (Qiagen) for two minutes at full speed. The lysates were then centrifuged at full speed for 10 minutes. Supernatants were harvested and quantified by BCA assay. Tissue lysates (30 or 50 μg) were incubated with Laemmli sample buffer at 95°C for 10 minutes and then resolved by SDS-PAGE. After transferring to polyvinylidenedifluoride (PVDF) membranes, Western blotting was performed by incubating with antibodies. The following antibodies were used for immunoblot analysis: LC3-II (1:1000), BECLIN-1 (1:1000), Caspase-1 (1:1000), HMGB1 (1:1000), phospho-NF-κB p65 (Ser536) (1:750), NF-κB p65 (1:750), GAPDH (1:2000) and β-actin (1:2000) from Cell Signaling; p62 (1:1000) from BD Transduction Laboratories; LCN2 (1:2000) from Antibody and Immunoassay Services; IL-1β (1:1000) from R&D systems. IL-18 (1:1000) from MBL Life science. After incubation with secondary antibodies, the immune complexes were detected with enhanced chemiluminescence (ECL) reagents from GE Healthcare (Uppsala, Sweden).
Quantitative reverse-transcription polymerase chain reaction
The custom fibrosis PCR array was purchased from SABiosciences-61 (QIAGEN Inc.). Total RNA was isolated from heart tissues or cultured mice and rat fibroblasts using TRIzol® Reagent according to the manufacturer’s instructions, and purified using the RNeasyMinElute Cleanup Kit to attain an A260/A280 ratio between 1.9 and 2.0. First-strand cDNA, synthesized from 0.5 μg RNA using the RT2 First Strand kit, was used in a custom PCR array comprising of 96-well plates pre-coated with primers listed in Table 1. Quantitative real-time PCR was conducted using a Chromo4™ Detection system (Bio-Rad Laboratories Canada Ltd., Mississauga, ON, CA) according to cycling conditions outlined by the PCR array manufacturer. Data were analyzed using RT2 Profiler PCR Array Data Analysis software (Version 3.5; QIAGEN Inc.) and normalized to GAPDH mRNA expression. For other genes detected listed in the Table 2, they were analyzed through real-time PCR using the following cycling conditions: 95°C/15 min, followed by 35 cycles of [95°C/30 sec, 55°C/30 sec, 72°C/30 sec], then 72°C/10 min. Melting curve analysis was used to ensure primer specificity. Data were then analyzed using the 2-ΔΔCt method.
Table 1.
Genes detected in fibrosis PCR array
| Col1a1 | Mmp8 |
| Col3a1 | Mmp9 |
| Col4a1 | Mmp13 |
| Timp1 | Mmp14 |
| Timp2 | Gapdh |
| Timp3 | Actb |
| Mmp1a | MGDC |
| Mmp2 | PPC |
Table 2.
Sequences of QPCR primers
| Gene Name | Primer Sequences |
|---|---|
| Murine | Forward 5’CAGAACATCATCCCCTGCATC3’ |
| Gapdh | Reverse 5’CTGCTTCACCACCTTCTTGA3’ |
| Murine | Forward 5’GGTCACCCATGGCACCATAA3’ |
| Nfκb1 | Reverse 5’AGCTGCAGAGCCTTCTCAAG3’ |
| Murine | Forward 5’GCCACCTTTTGACAGTGATGAG3’ |
| Il1β | Reverse 5’AAGGTCCACGGGAAAGACAC3’ |
| Murine | Forward 5’GACACGAGTCCTGGTGACTT3’ |
| Nlrp3 | Reverse 5’GTCCACACAGAAAGTTCTCTTAGC3’ |
| Murine | Forward 5’AACGCCATGGCTGACAAGA3’ |
| Casp1 | Reverse 5’TGATCACATAGGTCCCGTGC3’ |
| Murine | Forward 5’CTG CGC TTG CAG AGA TTA AA3’ |
| Tgfβ1 | Reverse 5’GAA AGC CCT GTA TTC CGT CT3’ |
| Rat | Forward 5’ATGTGCCGGACCTTGGAAG3’ |
| Gapdh | Reverse 5’CCTCGGGTTAGCTGAGAGATCA3’ |
| Rat | Forward 5’CACTGCTCAGGTCCACTGTC3’ |
| Nfkb1 | Reverse 5’CTGTCACTATCCCGGAGTTCA3’ |
| Rat | Forward 5’CACCTCTCAAGCAGAGCACAG3’ |
| Il1β | Reverse 5’GGGTTCCATGGTGAAGTCAAC3’ |
| Rat | Forward 5’CCAGGGCTCTGTTCATTG3’ |
| Nlrp3 | Reverse 5’CCTTGGCTTTCACTTCG3’ |
| Rat | Forward 5’AGGAGGGAATATGTGGG3’ |
| Casp1 | Reverse 5’AACCTTGGGCTTGTCTT3’ |
Cultured adult fibroblasts from mice, adult and neonatal rat fibroblasts from rats
Mice (male, 8 weeks) or rat heart (Wistar rats male, 6 months) was isolated from anesthetized animal and perfused with Ca2+ free K-H buffer for 6 minutes and then with LiberaseBlendyzme 4 (Roche) contained K-H buffer for 3 times (10 ml per heart for mice, 20 ml per heart for rat). The hearts were perfused with 5% BSA K-H buffer before mincing. After stabilization for 3 hours, fibroblasts were stimulated with LCN2 (1 μg/ml) for 24 hours. Hearts were collected from 1- to 2-day-old neonatal rat pups, promptly after euthanasia by decapitation, and primary cultures of neonatal rat cardiomyocytes were performed as described previously [17]. After stabilization for 24 hours, neonatal rat fibroblasts were stimulated with LCN2 (1 μg/ml) for 48 hours. Samples were collected to examine the expression levels of HMGB1 by western blot analysis and mRNA by quantitative RT-PCR.
Statistical analysis
All results were derived from at least three sets of repeated experiments. The statistical calculations were performed by one-way analysis of variance followed by Tukey’s multiple comparison post-test using Prism version 5 (GraphPadSoftware; San Diego, CA, USA). Independent t tests were performed when there are comparisons between two groups. All values are presented as means ± SD and where arbitrary and not absolute value were involved the final graph displays fold over control values. For all statistical comparisons, a P value less than 0.05 was accepted to indicate significant differences.
Results
PO-induced inflammasome activation is attenuated in LKO mice
We examined NLRP3 inflammasome in WT and LKO (±LCN2 restoration using L2AV) mice four weeks after transverse aortic banding surgery. Serum interleukin (IL)-1β measurement by ELISA demonstrated increased IL-1β in WT and LKO+L2AV with PO, although IL-1β was increased in LKO, the magnitude of change under PO was greatly reduced (Figure 1A). Inflammasome-priming, measured via expression levels of caspase-1, NLPR3, IL-1β and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) showed similar significant increases in WT and LKO+L2AV after PO at the mRNA level (Figure 1B) [18]. Having shown that LCN2 positively regulates inflammasome activation in PO, we sought to decipher the mechanism of this action. NLRP3 inflammasome activation was previously shown to be regulated by the release of HMGB1 (high mobility group box chromosomal protein 1) from cells, after HMGB1-BECLIN-1 complex dissociation, via a mechanism involving toll-like receptor (TLR)4-mediated signaling [19-21]. Both Western blotting of heart homogenates (Figure 1C and 1D) and immunofluorescent detection in tissue sections (Figure 1E) demonstrated that HMGB1 was significantly decreased in WT but not LKO mice after PO. Under PO, restoring LCN2 in LKO mice with L2AV decreased cellular HMGB1 to amount similar as WT mice (Figure 1E). After four weeks PO, a significant increase of LCN2 in both serum and heart tissue was detected in WT mice with PO (Figure 1F). In agreement with our mouse data, HMGB1 levels were significantly decreased in heart tissues of HF patients compared to healthy individuals (Figure 1G and 1H). Furthermore, cardiac content of LCN2, NF-κB and Caspase-1 were all significantly increased in HF patients.
Figure 1.
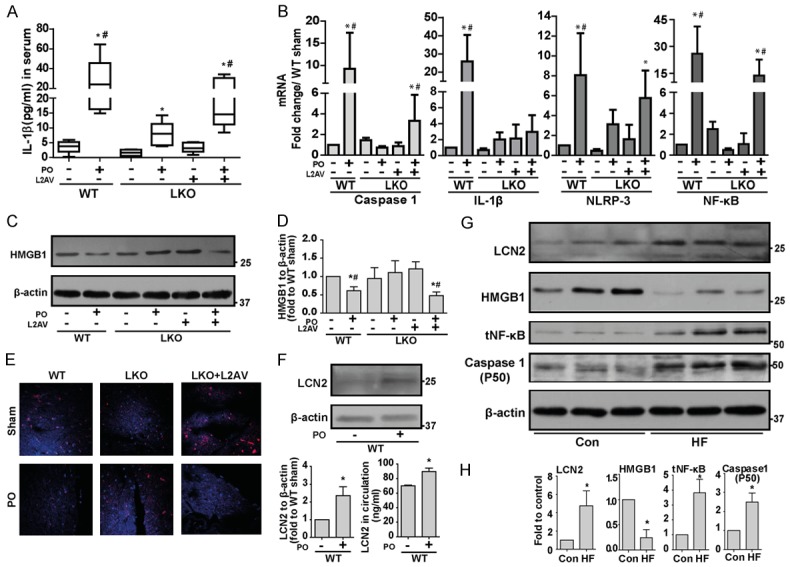
Inflammasome activation is attenuated in LKO mice challenged with PO. (A) IL-1β levels were determined by ELISA in serum collected from WT and LKO mice subjected to either sham or PO surgery. (B) qPCR was performed for gene expressions of inflammasome markers (Caspase-1, IL-1β, NLRP3 and NF-κB) in heart tissues collected from WT and LKO mice subjected to either sham or PO surgery. (C, D) Western blotting and densitometric quantifications was performed to analyze HMGB1 protein levels in the heart tissues from the above treated mice. (E) Immunofluorescence was performed for HMGB1 in heart tissue sections collected from WT and LKO mice subjected to either sham or PO surgery. Protein levels of LCN2 in heart tissue (F), densitometric quantification (F, left panel) and LCN2 level in circulation (F, right panel) were determined by Western blotting and ELISA, respectively. (G) Western blotting was performed to examine the protein levels of inflammation markers (LCN2, HMGB1, NF-κB and Caspase-1) in heart tissue lysates from healthy human subjects (Con) or patients with HF, with quantification shown in (H). *, P<0.05 vs. WT Sham or healthy human subjects and, #, vs. LKO PO, n=5-6.
LCN2 exacerbates mitochondrial deterioration and attenuates autophagic flux in response to PO
Transmission electron microscopy indicated LCN2 deficiency greatly attenuated PO-induced mitochondria damage in cardiomyocytes, as shown by the presence of clear well-structured cristae in LKO mice before and after PO (Figure 2A). This protective effect was partially lost with the administration of L2AV (Figure 2A). Transmission electron microscopy is a gold-standard approach for analysis of autophagic structures, and our data indicated induction of autophagy in mice lacking LCN2 after PO-treatment (Figure 2A, arrow), with less observed in WT mice. Next, autophagic flux was evaluated by examining cardiac protein expression levels of P62, LC3-II, BECLIN-1. Western analysis showed increased protein expression of P62 and decreased LC3-II and BECLIN-1, suggesting less flux in WT versus LKO mice after PO (Figure 2B and 2C). Analysis of cardiac biopsy tissue samples obtained from human subjects with or without HF indicated increased P62 plus decreased LC3-II and BECLIN-1 levels (Figure 2D and 2E), indicative of less autophagy when compared with control. It is conceivable that signals such as reactive oxygen species or mitochondria DNA released from damaged mitochondria might be responsible for priming or activation of NLRP3 inflammasome [22,23], whereas LCN-2 deficiency preserved mitochondria morphology and autophagic flux under PO.
Figure 2.
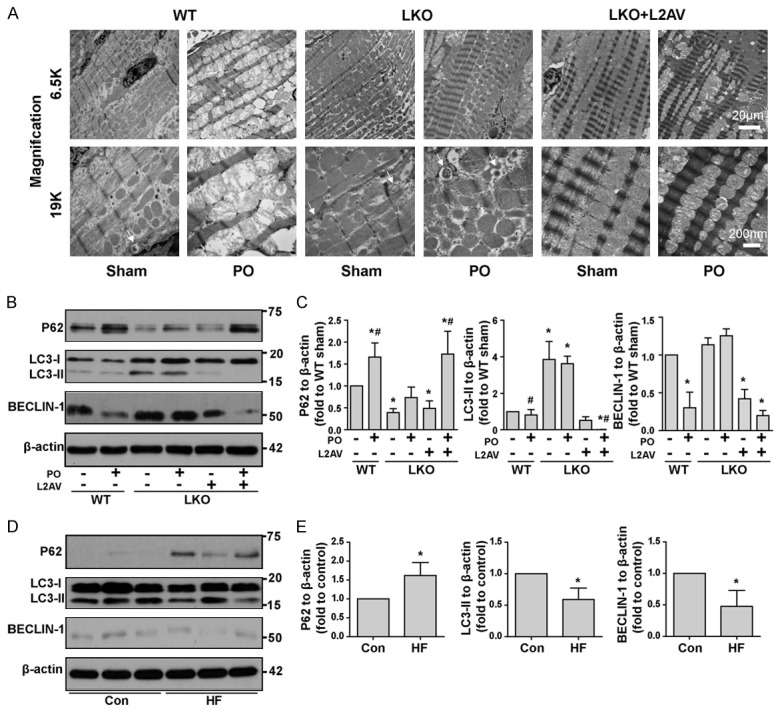
LCN2 deficiency protects against loss of mitochondrial morphology and autophagy flux disruption induced by PO. A: Mitochondria integrity was evaluated using TEM from heart tissue sections of WT and LKO (±L2AV) mice that had undergone either sham or pressure overload (PO) surgery. Autophagosomes were indicated by arrows. B: Autophagic flux was analyzed by examining the protein expression of P62, LC3-II and BECLIN-1 in heart tissue lysates from WT and LKO (±L2AV) mice that have undergone either sham or PO surgery. C: Densitometric quantifications of western blots in b. D: Autophagic flux was analyzed by examining the protein expression of P62, LC3-II, and BECLIN-1 in heart tissue lysates from healthy human subjects (Con) or patients with heart failure (HF). E: Densitometric quantifications of western blots in d. *, P<0.05 vs. WT Sham or healthy human subjects and, #, vs. LKO PO, n=5-6.
LCN2 deficiency protects mice from PO-induced development of fibrosis
Fibrosis is an established component of ventricular remodeling in HF patients [14,24]. Masson’s trichrome staining revealed enhanced accumulation of collagen after PO in WT and LKO+L2AV, but not in LKO mice (Figure 3A). Assessing mRNA levels of fibrosis-related genes showed matrix metalloproteinases (MMP)-1, MMP-8 and TGF-β1 increased significantly after PO in WT and LKO+L2AV mice but not in LKO mice (Figure 3B). MMP-9 levels increased after restoring LCN2 in LKO with L2AV under both sham, and to a greater extent, PO conditions (Figure 3B). Immunofluorescence staining showed that LKO mice were somewhat protected against PO-induced occurrence of vimentin-positive fibroblasts (Figure 3C). Administration of L2AV to LKO mice negated these protective effects; however, there was much less accumulation of fibroblasts compared to WT mice with PO (Figure 3C). Immunostaining for smooth muscle actin (SMA) showed that there was no interstitial activation of myofibroblast after PO in all six groups, as expected from this model of mild PO (Figure 3C).
Figure 3.

Development of PO-induced fibrosis is attenuated in LKO mice. A: Masson’s trichrome staining of heart tissue taken from WT and LKO (±L2AV) mice four weeks following PO or sham surgery. Cardiomyo-cytes are stained red, nuclei are stained black, and collagen is stained blue. B: Fibrosis PCR array was performed using heart tissues. Genes detected in this array are shown in Table 1. MMP-1, MMP-8, MMP-9 are present. C: Fibroblast to myofibroblast differentiation was assessed by co-staining with vimentin, α-SMA, and desmin in heart tissue sections from WT and LKO (±L2AV) mice that have undergone either sham surgery or PO. *, P<0.05 vs. WT Sham and, #, vs. LKO PO, n=4.
LCN2 deficiency protects mice from PO-induced development of cardiac dysfunction
Cardiac function of WT and LKO (±LCN2 restoration using L2AV) mice was evaluated by echocardiography four weeks after transverse aortic banding surgery. Consistent with previous reports [9], LKO mice demonstrated a higher ejection fraction (EF) and fractional shortening (FS) compared to age-matched WT mice that showed significant PO-induced cardiac dysfunction (Figure 4A and 4B). This heart protective effect in LKO mice was abolished by administration of LCN2 (Figure 4A and 4B) as the basal level of EF and ES in LKO mice treated with L2AV was similar to WT mice (Figure 4A and 4B). Importantly, we validated the effectiveness of adenoviral LCN2 delivery by showing that circulating LCN2 produced by L2AV reached a peak above normal level observed in WT mice one week after virus injection (Supplementary Figure 1) [7]. An inherent limitation of this approach is that levels gradually decline after 2 and 4 weeks yet we did not perform a second adenovirus injection to avoid confounding effects from immune system activation and inflammation. As visualized in representative short axis images of M-mode and speckle tracking echocardiography (Supplementary Figure 2), ventricular wall constriction and both systole (Figure 4C) and diastole (Figure 4D) circumferential strain rate in WT and LKO+L2AV mice under PO dropped significantly four weeks after PO, but were preserved in LKO mice (Figure 4E; Supplementary Table 1). PO induced increased LV mass and the response was similar in both genotypes and in LKO with LCN2 replenishment (Supplementary Figure 3). In summary, these data indicate a detrimental role of LCN2 in the development of cardiac dysfunction.
Figure 4.
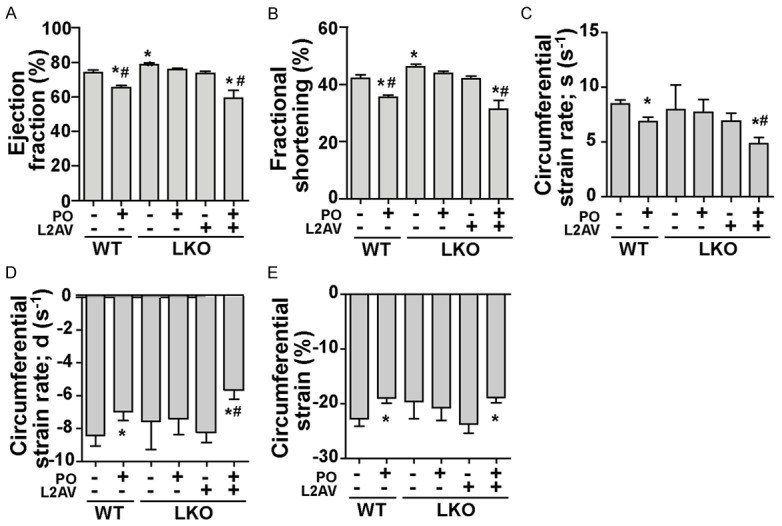
Temporal analysis of cardiac dysfunction using echocardiography after thoracic aorta banding or sham surgery. A: Ejection Fraction (EF); B: Fractional Shortening (FS); C: Circumferential systole strain rate; D: Circumferential diastole strain rate; E: Circumferential strain change percentage. *, P<0.05 vs. WT Sham and, #, vs. LKO PO, n=5-7. L2AV, lipocalin-2 adenovirus. LKO PO, lipocalin-2 knock-out mice with pressure overload surgery.
LCN2 acts directly on primary cardiac fibroblasts to decrease intracellular HMGB1 levels, induce NLRP3 inflammasome activation and attenuate autophagy
During pathological conditions, cardiac fibroblasts play a crucial role in maintaining normal cardiac function by modulating the synthesis not only via deposition of extracellular matrix, but also upon autocrine and paracrine cell-to-cell communication [25]. We observed accumulation of fibroblasts after PO model and these may play a role in NLRP3 inflammasome complex formation. To test this possibility, we isolated primary cardiac fibroblasts from neonatal rats and treated with recombinant LCN2 for 48 hours. Immunofluorescent staining indicated increased LCN2 signal in the cytosol and reduced cytosolic HMGB1 levels in cells treated with LCN2 (Figure 5A). Western blotting data also confirmed that total cellular HMGB1 levels were decreased after LCN2 treatment (Figure 5B, lower). Meanwhile, HMGB1 was detected in concentrated conditioned medium after LCN2 treatment (Figure 5B, top). Protein levels of IL-1β and IL-18 were also increased in this medium after LCN2 treatment (Figure 5B, top). Gene expressions of Caspase-1, IL-1β, NLRP3 and NF-κB in fibroblasts (Figure 5C) were induced by LCN2 treatment. We found that LCN2 also had direct effects on protein markers of autophagy with decreased LC3-II and elevated P62 levels and a reduction in BECLIN-1 (Figure 5D and 5E). HMGB1 also regulates autophagy by competing for BECLIN-1 binding with Bcl-2. To determine whether the interruption of autophagy is due to decreased binding between HMGB1 and BECLIN-1 after LCN2 treatment of cells, we immunoprecipitated BECLIN-1 and observed less association of HMGB1 after LCN2 treatment (Figure 5F and 5G).
Figure 5.

LCN2 treatment of rat neo-fibroblasts interrupts autophagic flux and induces inflammasome activation by releasing HMGB1. Rat neo-fibroblasts were isolated and treated with recombinant LCN2 (1 μg/ml) or control for 48 hours. Immunofluorescence (A) and Western blotting (B) were performed to examine the localization and protein expression levels of HMGB1 fibroblasts and IL-18 and IL-1β in medium. Expression levels of inflammasome markers showed in the graph (C) were examined by qPCR. (D) Western blotting was performed to examine the protein levels of autophagic markers (LCN2, LC3-II, P62 and BECLIN-1) with quantification shown in (E). (F, G) Co-immunoprecipitation (Co-IP) was performed in neonatal fibroblast cells to evaluate the binding status between BECLIN-1 and HMGB1. *, P<0.05, **, P<0.01 vs. corresponding control, n=3.
LCN2 induced inflammasome activation is attenuated by NLRP3 deficiency and TLR4 inhibition
Primary cardiac fibroblasts from WT and NLRP3 knockout (NLRP3-/-) adult mice were treated in culture with recombinant LCN2 or NLRP3 inflammasome activator lipopolysaccharide/adenosine triphosphate (LPS/ATP) for 24 h [26]. LCN2 treatment increased mRNA levels of inflammasome related genes Caspase-1 and IL-1β in WT mice but not in NLPR3-/- mice (Figure 6A-D). When treated with LCN2 and LPS/ATP in the presence or absence of NLRP3 inhibitor (MCC-950) or TLR4 inhibitor (Cli-095), mRNA levels of Caspase-1 and IL-1β were significantly attenuated (Figures 6F, 6G, 7B and 7C). MCC-950 and Cli-095 also attenuated the level of LCN2 induced IL-1β release into the media, as well as Caspase-1 and phospho-NF-κB p65 (Ser536) content in rat adult fibroblasts (Figures 6E and 7A). Consistently, the protein expression level of HMGB1 decreased after LCN2 treatment, but not LPS/ATP treatment, indicating the role of LCN2 in releasing HMGB1 from the cytosol (Figures 6E and 7A).
Figure 6.
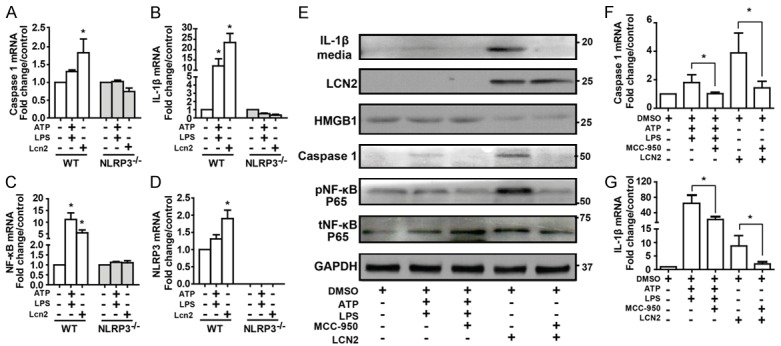
LCN2 induced inflammasome activation is attenuated by NLRP3 deficiency. Wild type and NLRP3-/- mice heart fibroblasts were isolated and treated with recombinant LCN2 (1 μg/ml) or ATP plus LPS for 24 hours. Expression levels of inflammasome markers, Caspase-1 (A), IL-1β (B), NF-κB (C) and NLRP3 (D) were examined by qPCR. Adult rat heart fibroblasts were isolated and treated with recombinant LCN2 (1 μg/ml) or ATP (5 μM) plus LPS (2.5 μg/ml) for 24 hours in the presence or absence of NLRP3 inhibitor MCC-950 (0.1 μM). (E) Western blotting was performed to examine the protein levels of LCN2 and inflammatory markers in medium (IL-1β) and fibroblasts (HMGB1, Caspase-1, pNF-κB, tNF-κB). Caspase-1 (F) and IL-1β (G) mRNA levels were checked by qPCR. *, P<0.05, **, P<0.01 vs. corresponding control, n=3.
Figure 7.

LCN2 induced inflammasome activation is attenuated by TLR4 inhibition. Adult rat heart fibroblasts were isolated and treated with recombinant LCN2 (1 μg/ml) or ATP (5 μM) plus LPS (2.5 μg/ml) for 24 hours in the presence or absence of TLR4 inhibitor Cli-095 (1 μg/ml). (A) Western blotting was performed to examine the protein levels of LCN2 and inflammatory markers in medium (IL-1β) and fibroblasts (HMGB1, Caspase-1, pNF-κB, tNF-κB). Caspase-1 (B) and IL-1β (C) mRNA levels were checked by qPCR. *, P<0.05, **, P<0.01 vs. corresponding control, n=3.
Discussion
Our analysis of the contribution by LCN2 to cardiac dysfunction, and in particular the discovery of novel mechanisms of LCN2 action has wide-ranging diagnostic and therapeutic implications, not only in heart failure but also other conditions such as kidney dysfunction. A principal novel discovery of this study was that LCN2 induced NLRP3 inflammasome activation via release of the danger-associated molecular pattern (DAMP) protein HMGB1 and subsequent TLR4-dependent signaling. Our results revealed that lack of LCN2 in mice mitigates maladaptive fibrotic remodeling and facilitates adequate induction of autophagic flux in response to PO, in keeping with our previous work indicating that LCN2 directly attenuated autophagic flux [13]. Thus, we have validated the functional consequences of cardiac LCN2 actions and provided new insight into the mechanisms involved (Figure 8).
Figure 8.
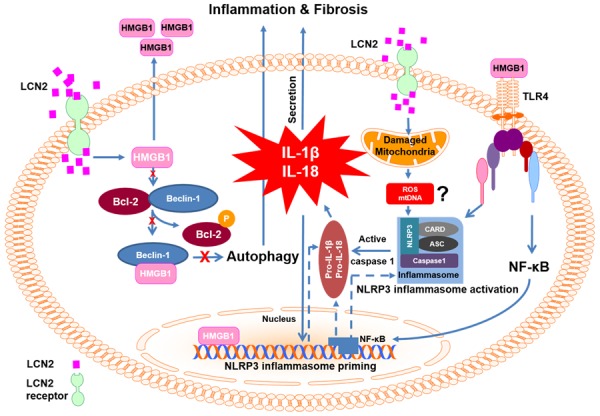
Schematic model depicting mechanisms via which LCN2 regulates NLRP3 inflammasome. LCN2 disrupts the association of BECLIN-1 and HMGB1, one consequence of which is altered autophagic flux (left side). Another is HMGB1 release from cells which can then induce an inflammatory response by binding to TLR4 receptors (right side). LCN2 also induced mitochondrial dysfunction and we propose that consequent increases in ROS or mtDNA may contribute to priming and activation of the NLRP3 inflammasome. The end result of NLRP3 inflammasome activation is production and cleavage of proinflammatory cytokines (IL-1 β and IL-18) via Caspase-1 activation. Together these effects may dictate cardiac remodeling events such as fibrosis. ASC, apoptosis-associated speck-like protein; Bcl-2, B-cell lymphoma 2; CARD, caspase activation and recruitment domain. IL-1β, interleukin-1β; IL-18, interleukin-18; NF-κB, transcription factor nuclear factor kappa-light chain-enhancer of activated B cells; NLRP3, NACHT, LRR and PYD domains-containing protein 3; TLR-4, toll-like receptor 4.
An important impact of NLRP3 inflammasome activation in diabetes and cardiovascular disease is well documented [11,27,28]. Previous studies showed that serum LCN2 levels correlate positively with various cardiovascular diseases including HF [2]. Basal expression of LCN2 in heart tissue is low and we found that direct administration of LCN2 accelerated the progression of HF [6]. One of the best characterized DAMPs is HMGB1 which promotes NLRP3 inflammasome and NF-κB activation [29]. Effects of HMGB1 can be mediated via several mechanisms, including HMGB1 binding to receptor for advanced glycation end products (RAGE) and toll-like receptors TLR2 and TLR4 [21,29]. In astrocytes, the inhibition of TLR4, rather than RAGE or TLR2 attenuated HMGB1-induced IL-18 production [30]. We found decreased intracellular HMGB1 levels in rat adult and neonatal fibroblasts treated with LCN2, suggesting that LCN2 induces release of HMGB1 into the extracellular space by an as yet unknown mechanism. Furthermore, we found decreased HMGB1 levels in WT and LKO+L2AV, but not LKO mice after PO as well as in cardiac biopsy samples of human HF patients. Our data indicated that NLRP3 inflammasome was initiated by LCN2 via the release of HMGB1 followed by TLR-4 activation and mitochondria damage, thus assembling various lines of evidence in the literature into a detailed cohesive mechanism of LCN2 action. The induction of the NLRP3 inflammasome complex and subsequent inflammatory effects are then reasonably considered to contribute to cardiac dysfunction.
Reciprocal crosstalk between NLRP3 inflammasome activation and LCN2 action was suggested by studies showing that stimulation of TLR4 induces the formation of the inflammasome complex and production of LCN2 [31]. Many studies have now indicated that the degree of autophagy changes in the failing heart although the functional significance is still contentious [32]. Disruption of autophagy by cardiac-specific knockdown of Atg5 in mice leads to cardiomyopathy [33]. We believe that LCN2 is an important suppressor of cardiac autophagy and have recently shown reduced autophagic flux in cardiomyoblast cells treated with LCN2 [13]. Our current study provides evidence that the increase in cardiac autophagy normally seen in response to PO, and which is generally regarded as a beneficial adaptive response, is enhanced in LKO mice and attenuated by adenoviral delivery of LCN2 to these mice. Together, these data indicate that LCN2 acts to suppress cardiac autophagic flux and this likely contributes to cardiac dysfunction. Interestingly, sustained autophagy upon transgenic Atg7 overexpression in mice decreased cardiac fibrosis and dysfunction [34]. We showed that LKO mice were somewhat protected against PO-induced induction of vimentin-positive fibroblasts and fibrosis, while administration of L2AV to LKO mice negated these protective effects. In addition to the classic role of extracellular matrix regulation, MMP-9 activity can also contribute to inflammation in the failing heart via activation of inflammatory cytokines such as IL-1β, IL-8 and tumor necrosis factor alpha (TNF-α) [35].
In conclusion, our study confirms the detrimental role of LCN2 in development of cardiac dysfunction and provides new mechanistic insight on mechanisms of LCN2 action. Indeed, the effects which we have observed in this study could well be at least in part mediated via systemic effects of LCN2 in other tissues which causes crosstalk with the heart. A principal novel discovery of this study was that LCN2 induced NLRP3 inflammasome activation via release of the danger-associated molecular pattern (DAMP) protein HMGB1 and subsequent TLR4-dependent signaling. This study also provided further evidence that LCN2 acts to suppress stress-induced autophagic flux. Our focus was on the heart yet this data has potentially wide-ranging diagnostic and therapeutic implications, since the physiological significance of LCN2, also commonly referred to as NGAL, is perhaps most well established in kidney. Nevertheless, potential for suppressing neutrophil-derived LCN2-mediated inflammation must be balances by potential adverse effects of over-suppression due to the beneficial role of neutrophils in cardiac repair by polarizing macrophages towards a reparative phenotype [36]. This work will also likely stimulate interest in new avenues of research on the regulation of NLRP3 inflammasome, autophagy and fibrosis by LCN2 in other target tissues. In summary, we have characterized the cardiac functional consequences of LCN2 deletion, with or without readministration, in mice and provided new insight into the mechanisms involved.
Acknowledgements
This work was supported by Grant-in-aid and Career Investigator Award to GS from Heart & Stroke Foundation of Canada/Ontario. BH was supported by the Canadian Institutes of Health Research (grant #286920) and the E-Rare Joint Transnational Call ‘Development of Innovative Therapeutic Approaches for Rare Diseases’. MC/AR were supported by NHMRC Project Grant APP1086786.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. doi: 10.1373/clinchem.2006.075614. [DOI] [PubMed] [Google Scholar]
- 3.Chan YK, Sung HK, Sweeney G. Iron metabolism and regulation by neutrophil gelatinase-associated lipocalin in cardiomyopathy. Clin Sci (Lond) 2015;129:851–862. doi: 10.1042/CS20150075. [DOI] [PubMed] [Google Scholar]
- 4.Jang Y, Lee JH, Wang Y, Sweeney G. Emerging clinical and experimental evidence for the role of lipocalin-2 in metabolic syndrome. Clin Exp Pharmacol Physiol. 2012;39:194–199. doi: 10.1111/j.1440-1681.2011.05557.x. [DOI] [PubMed] [Google Scholar]
- 5.Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, Fong HE, Cheung CC, Mak TW. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song E, Fan P, Huang B, Deng HB, Cheung BM, Feletou M, Vilaine JP, Villeneuve N, Xu A, Vanhoutte PM, Wang Y. Deamidated lipocalin-2 induces endothelial dysfunction and hypertension in dietary obese mice. J Am Heart Assoc. 2014;3:e000837. doi: 10.1161/JAHA.114.000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Law IK, Xu A, Lam KSL, Berger T, Mak TW, Vanhoutte PM, Liu JT, Sweeney G, Zhou M, Yang B, Wang Y. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes. 2010;59:872–882. doi: 10.2337/db09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu JT, Song E, Xu A, Berger T, Mak TW, Tse HF, Law IK, Huang B, Liang Y, Vanhoutte PM, Wang Y. Lipocalin-2 deficiency prevents endothelial dysfunction associated with dietary obesity: role of cytochrome P450 2C inhibition. Br J Pharmacol. 2012;165:520–531. doi: 10.1111/j.1476-5381.2011.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang B, Fan P, Xu A, Lam KS, Berger T, Mak TW, Tse HF, Yue JW, Song E, Vanhoutte PM, Sweeney G, Wang Y. Improved functional recovery to I/R injury in hearts from lipocalin-2 deficiency mice: restoration of mitochondrial function and phospholipids remodeling. Am J Transl Res. 2012;4:60–71. [PMC free article] [PubMed] [Google Scholar]
- 10.Matsa R, Ashley E, Sharma V, Walden AP, Keating L. Plasma and urine neutrophil gelatinase-associated lipocalin in the diagnosis of new onset acute kidney injury in critically ill patients. Crit Care. 2014;18:R137. doi: 10.1186/cc13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butts B, Gary RA, Dunbar SB, Butler J. The Importance of NLRP3 Inflammasome in Heart Failure. J Card Fail. 2015;21:586–593. doi: 10.1016/j.cardfail.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan YK, Sung HK, Jahng JW, Kim GH, Han M, Sweeney G. Lipocalin-2 inhibits autophagy and induces insulin resistance in H9c2 cells. Mol Cell Endocrinol. 2016;430:68–76. doi: 10.1016/j.mce.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Dadson K, Kovacevic V, Rengasamy P, Kim GH, Boo S, Li RK, George I, Schulze PC, Hinz B, Sweeney G. Cellular, structural and functional cardiac remodelling following pressure overload and unloading. Int J Cardiol. 2016;216:32–42. doi: 10.1016/j.ijcard.2016.03.240. [DOI] [PubMed] [Google Scholar]
- 15.Jahng JW, Turdi S, Kovacevic V, Dadson K, Li RK, Sweeney G. Pressure overload-induced cardiac dysfunction in aged male adiponectin knockout mice is associated with autophagy deficiency. Endocrinology. 2015;156:2667–2677. doi: 10.1210/en.2015-1162. [DOI] [PubMed] [Google Scholar]
- 16.Castillero E, Akashi H, Pendrak K, Yerebakan H, Najjar M, Wang C, Naka Y, Mancini D, Sweeney HL, J DA, Ali ZA, Schulze PC, George I. Attenuation of the unfolded protein response and endoplasmic reticulum stress after mechanical unloading in dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2015;309:H459–470. doi: 10.1152/ajpheart.00056.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dadson K, Turdi S, Boo S, Hinz B, Sweeney G. Temporal and molecular analyses of cardiac extracellular matrix remodeling following pressure overload in adiponectin deficient mice. PLoS One. 2015;10:e0121049. doi: 10.1371/journal.pone.0121049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 20.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial ROS induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013;191:1–19. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173–185. doi: 10.1007/s10741-012-9365-4. [DOI] [PubMed] [Google Scholar]
- 25.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henriksbo BD, Schertzer JD. Is immunity a mechanism contributing to statin-induced diabetes? Adipocyte. 2015;4:232–238. doi: 10.1080/21623945.2015.1024394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, Tarnopolsky MA, Steinberg GR, Schertzer JD. Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes. 2014;63:3742–3747. doi: 10.2337/db13-1398. [DOI] [PubMed] [Google Scholar]
- 29.Pisetsky DS, Erlandsson-Harris H, Andersson U. High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. 2008;10:209. doi: 10.1186/ar2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banjara M. Lipocalin-2: a new regulator of nonpathogen-associated neuroinflammation. Int J Clin Exp Neurol. 2014;2:8–15. [Google Scholar]
- 31.Wang Y, Song E, Bai B, Vanhoutte PM. Tolllike receptors mediating vascular malfunction: Lessons from receptor subtypes. Pharmacol Ther. 2016;158:91–100. doi: 10.1016/j.pharmthera.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Gottlieb RA, Mentzer RM Jr. Autophagy: an affair of the heart. Heart Fail Rev. 2013;18:575–584. doi: 10.1007/s10741-012-9367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 34.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. doi: 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.