

Abstract
Cervical cancer is the second most common malignancy in women worldwide. HPV infections are the leading cause of cervical cancer. Although progress has been made in understanding cervical cancer, knowledge of oncogenic gene clusters that participate in squamous-cell mitosis is still lacking. We performed a computational analysis with qRT-PCR validation of gene expression profiles of cervical cancer tissues. Genes involved in muscle contraction and development were downregulated in cervical cancer tissues, suggesting decreased muscle function in cervical cancer. Among the genes that were upregulated in cervical cancer tissues, several groups of genes were found to interact with each other and synergistically participate in multiple stages of mitosis including DNA replication, cell cycle progression, and cell division. An analysis of gene regulatory networks showed that replicative helicase proteins (MCM2, MCM4, MCM5, MCM6, and MCM10) and DNA polymerases (PLOA1/E2/E3/Q) have enhanced DNA replication in cervical cancer. A group of kinases, cyclins, and transcriptional factors were found to promote cell cycle transitions from G1 phase to S phase and from G2 phase to M phase. Those proteins included CDK1, CCNA2, CCNB2, and TFDP2. Moreover, a set of motor proteins (KIF11, KIF14 and KIF4A) and their partner PRC1 were found to mediate cytokinesis during cervical cancer progression. Those findings present a better understanding of the mechanism of mitosis in cervical cancer from an interactomic perspective and provide potential targets for anticancer therapies.
Keywords: Cervical cancer, computational analysis, mitosis, DNA replication, cell cycle
Introduction
Cervical cancer is the second most common malignancy in women worldwide and is the third leading cause of cancer deaths among females in less developed countries [1,2]. It was estimated that there were 527,600 new cases of cervical cancer and 265,700 deaths caused by cervical cancer in 2012 [1]. Although vaccination and screening are efficient ways to prevent the disease, poor prognosis is observed in patients with bulky tumors or adenocarcinoma. It is widely recognized that persistent infection by human papillomaviruses (HPVs) is the leading cause of cervical cancer [3]. More than 120 HPV types have been identified, and HPV16 and HPV18 are the types that most frequently infect women [4,5]. HPV16 is reported to have the greatest oncogenic potential, and women who are persistently infected with HPV16 are at high risk of developing cervical intraepithelial carcinoma [6,7].
The pathogenesis of HPV-related cervical cancer involves the overexpression of viral oncoproteins E5, E6, and E7, which inhibit a variety of cellular proteins including p53, pRb, p21, and p27 and affect a series of biological processes including cell proliferation, cell cycle, and apoptosis [8-10]. The HPV E7 protein binds to Rb family members and marks them for degradation, which causes the release and activation of E2F transcription factors and drives S-phase gene expression [11,12]. Moreover, E7 can control cell cycle progression through interaction with key regulators. E7 can target cyclin-dependent kinase 2 (CDK2) inhibitors and maintain CDK2 activity, which is important for the transition from G1 phase to S phase [13,14]. In addition, E6 proteins from high-risk HPV strains can promote the degradation of tumor suppressor p53 through the ubiquitin pathway, which prevents the inhibition of cell growth in both undifferentiated and differentiated cells [15]. A previous study reported that E6 can also bind to and inhibit the transcriptional activity of p53 [16]. In addition to p53, E6 is able to bind to several other proteins including E6BP, E6TP1, and MCM7 [17]. High-risk E6 and E7 proteins work in synergy with each other to maintain S-phase competence by targeting factors that promote cell cycle progression. It was reported that the combination of E5, E6, and E7 could promote the hyperproliferation of HPV-infected cells and facilitate malignant progression [18].
Comprehensively, the aberrant expression of multiple genes in cervical cancer leads to the maintenance of cervical epithelial-cell hyperproliferation. To better understand the mechanism of cervical cancer progression, we performed a comparative analysis of the gene expression profiles of cervical cancer cells. We employed a series of bioinformatic strategies to identify the key genes that are dysregulated in cervical cancer. We also performed validation assays to examine the genes that may contribute to carcinogenesis.
Material and methods
Cervical cancer samples
We collected 12 cervical cancer samples and nine non-cancerous hysteromyoma control samples from the biological specimen bank of the Second Affiliated Hospital of Wenzhou Medical University. The samples were fresh-frozen and stored in liquid nitrogen. The Bioethics Committee of the Second Affiliated Hospital of Wenzhou Medical University reviewed and approved the study and protocol. All patients included in the study gave written informed consent. A pathologist in the Department of Pathology in the Second Affiliated Hospital of Wenzhou Medical University who was blind to the study performed histological examinations of the clinical samples.
Cervical cancer gene expression dataset
We downloaded the genome-wide transcriptional expression data (GSE63678) of five human cervical tumors and five normal cervix tissue samples from the Gene Expression Omnibus (GEO) database. The mRNA expression levels in the dataset were measured using the Affymetrix Human Genome U133A 2.0 Array.
Identification of differentially expressed genes (DEGs)
We used the R limma package to normalize the GEO data and performed a three-dimensional principle component analysis (PCA) to identify the DEGs in the cervical cancer tissues and control tissues [19]. We used the Benjamini-Hochberg test to calculate the False Discovery Rate (FDR) for p-value correction. We selected the DEGs based on the following criteria: P<0.05, FDR<0.05, and Log2FC (Fold Change) <-1 or Log2FC>1.
Gene ontology (GO) and two-dimensional view analysis
We applied GO analysis to identify the significant functions of the DGEs according to the Gene Ontology Database, which is the key functional classification of NCBI [20]. We used Fisher’s exact test with a significance threshold of P<0.05 to determine the significance of the GO terms linked to the DEGs. We used the DAVID gene functional annotation tool to perform a two-dimensional view analysis [21]. We input all of the DGEs and picked the GO terms with the highest enrichment scores. Then, we extracted the associations between the genes and GO terms and visualized them on a heatmap.
Pathway analysis
We performed a pathway analysis using KEGG annotations to discover the significant pathways linked to the DEGs [22]. We employed Fisher’s exact test to identify the significant pathways. We built a pathway interaction network based on the relationships among the pathways from the KEGG database. The DGEs that we identified could be involved in multiple pathways, so there was some overlap among the pathways. We identified the significant pathways that shared the same DGEs and used Cytoscape to construct graphical representations of the interactive relationships among the pathways.
Gene regulatory network
We used STRING (Version10.0), a tool for functional protein-association analysis, to build a gene regulatory network to elucidate the genes that may play key roles in carcinogenesis [23]. We entered the DEGs into the STRING database and extracted the regulatory relationships including activation, expression, binding, post-transcriptional modification, inhibition, and catalysis to construct the gene interaction network. We used a highest confidence of 0.900 as the minimum required interaction score.
Co-expression network analysis
We calculated the Pearson correlation coefficient for the expression levels of each pair of DEGs in the GEO data. We defined 0.99 as the threshold to build a gene co-expression network. In network analysis, the degree of association is an important factor to determine the relative importance of DGEs. We marked the gene nodes with different colors and sizes to highlight the degree of the associations with other nodes. We constructed the co-expression network using Cytoscape.
qRT-PCR
We used a Trizol-based method to extract RNA from the cervical cancer and non-cancerous hysteromyoma control samples. We then adjusted the RNA concentration of each sample to 500 ng. We synthesized the first strand of cDNA using the SuperScript IV First-Strand Synthesis System. We amplified the selected genes using 480 SYBR Green I Master kit and the LightCycler 480 platform (Roche). We normalized the expression level of each gene to that of β-actin and calculated the relative fold changes in expression between the cancer and control samples.
Results
Overview of the GEO microarray data and identification of DEGs
The results of the PCA indicated that the samples of cervical cancer and normal control tissues were closely associated (Figure 1A). We identified 190 genes with differential expression between the cancer and control tissues. Among the DEGs, 78 were downregulated in the cancer tissues, and 112 were upregulated in the cancer tissues (Figure 1B). The results of the hierarchical cluster analysis of the expression profiles of the DGEs are shown in Figure 1C.
Figure 1.

Overview of the expression profiles of samples of cervical cancer tissues and healthy control tissues. A: Three-dimensional principal component analysis. The N and C represent normal controls and cervical cancer, respectively. The numeric labels represent different samples. B: Volcano plot to highlight the differentially expressed genes (DEGs) among all the transcripts. The red dots represent significant DEGs between tumor and normal tissues, and black dots represent DEGs with no significance. C: A heatmap showing clusters of DEGs.
GO and two-dimensional view analysis
The results of the GO analysis indicated that DNA replication, DNA metabolic process, mitotic cell cycle, cell division, and cell cycle were the top five significant biological processes assigned to the upregulated genes (Figure 2A). All of the significant molecular functions of the upregulated genes were related to nucleotide binding and DNA polymerase activity (Figure 2A). Furthermore, the results indicated that the upregulated DNA synthesis was mainly located on chromosomes (Figure 2A). Tube development was the most significant biological process assigned to the downregulated genes. The functions of the downregulated genes were related to contractile fiber and smooth muscle, indicating that cytoskeletal protein binding function was decreased in the cervical cancer tissues (Figure 2B).
Figure 2.
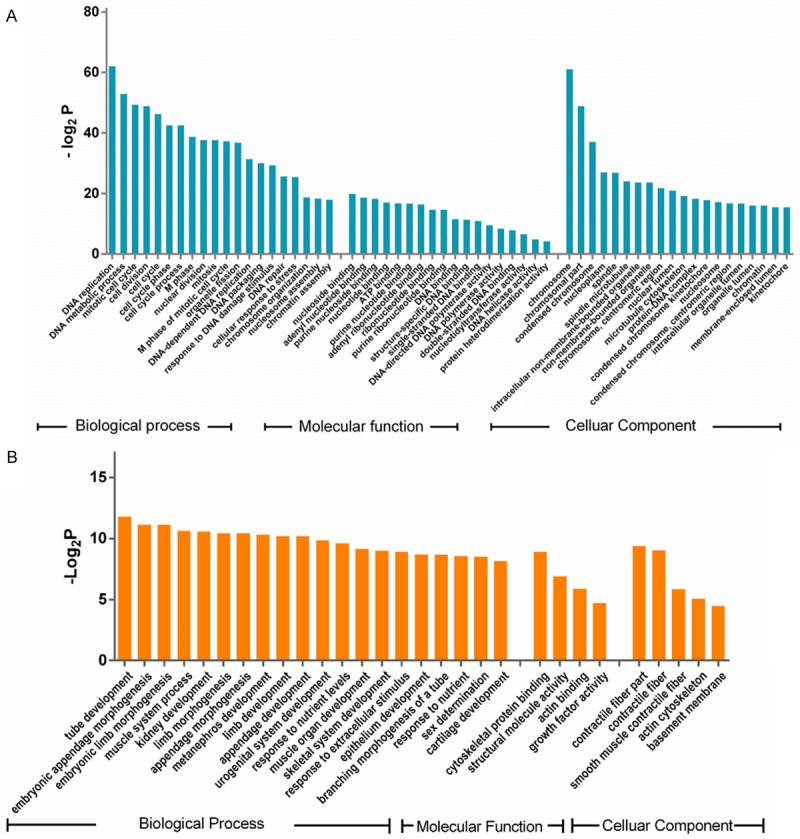
The Gene Ontology (GO) annotations of the differentially expressed genes (DEGs). A: Significant GO terms of the upregulated DEGs in cervical cancer. B: Significant GO terms of the downregulated DEGs in cervical cancer. All of the GO terms included biological process, molecular function, and cellular components.
The two-dimensional view analysis showed that GO terms involved in microtube function and cell cycle progression were clustered together (Figure 3A). G2/mitotic-specific cyclin-B1 (CCNB1) and Cyclin-dependent kinase 1 (CDK1) were positively associated with all of the mitotic GO terms (Figure 3A). The analysis also identified a group of DEGs that were either components of or functionally required by the mitotic spindle, including CEP55, KIF11, KNTC1, ASPM, NUSAP1, MAD2L1, NEK2, and SPAG5 (Figure 3A).
Figure 3.
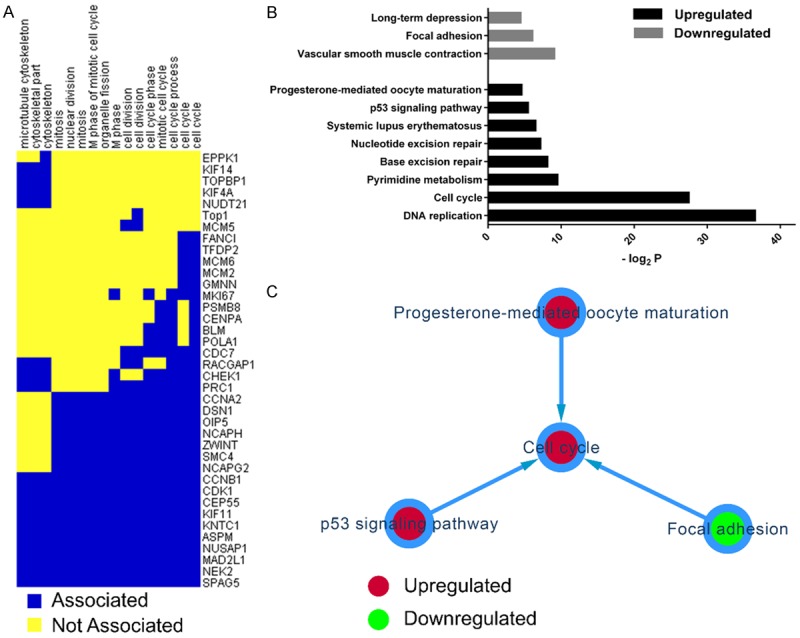
Two-dimensional view and pathway analysis of differentially expressed genes (DEGs). A: Two-dimensional view analysis of upregulated DEGs in cervical cancer. B: Significant pathway analysis based on the KEGG annotations. C: Pathway interaction analysis.
Pathway analysis
In the KEGG pathway analysis, the most significant of the upregulated pathways were DNA replication and the cell cycle (Figure 3B). Additionally, we identified the P53 signaling pathway as an upregulated pathway. The significant downregulated pathways included vascular smooth muscle contraction, focal adhesion, and long-term depression (Figure 3B). The pathway interaction network based on the relationships recorded in the KEGG database indicated three one-way interactive links focused on the cell cycle pathway.
Gene regulatory network
The gene regulatory network based on the STRING database was composed of five subnets and included eight types of interactive relationships. The gene regulatory analysis identified a cluster of DEGs that participate in mitosis and the cell cycle (Figure 4). Most of the DEGs were upregulated in cervical cancer; however, NES and TUBA1A were downregulated in cervical cancer. In the mitosis and cell cycle module, it was obvious that CDK1 was the master DEG, showing the most interactive relationships with the other DEGs. Moreover, we found that components of the MCM complex (MCM2, MCM4, MCM5, MCM6, and MCM10), which acts as a DNA replicative helicase, bound to each other. The network also demonstrated a module composed of HIST1H2BI, HIST1H2B, H2AFZ, and HIST1H2BH that functioned in nucleosome assembly. In contrast, a subnet composed of the downregulated genes MYL9, MYH11, LMOD1, ACTA2, and MYLK was shown to be related to muscle contraction. Those results indicated that the function of muscle contraction was decreased in the cervical cancer tissues.
Figure 4.

Gene regulatory network analysis of differentially expressed genes. The red and green circles represent genes that were upregulated and downregulated, respectively, in cervical cancer. The size of each node represents the degree of the associations with the nodes around.
Co-expression network
We constructed a co-expression network to disclose gene clusters that may work together to promote mitosis. The co-expression network identified three DEG clusters that promote DNA replication, the cell cycle, and cell division (Figure 5). The first cluster included TFDP2, PRC1, KIF14, NEK2, and MELK and may control cell cycle transition from G1 phase to S phase and from G2 phase to M phase (Figure 5). The second cluster included HMMR, POLQ, RRM2, NCAPG2, KIF4A, and NCAPH and mainly promotes DNA synthesis, the formation of mitotic chromosomes, and spindle organization (Figure 5). The third cluster included MCM2, CCNA2, CENPA, and OIP5 and is responsible for DNA replication, cell cycle transition, mitotic spindle assembly, and cell division (Figure 5). The nucleosome compacting proteins HIST1H2BK and H2BF also showed potential co-expression (Figure 5).
Figure 5.
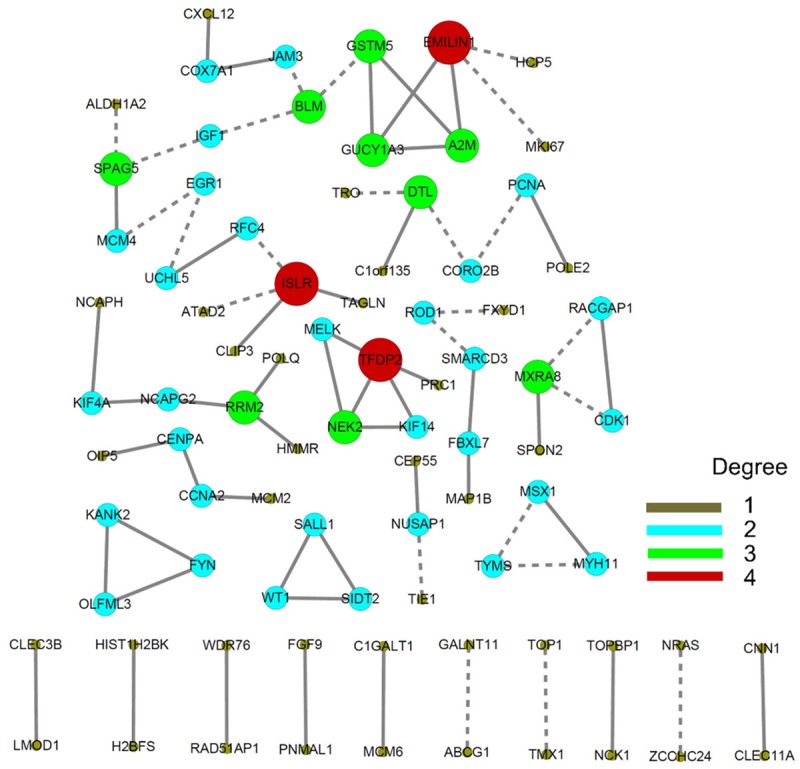
Co-expression network analysis of expression profiles in cervical cancer. Different sizes and colors were employed to discriminate the degrees of each gene.
Expression profiles of selected genes measured by qRT-PCR
The qRT-PCR results showed that MCM2, MCM4, and MCM5, but not MCM6, were expressed differentially between the cervical cancer tissues and the hysteromyoma control tissues (Figure 6). The cyclin proteins CCNA2 and CCNB1 and their kinases CHEK1 and CDK1 were significantly upregulated in the cervical cancer tissues. Furthermore, TFDP2 and PCNA were also upregulated in the cervical cancer tissues. There was no significant difference in CDC7 between the tumor and control tissues.
Figure 6.

Validation of the protein expression of important genes involved in mitosis. qRT-PCR analysis of gene expression profiles of selected genes (*P<0.05; NS: no significance).
Discussion
Although the etiology of cervical cancer is multifactorial, including genetic and environmental factors as well as combinations of those factors, infections by the oncogenic viruses HPV and HSV-2 are the dominant cause of the disease [24,25]. Other risk factors for cervical cancer include smoking, immunosuppression caused by HIV, low consumption of fruits and vegetables, high BMI, and multiple full-term pregnancies. It was reported that a family history of cervical cancer is associated with an approximately twofold increased risk of cervical cancer [26]. Moreover, a number of studies suggested that SNPs in some genes are associated with the occurrence of cervical cancer [27-29]. Whole-exome sequencing of cervical carcinomas has identified somatic mutations in nine genes: MAPK1, HLA-B, EP300, FBXW7, NFE2L2, TP53, ERBB2, ELF3, and CBFB [30]. At the transcriptional level, several protein-coding genes, long noncoding RNAs, and miRNAs were reported to be downregulated or upregulated in cervical cancer and to show relationships with clinical characteristics such as tumor size, lymphatic metastasis, and invasion [31-34]. TCL1A expression in B-cells in cervical tumors was correlated with patient survival [35]. Although some important genes have been identified as being dysregulated in cervical cancer, the data should be analyzed further using computational strategies to better elucidate the significance of such genes in the initiation and progression of cervical cancer. We performed a bioinformatic analysis of a transcriptomic microarray dataset comprising five cervical cancer tissues and five normal control tissues. We employed GO and pathway analyses to explore the functions of the DEGs identified based on the microarray data. Both the GO analysis and the KEGG pathway analysis indicated that the genes that were upregulated in the cancer tissues were enriched with functions linked to DNA replication and the cell cycle, which are key steps in mitosis. The genes that were downregulated in the cancer tissues were enriched with functions linked to muscle development, indicating that the physiological function of the cervix was weakened.
The cervix is the lower part of the uterus. It is divided from the upper part of the uterus by a fibromuscular junction, which joins the muscular corpus to the fibrous cervix. The cervix has an inner mucosal layer, a thick layer of smooth muscle, and a supravaginal portion. The function of the smooth muscle in the cervix has been recognized as the reason that the cervix is refractory to stimuli from the uterus, which produce growth and contraction. The cervical smooth muscle is also able to act as a barrier to hold the products of conception [36]. Our results indicate that genes involved in muscle development and contraction are downregulated in cervical cancer cells. The gene regulatory analysis showed that genes involved in muscle contraction (MYL9, MYH11, LMOD1, ACTA2, and MYLK) can bind, modify, and activate each other; however, further evidence is needed to demonstrate that muscle formation and contraction are decreased in patients with cervical cancer.
Enhanced cell proliferation capacity is the essential characteristic of cancer. We observed that the biological processes of DNA replication and cell cycle transition, which are critical events for mitosis, were increased in the cervical cancer tissues. In the cell cycle, transition from G1 phase to S phase is the first step of mitosis. The two-dimensional view analysis and gene interaction network identified CDK1 as the most important gene that regulates the other DEGs, indicating that CDK1 plays multiple roles in cervical cancer-cell proliferation. According to the regulatory network, CDK1 can interact with MCM family components and cyclins and kinesin-like proteins, which modulate DNA replication, cell cycle progression, and cell division [37-39]. Consistently, several studies have found that the expression of CDK1 was increased in cervical cancer and that CDK1 plays a key role in tumor progression and can serve as a therapeutic target [40].
In the co-expression network, we found two crucial gene subnets that may control cell cycle progression. TFDP2 and CCNA2 were the master nodes in the two subnets, which control cell cycle progression from G1 phase to S phase. Previous studies have reported that TFDP2 was upregulated in cervical cancer [41]. We found that CCNA2 was significantly upregulated in cervical cancer, implicating a potential mechanism of abnormal mitosis in cervical cancer.
To maintain a hyperproliferative state, cervical cancer cells upregulate a group of genes that control multiple steps of DNA replication. Among the upregulated genes, we identified five components of the MCM protein complex: MCM2, MCM4, MCM5, MCM6, and MCM10. MCM2-7 form a replicative helicase complex that unwinds the DNA during replication [42]. MCM10 is a replication-initiation factor that recruits MCM2-7 helicase and DNA polymerase complexes to start DNA replication [43]. Consistently, studies have reported that the expression of MCM proteins was increased in a variety of cancers including breast cancer, cancer of the uterine cervix, and gliomas. The MCM proteins could be used as prognostic biomarkers for patients with cancer. Our analysis indicated that the expression levels of a group of DNA polymerases were increased in cervical cancer, including PLOA1, PLOE2, PLOE3, and PLOQ. PLOA1 is the DNA polymerase alpha catalytic subunit, which initiates DNA replication at replicative forks through interaction with MCM10. PLOE2 and POLE3 are DNA polymerase epsilon subunits responsible for DNA replication and repair. PLOQ encodes DNA polymerase theta, which mediates the repair of double-stranded DNA breaks. We did not identify any direct evidence to elucidate the roles of DNA polymerases in cervical cancer. The associations between PLOA1/E2/E3/Q and cervical cancer are not understood. However, it is possible that the upregulation of the DNA polymerases group with similar expression profile may facilitate the DNA replication which is required for mitosis.
We found that the expression levels of three kinesin-like proteins (KIF11, KIF14, and KIF4A) were increased in cervical cancer. In the gene regulatory network, PRC1 can bind to those 3 kinesin-like proteins and can be modified by CDK1. The binding of those protein mediates cytokinesis during mitosis.
In conclusion, we used a computational bioinformatic strategy to identify groups of genes that promote mitosis in cervical cancer. We found that some genes involved in muscle contraction and development were downregulated in cervical cancer cells, which suggests decreased muscle function in cervical cancer. We identified several groups of genes that synergistically participate in multiple stages of mitosis, including DNA replication, cell cycle progression, and cell division. Most of the genes that were upregulatedin cervical cancer were clustered together and displayed similar functions. The regulatory network analysis showed that replicative helicase proteins (MCM2, MCM4, MCM5, MCM6 and MCM10) and DNA replication polymerases (PLOA1/E2/E3/Q) have enhanced expression in cervical cancer. A group of kinases, cyclins, and transcriptional factors (CDK1, CCNA2, CCNB2, and TFDP2) promote cell cycle transitions from G1 phase to S phase and from G2 phase to M phase in cervical cancer. Moreover, the expression of a group of motor proteins (KIF11, KIF14 and KIF4A) and their partner PRC1 were increased in cervical cancer and were found to mediate cytokinesis during cervical cancer progression. Our findings present a better genome-wide understanding of the mechanism of mitosis in cervical cancer and provide potential targets for anticancer therapies.
Acknowledgements
This study was supported by the Natural Sciences Fund of Zhejiang Province (LY13C060003) and, in part, by grants from the Public Welfare Projects, Department of Science, Zhejiang Province (2014C37003), China.
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 4.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 5.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kjaer SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst. 2010;102:1478–1488. doi: 10.1093/jnci/djq356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiffman M, Herrero R, Desalle R, Hildesheim A, Wacholder S, Rodriguez AC, Bratti MC, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Wright TC, Solomon D, Chen Z, Schussler J, Castle PE, Burk RD. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 9.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 10.Demers GW, Halbert CL, Galloway DA. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology. 1994;198:169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- 11.Gage JR, Meyers C, Wettstein FO. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J Virol. 1990;64:723–730. doi: 10.1128/jvi.64.2.723-730.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones DL, Alani RM, Munger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997;11:2101–2111. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- 15.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lechner MS, Laimins LA. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol. 1994;68:4262–4273. doi: 10.1128/jvi.68.7.4262-4273.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiMaio D, Mattoon D. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene. 2001;20:7866–7873. doi: 10.1038/sj.onc.1204915. [DOI] [PubMed] [Google Scholar]
- 19.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gene Ontology Consortium. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones C. Cervical cancer: is herpes simplex virus type II a cofactor? Clin Microbiol Rev. 1995;8:549–556. doi: 10.1128/cmr.8.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 26.Zelmanowicz A, Hildesheim A. Family history of cancer as a risk factor for cervical carcinoma: a review of the literature. Papillomavirus Report. 2004;15:113–120. [Google Scholar]
- 27.Reshmi G, Surya R, Jissa VT, Babu PS, Preethi NR, Santhi WS, Jayaprakash PG, Pillai MR. C-T variant in a miRNA target site of BCL2 is associated with increased risk of human papilloma virus related cervical cancer--an in silico approach. Genomics. 2011;98:189–193. doi: 10.1016/j.ygeno.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Wu HH, Liu YF, Yang SF, Lin WL, Chen SC, Han CP, Wang HL, Lin LY, Wang PH. Association of single-nucleotide polymorphisms of highmobility group box 1 with susceptibility and clinicopathological characteristics of uterine cervical neoplasia in Taiwanese women. Tumour Biol. 2016 doi: 10.1007/s13277-016-5408-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Sousa H, Oliveira S, Santos AM, Catarino R, Moutinho J, Medeiros R. Tumour necrosis factor alpha 308 G/A is a risk marker for the progression from high-grade lesions to invasive cervical cancer. Tumour Biol. 2014;35:2561–2564. doi: 10.1007/s13277-013-1337-3. [DOI] [PubMed] [Google Scholar]
- 30.Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, Guadarrama AS, Wright AA, Rosenberg MW, Duke F, Kaplan B, Wang R, Nickerson E, Walline HM, Lawrence MS, Stewart C, Carter SL, McKenna A, Rodriguez-Sanchez IP, Espinosa-Castilla M, Woie K, Bjorge L, Wik E, Halle MK, Hoivik EA, Krakstad C, Gabino NB, Gomez-Macias GS, Valdez-Chapa LD, Garza-Rodriguez ML, Maytorena G, Vazquez J, Rodea C, Cravioto A, Cortes ML, Greulich H, Crum CP, Neuberg DS, Hidalgo-Miranda A, Escareno CR, Akslen LA, Carey TE, Vintermyr OK, Gabriel SB, Barrera-Saldana HA, Melendez-Zajgla J, Getz G, Salvesen HB, Meyerson M. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng G, Dan W, Jun W, Junjun Y, Tong R, Baoli Z, Yang X. Transcriptome profiling of the cancer and adjacent nontumor tissues from cervical squamous cell carcinoma patients by RNA sequencing. Tumour Biol. 2015;36:3309–3317. doi: 10.1007/s13277-014-2963-0. [DOI] [PubMed] [Google Scholar]
- 32.Safari A, Seifoleslami M, Yahaghi E, Sedaghati F, Khameneie MK. Upregulation of miR-20a and miR-10a expression levels act as potential biomarkers of aggressive progression and poor prognosis in cervical cancer. Tumour Biol. 2015 doi: 10.1007/s13277-015-4064-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Jing L, Yuan W, Ruofan D, Jinjin Y, Haifeng Q. HOTAIR enhanced aggressive biological behaviors and induced radio-resistance via inhibiting p21 in cervical cancer. Tumour Biol. 2015;36:3611–3619. doi: 10.1007/s13277-014-2998-2. [DOI] [PubMed] [Google Scholar]
- 34.Reshmi G, Chandra SS, Babu VJ, Babu PS, Santhi WS, Ramachandran S, Lakshmi S, Nair AS, Pillai MR. Identification and analysis of novel microRNAs from fragile sites of human cervical cancer: computational and experimental approach. Genomics. 2011;97:333–340. doi: 10.1016/j.ygeno.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Punt S, Corver WE, van der Zeeuw SA, Kielbasa SM, Osse EM, Buermans HP, de Kroon CD, Jordanova ES, Gorter A. Whole-transcriptome analysis of flow-sorted cervical cancer samples reveals that B cell expressed TCL1A is correlated with improved survival. Oncotarget. 2015;6:38681–38694. doi: 10.18632/oncotarget.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motta PM. Ultrastructure of smooth muscle. Boston: Kluwer Academic Publishers; 1990. [Google Scholar]
- 37.Henneke G, Koundrioukoff S, Hubscher U. Multiple roles for kinases in DNA replication. EMBO Rep. 2003;4:252–256. doi: 10.1038/sj.embor.embor774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 40.van Dam PA, van Dam PJ, Rolfo C, Giallombardo M, van Berckelaer C, Trinh XB, Altintas S, Huizing M, Papadimitriou K, Tjalma WA, van Laere S. In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget. 2016;7:2780–2795. doi: 10.18632/oncotarget.6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, Chen Q, Brandwein-Gensler M, Prystowsky MB, Childs G, Smith RV, Belbin TJ. Gene expression profiles in HPVinfected head and neck cancer. J Pathol. 2007;213:283–293. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 42.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci U S A. 2009;106:20240–20245. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thu YM, Bielinsky AK. Enigmatic roles of Mcm10 in DNA replication. Trends Biochem Sci. 2013;38:184–194. doi: 10.1016/j.tibs.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]