

Abstract
In this paper, we describe developments in adaptive design methodology and discuss implementation strategies and operational challenges in early phase adaptive clinical trials. The BATTLE trial – the first completed, biomarker-based, Bayesian adaptive randomized study in lung cancer – is presented as a case study to illustrate main ideas and share learnings.
Keywords: Adaptive design, early phase designs, clinical trial management, data monitoring committee, modeling and simulation
Introduction
The number of publications describing adaptive and flexible designs has increased significantly in recent years, and the adoption of many of these designs is accelerating rapidly. The Bayesian approach is becoming more popular in clinical trial design to monitor efficacy and toxicity simultaneously, and in data analysis, due to its flexibility and ease of interpretation (1-4). High-performance computers have facilitated widespread advances in the development of computational algorithms, statistical modeling, and simulations. These advancements have enhanced the use of Bayesian and hybrid designs in clinical trials, specifically in early phase trial development. Two Bayesian Phase II adaptive trials that gained public and media attention focused clinical development on pairing oncology therapies and biomarkers. These trials are the Biomarker-integrated Approaches of targeted Therapy for Lung Cancer Elimination (BATTLE) (5, 6) and I-SPY 2 trial which takes its name from the phrase “Investigation of Serial studies to Predict Your Therapeutic Response with Imaging And moLecular analysis” (7).
Execution of adaptive designs at the operational level adds complexities and can be challenging, especially in studies involving multiple drugs, doses, biomarkers and populations, as in the BATTLE and I-SPY 2 trial. The additional time necessary for upfront planning and cross-functional coordination means that various stakeholders should be involved in the early planning of adaptive studies. Information on the risks and benefits of applying adaptive designs should be provided to all operational staff supporting the trial. Detailed statistical design simulations and operational simulation models are required for study planning to evaluate operating characteristics of the design under a range of assumptions and to ensure effective execution. At the planning stage, various factors should be considered, including the randomization scheme, recruitment rate, treatment duration, timing of treatment read-outs, endpoints, planned patient and site enrollment, likely drop-outs, study drug formulation, route of drug administration, and drug supply. Timely data capture is an important enabler for adaptive designs. Electronic Data Capture (EDC) should be used for studies with adaptive designs, especially for those with decision-critical data. The quality of data, effective data flow, and transfer processes should be discussed and pre-planned prior to interim analyses, which can introduce statistical and operational biases due to the feedback of their results. To minimize operational bias, interim analyses are performed and reviewed by an Independent Statistical Centre (ISC) – often a Contract Research Organization (CRO) – and a Data Monitoring Committee (DMC) convened by the sponsor, but independent of the sponsor in terms of financial and professional interests. It is critical that the ISC have extensive experience in performing interim analyses, appropriate firewalls and Standard Operating Procedures (SOP) that guard the unblinded processes. The team should include a designated data analysis center biostatistician with expertise in adaptive designs, to perform necessary adaptations and analysis and serve as a link between the study sponsor and the DMC. Several recent publications discuss the elements that are important for successful execution of adaptive designs in general (9-10).
In this paper, we describe developments in adaptive design methodology and further discuss the implementation strategy and operational challenges in early phase adaptive clinical trials. The BATTLE trial is presented as a case study to illustrate main ideas and share learnings.
Developments in Adaptive Design Methodology
An adaptive design is defined as “a multistage study design that uses accumulating data to decide how to modify aspects of the study without undermining the validity and integrity of the trial” (11). Maintaining study validity requires correct statistical inference and minimal operational bias, and maintaining study integrity means providing convincing results, preplanning, and maintaining blinded interim analysis results. Trial procedures that may be modified potentially include eligibility criteria, study dose or regimen, treatment duration, study endpoints, laboratory testing procedures, diagnostic procedures, criteria for evaluation and assessment of clinical responses. Statistical procedures might include randomization, number of treatment arms, study design, study hypotheses, sample size, data monitoring and interim analysis rules, statistical analysis plan and/or methods for data analysis
However, flexibility does not mean that the trial can be modified any time. Modifications and adaptations must be pre-planned and based on data collected during the course of the study. Accordingly, the U.S. Food and Drug Administration's draft guidance for industry on adaptive design clinical trials (8) defines such a trial as “a study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (usually interim data) from subjects in the study.” Analyses of the accumulating study data are performed at pre-planned time points, with or without formal statistical hypothesis testing. Ad hoc, unplanned adaptations may increase the chance of misuse or abuse of an adaptive design trial and should therefore be avoided. Operational teams must have a general understanding of adaptive design methods to proceed to the planning and design stage. The adaptive design methods commonly used in early phase trials include:
1) Adaptive Randomization Designs
Here, alterations in the randomization schedule are allowed depending upon the varied or unequal probabilities of treatment assignment. Adaptive randomization categories include restricted randomization, covariate-adaptive randomization, response-adaptive (or outcome-adaptive) randomization, and covariate-adjusted response-adaptive randomization (54). Restricted randomization procedures are preferred for many clinical trials because it is often desirable to allocate equal number of patients to each treatment. This is usually achieved by changing the probability of randomization to a treatment according to the number of patients that have already been assigned. Examples of restricted randomization procedures include the random allocation rule, the truncated binomial design, Efron's biased coin design, and Wei's urn design (12). Covariate-adaptive randomization is used to ensure the balance between treatments with respect to certain known covariates. Several methods exist to accomplish this, including Zelen's rule, the Pocock-Simon procedure, and Wei's marginal urn design. Response-adaptive randomization is used when ethical considerations make it undesirable to have an equal number of patients assigned to each treatment. Adaptive assessment is made sequentially, updating the randomization for the next single patient or a cohort of patients using treatment estimates calculated from all available patient data received so far. Covariate-adjusted response-adaptive randomization combines covariate-adaptive and response-adaptive randomization. These methods are reviewed by Hu and Rosenberger (54) and more recently, by Rosenberger et al. (13).
The Bayesian probit model described by Albert and Chib (57) is used to define the response for each treatment by marker group in the BATTLE trial. With this model, all cumulative data are used in computing the posterior of the response variable and in determining the randomization ratio. More on Bayesian adaptive randomization can be found in Berry and Eick (22), Thall and Wathen (23), Wathen and Cook (24), and Berry et al. (21). Wathen and Cook (24) summarize extensive simulations and give recommendations for the implementation of Bayesian adaptive randomization.
2) Adaptive Group Sequential Designs
Here, a trial can be stopped prematurely due to efficacy or futility at the interim analysis. The total number of stages (the number of interim analyses plus a final analysis) and stopping criterion to reject or accept the null hypothesis at each interim stage is defined, in addition to critical data values and sample size estimates for each planned interim stage of the trial. At each interim stage, all the data are collected up to the interim data cutoff time point. Data are then analyzed to confirm whether the trial should be stopped or continued. Staged interim analyses are preplanned during the course of the trial and must be carefully managed by the operational teams. The opportunity to stop the trial early and claim efficacy increases the probability of an erroneous conclusion regarding the new treatment (Type I error). For this reason, it is important to choose the significance levels for interim and final analyses carefully so that the overall Type I error rate is controlled at the pre-specified level. The stopping rules can be based on rejection boundaries, on a conditional power, or on a predictive power/predictive probability in a Bayesian setting. The boundaries determine how conclusions will be drawn following the interim and final analyses, and it is important to pre-specify which type of boundary and spending function (if applicable) will be employed. The conditional power approach is based on an appealing idea of predicting the likelihood of a statistically significant outcome at the end of the trial, given the data observed at the interim and some assumption of the treatment effect. If the conditional power is extremely low, it is wise to stop the trial early for both ethical and financial reasons. While it is possible to stop the trial and claim efficacy if the conditional power is extremely high, the conditional power is mostly used to conclude futility. More details on sequential designs can be found in Ghosh and Sen (14), Jennison and Turnbull (15), and Proschan, Lan and Wittes (16). More information on Bayesian sequential stopping rules can be found in Thall et al. (42, 43), Lee and Liu (44), and Berry et. al (21).
3) Adaptive Dose Ranging Designs
Insufficient exploration of a dose-response relationship often leads to a poor choice of the optimal dose used in the confirmatory trial, and may subsequently lead to the failure of the trial and the clinical program. Understanding of a dose-response relationship with regard to efficacy and safety prior to entering the confirmatory stage is a necessary step in drug development. During an early development phase, limited knowledge about the compound opens more opportunities for adaptive design consideration. Adaptive dose-finding designs allow fuller and more efficient characterization of the dose response by facilitating iterative learning and decision-making during the trial.
Adaptive dose-ranging designs can have several objectives. For example, they can be used to establish the overall dose-response relationship for an efficacy parameter or efficacy and safety parameters, estimate the therapeutic window, or help with the selection of a single target dose. The allocation of subjects to the dose currently believed to give best results, or to doses close to the best one, has become very popular in clinical dose-finding studies – for example, when the intention is to identify the maximum tolerated dose (MTD), the minimum efficacious dose (MED), or the most efficacious dose. Examples are cited by Wetherill (26), Lai and Robbins (27), Q'Quigley, Pepe and Fisher (17), Li, Durham and Flournoy (28), and Thall and Cook (19). This type of design – dubbed ‘best intention’ designs – are promoted as being ethically sound, and as taking into account the interests of the subjects. However, doubts about the convergence and informativeness of best intention designs were raised long ago, and cases were found in which such designs led to allocations converging to the wrong point. Examples are provided by Azriel (29), Mandel and Rinott (25), Bozin and Zarr (30), Chang and Ying (31), Ghosh, Mukhopadhyay and Sen (32), Lai and Robbins (33), Oron, Azriel and Hoff (34), and Pronzato (18). The major remedy to ensure convergence and increase informativeness of best intention designs is to introduce the intentional variability of allocations around the dose that is currently viewed as being the best. More rigorous approaches are based on the introduction of utility functions, which quantify the “effectiveness” of a particular dose, and penalty functions, which quantify potential harm due to exposure to toxic or non-efficacious doses. In the case of multiple responses (for instance, in which efficacy and different types of adverse events are of interest and can be measured, see Figure 1), examples are provided by Gooley, et al. (35), Li, Durham, and Flournoy (28), Fan and Chaloner (36), Rabie and Flournoy (37), Thall and Cook (19), and Dragalin and Fedorov (38). Various utility functions can be found in Gooley et al. (35), Thall (39), Dragalin, Fedorov and Wu (40), and Fedorov, Wu and Zhang (41). Examples of designs and references for the Bayesian setting can be found in Berry (55, 56), Thall and Cook (19), and Berry, Carlin, Lee and Muller (21).
Figure 1.

Applications of adaptive dose-ranging designs can be particularly relevant for specific indications. For example, an oncology study might use an adaptive approach to find a MTD during Phase I trials. Conventional Phase I designs are algorithm-based, and the widely used standard Phase I design uses the “3+3 design.” Lin and Shih (45) discussed the properties of the traditional (3+3) and modified algorithm-based designs in a general setting (A+B). Although the 3+3 method has been criticized for its tendency to include too many patients at suboptimal dose levels and to give an inaccurate estimate of the MTD, it is still widely used in practice because of its ease of implementation for clinical investigators. Other algorithm-based designs include accelerated titration designs and group up-and-down designs. The most widely used model-based approaches include the Continual Reassessment Method (CRM) proposed by O'Quigley, Pepe and Fisher (17) and the Escalation With Overdose Control (EWOC) method proposed by Babb, Rogatko and Zacks (46). More details on these and other dose-finding methods used in cancer research are described by Chevret (20). Recent extensions of the CRM are discussed by Cheung (47).
4) Biomarker Adaptive Designs
This type of design allows for adaptation using biomarker information. Modifications can be made to an ongoing trial based on the response of a biomarker that can predict a primary endpoint outcome, or one that helps select or change a treatment (e.g., BATTLE, I-SPY 2). Biomarkers can be used to select a subpopulation with an enhanced benefit from the study treatment. Wang et al. (48) describe approaches to evaluation of treatment effect in randomized trials with a genomic subset of the population. Designs that can be used to perform the subgroup search and identifications based on biomarkers are discussed in Lipkovich et al (49) and Lipkovich and Dmitrienko (50). Stallard (51) describes a seamless Phase II/III design based on a selection using a short-term endpoint; Jenkins et al. (52) present an adaptive seamless Phase II/III design with subpopulation selection using correlated endpoints, and Friede et al. (53) introduce a conditional error function approach for subgroup selection. Statistical designs that are used to screen biomarkers, validate biomarkers, and enrich the study population based on a biomarker or several biomarkers are of great interest to our industry and society. It should be kept in mind that there is still a gap in clinical development between identifying biomarkers associated with clinical outcomes and establishing a predictive model between relevant biomarkers and clinical outcomes.
Implementation Strategy and Operational Challenges
The use of adaptive clinical trial designs for a drug development program has clear advantages over traditional methods, given the ability to identify optimal clinical benefits and make informed decisions regarding safety and efficacy earlier in the clinical trial process. However, operational execution can be challenging given the additional complexities of implementing adaptive designs. These complexities deserve additional attention. Key operational challenges occur in several areas: the availability of statistical simulation tools for clinical trial modeling at the planning stages; the use of trial simulation modeling approaches to ensure the trial is meeting expected outcomes; and challenges regarding rapid data collection, clinical monitoring, resourcing, minimization of data leakage, IVRS, drug supply management, and systems integration. This section highlights several operational challenges that must be taken into consideration in conducting an adaptive clinical trial.
Planning and Design
The planning stages for an adaptive clinical trial must be completed prior to finalizing the decision to proceed. Adaptive designs should be considered only if they add benefit to the overall drug development process, allow for effective operational implementation, and provide efficiency gains, thus ensuring increased probability of success for a given compound. Adaptive designs are not a one-size-fits-all approach and should be carefully considered prior to implementation. Adequate planning can take 3-12 months, depending on clinical trial complexities. We recommend that the planning stage consist of three components – statistical design simulations, and operational simulation, followed by systems integration approaches – to ensure that all specified design requirements can be executed at the operational level.
The planning and design phase requires cross-functional collaboration and should include areas such as clinical research, biostatistics, pharmacology, regulatory, and clinical operations. Planning and executing an adaptive design study challenges the traditional approach to clinical trial conduct and requires a fully integrated team, nontraditional resourcing, and integrated informatics approaches.
Clinical Trial Modeling and Simulation
An important aspect of the planning process prior to finalizing the design is to complete the appropriate clinical trial simulations to optimize an individual trial and assess relative impact on overall development. Clinical trial simulation is a key step in evaluating potential clinical outcomes using various design scenarios and clinical trial assumptions to validate the design, ensuring effective execution at the operational level. Simulation models are used to predict the relationships between certain inputs, such as patient recruitment, dosing arms, clinical event rates (such as endpoints, Adverse Events (AEs), and Serious Adverse Events (SAEs)), sample size, interim analysis time points, and other inputs that must occur within the study domain. Simulation tools can also be used to monitor clinical trial outcomes during the course of the study, within trial simulation, to ensure that the study is meeting expectations. Clinical trial simulations utilize computer programs to mimic actual conduct of the trial in a virtual capacity, and can be used to reforecast predicted outcomes; simulations might also include an analysis of project cost, and cost management.
Patient Recruitment
Patient recruitment rates are a critical design element, as the rate of randomization dictates the rate at which treatment data can be collected and analyzed, allowing for appropriate decision making. Recruitment rates are specific to the therapeutic area, indication, protocol requirements, and standards of care for the country in which the study is conducted. Initial patient recruitment assumptions should utilize reliable data sources from historical trials, estimations using data mining techniques, data derived from full feasibility assessments, or a combination of the above. The rate at which patients are recruited determines the treatment data capture rates required for statistical analysis and decision making. As a result, the rate at which the trial recruits must compliment the desired adaptive design: faster is not necessarily better. Instead, recruitment rates must be optimized to meet the desired preplanned analysis within the specified time period. As one example, for dose response designs, slower recruitment is preferable so that a dose adjustment can effectively be implemented for the next patient or the group of patients. Recruiting too quickly may not allow effective dose adjustment to occur during the specified randomization period. Optimizing recruitment rates based on the unique design requirements has a positive impact on the quality, length, and cost of the clinical trial. The speed of randomization also has a direct impact on key operational components – all of which need to be simulated during the planning stages – such as total number sites required for study conduct, the rate of study start-up, site initiations, drug packaging and supply chain management.
Treatment Data and Data Collection
Careful consideration should be given to the types of data used for an adaptive design and the method for data collection. Preplanned statistical analysis must include a detailed assessment of all data that are required to perform an adaptation, in addition to when the data will be available and how they will be collected. Adaptive designs are better suited to the use of early outcome measurements as opposed to delayed ones. Early measures of clinical endpoints, biomarkers, or other efficacy endpoints allow for revised dosing allocations (response-adaptive designs), adaptive randomization (based on specific biomarkers), or other forms of design adaptations. Case report forms should focus on collection of key safety and efficacy data, and not on the collection of non-essential data elements, which can significantly increase trial costs and drive operational inefficiencies in an already complex study design.
Consideration must be given to those data elements that require cleaning rather than full source document verification, as source verification impacts the speed at which data can be utilized for decision making and increases operational complexity and cost. EDC systems are widely used today, which speed the data collection and cleaning process. However, fully integrated clinical trial platforms – allowing for accelerated data capture, remote data monitoring and cleaning, seamless data transfer, and statistical analysis for DMC decision-making – are not yet mainstream. As a result, clinical systems need to be tightly integrated to manage the complexities of an adaptive trial, ensuring minimization of data leakage and protection of the data and preserving blinded trial status. As technology improves, it is conceivable that informatics platforms will be available that allow for real time data capture, interoperability with EMRs, e-Source archives, reduced dependencies on clinical monitoring, and the provision of fully integrated statistical analysis tools used for decision making for adaptive trials. However, use of existing systems, along with integrated approaches, allow for conduct of an adaptive clinical trial, but require additional upfront planning time.
Centralized Remote Clinical Monitoring
A nontraditional clinical monitoring approach should be utilized for adaptive design trials, including a hybrid clinical monitoring approach consisting of Centralized Remote Monitoring in addition to on-site Source Data Verification (SDV). Centralized Remote Monitoring provides for continuous cleaning across key data elements in near real time, allowing for more immediate data transfers, statistical analysis, and decision making. The onsite clinical monitoring effort should take a risk-based monitoring approach, requiring minimal on site time and source verification only for key data elements. SDV activities will typically lag behind remote data cleaning, so it is important that decision-makers carefully consider those data elements that do not require SDV versus those that do. In general, most planned analyses that result in adaptations rely on data elements that do not require SDV. However, for some design elements, SDV may be required; in these cases, the timing of the data transfer and interim analysis must be carefully planned.
Risk based monitoring techniques using advanced analytics and signal detection methodologies can also improve data quality by highlighting potential quality issues that need to be addressed during the trial. Some risk based monitoring analytics utilize statistical analysis and variance around key risk areas that must be mitigated, such as AEs, SAEs, enrollment rates, protocol violations, and missing data.
In conclusion, nontraditional monitoring methods should be employed for adaptive clinical trials, taking account of data flow, timing of data entry, types of data collected, risk, data cleaning requirements and clinical resource allocations. This will enable study requirements to be met and decision making to be based on the specified design parameters.
IVRS and Drug Supply
Interactive Voice Randomization Systems (IVRS), used to manage patient randomization and assignment to treatment arms, must be fully integrated into the clinical trial's operational processes. Statistical analysis outputs used for an adaptive randomization or dose response designs are directly integrated into the IVRS, ensuring appropriate subject randomization. IVRS' must be tightly integrated with the EDC platform. A typical IVRS data set may contain the following for newly randomized subjects: Country, Site, Subject ID, Birth Date, Gender, Randomization Code, Randomization Date, Core Study or Sub Study, Enrollment Status, Drug Interruption(s), and Drug Re-start. Study coordinators will call the IVRS to notify the system of patient status, allowing data to be tracked in real time. This data is extremely valuable when managing patient enrollment and trial operations.
The IVRS must also integrate directly with the drug supply chain mechanism. Drug supply requirements need to be simulated during the planning stage as part of the Clinical Supply Optimization Process to ensure appropriate production, labeling, and inventory management. Clinical drug supply optimization parameters typically include: simulation and demand forecasting, regulatory strategy for submission and approvals, packaging and labeling strategy, distribution strategy, drug supply plan with trigger methodology, GMP/GDP regulatory review, IVRS specification requirements, and systems integration strategy.
Appropriate drug formulations, dosing regimens and routes of administration also need to be identified. For example, various dose levels can be produced by combining two or more tablets of specific doses. For intravenous drugs, varying dose levels can be achieved by requiring drug preparation to be conducted on site, using vials of equal volume dispensed in several dose strengths, and providing instructions as to how much should be removed from each vial to prepare a new dose.
Data Monitoring Committee
DMCs are an important component in adaptive design trials, proactively assessing the risk benefit of the treatment, often at several time points during the trial, and making recommendations for protocol changes based on adaptive rules specified in the protocol. In order to maintain trial integrity and minimize bias, an ISC should be utilized to prepare the data for DMC review and decision making. The DMC's charter outlines roles and responsibilities and summarizes statistical methods and necessary adaptations; the charter should be drafted at the planning stage and finalized prior to the first look/ interim analysis.
Clinical Trial Management and Communication
Given the increased operational complexities of an adaptive design, effective project management and communication is a critical component to success. Adaptive design project teams must work in a non-traditional environment, be tightly integrated, and have the proper resources, methods and tools to manage the clinical trial.
Cross functional collaboration is paramount when designing an adaptive design clinical trial, and the planning and design phase is a critical element that must be implemented to ensure success. How can the success of an adaptive clinical trial be measured? This is an important step, since a successful adaptive trial provides benefit to the overall drug development process, allows for effective implementation at the operational level, and provides efficiency gains from the standard model. In addition, we can use existing technologies to develop and operationalize an adaptive design trial, and can leverage common best practices across all unique designs.
In the next section, the BATTLE trial design is presented using some of the approaches introduced in this and previous sections. The trial employs the Bayesian adaptive design with a response adaptive randomization and a futility stopping rule.
A Case Study: Battle Trial
The Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial was the first prospective, biopsy-mandated, biomarker-based, adaptively randomized study in patients with heavily pre-treated lung cancer (5,6). This Phase II trial, involving patients with advanced non-small cell lung cancer (NSCLC), evaluated four treatments in individuals stratified into five biomarker subgroups.
From 2006 to 2009, 341 patients were enrolled in the BATTLE trial and 255 were randomized. Upon biopsy, tissue samples were analyzed for their biomarker profiles. Patients were assigned to one of the five biomarker groups according to the rank order of the estimated predictive value as follows: [1] EGFR mutation, amplification, or high polysomy; [2] KRAS or BRAF mutation; [3] VEGF or VEGFR-2 overexpression; [4] RXR α, β, or γ overexpression and/or Cyclin D1 overexpression/amplification; or [5] no study biomarkers. Following an initial equal randomization period (N=97), patients were adaptively randomized (N=158) to one of the four study arms: erlotinib, vandetanib, erlotinib plus bexarotene, or sorafenib, based on each patient's relevant molecular biomarkers.
The primary endpoint of the study is the 8-week disease control rate (DCR), which has been shown to be a good surrogate for the overall survival in this patient population (58). Based on the preliminary data, the 8-week DCR was considered to be 30% as the null hypothesis and 50% as the alternative hypothesis.
Statistical Methodology
The statistical design of the BATTLE trial was based on adaptive randomization using a Bayesian hierarchical model that would assign more patients into more effective treatments, with the randomization probability proportional to the observed efficacy based on patients' individual biomarker profiles (6). The primary endpoint was reported as a binary response: patients' disease is controlled (no disease progression) or not controlled at 8 weeks (i.e, disease progression or death within 8 weeks). The design adaptively randomized patients into one of the four treatments based on their biomarker profile and the cumulative response data.
Let γjk denote the current posterior mean probability of disease control for a patient in biomarker group k (k = 1… 5) under treatment j (j = 1… 4). The next patient in biomarker group k is assigned to treatment t with probability proportional to . The probabilities of the DCR are calculated with respect to a hierarchical probit model (57). The probit model is written in terms of a latent probit score zijk for patient i under treatment j in biomarker group k as zijk ∼ N(μijk, 1) for i = 1,…, njk where μjk ∼ N(ϕj, σ2) for k = 1, …, 5 and ϕj ∼ N(0, τ2) for j = 1, …, 4. The model assumes a hierarchical normal/normal model which allows borrowing strength across related subpopulations. The disease control status yijk = 1 if zijk > 0 and yijk = 0 if zijk ≤ 0. With normal distributions and conjugate priors, the posterior probability can be computed via Gibbs sampling from the following full conditional distributions:
An early futility stopping rule was added to the trial design. If the current data indicate that a treatment is unlikely to be beneficial to patients in a certain marker group, randomization to that treatment is suspended for these patients. Specifically, the target DCR is denoted by θ1 and the critical probability for early stopping (i.e., suspension of randomization due to futility) by θL. The trial will be suspended for treatment j and marker k if Pr(γjk ≥ θ1 | Data) ≤ θL; θ1 = 0.5 and θL = 0.1, respectively were chosen. Hence, randomization in a marker subgroup for a treatment is suspended when the probability of achieving a target DCR is unlikely based on the observed data. At the end of trial, the treatment will be considered a success if Pr(γjk ≥ θ0 | Data) > θU. In this study, we chose θ0 = 0.3 and θU = 0.8 to correspond to the probability of a treatment in a biomarker subgroup is very likely to be better than the null DCR. The trial design had no early stopping rule for effective treatments. If a treatment shows early signs of efficacy, more patients will continue to be enrolled to that treatment under the adaptive randomization scheme and the declaration of efficacy will occur at the end of the trial.
Patient Randomization
Prior to the adaptive randomization, patients were equally randomized to the four treatments to calibrate the model for calculating the randomization probability. The trial design also allowed the suspension of underperforming treatments in marker groups. At the end of the trial, a treatment is considered efficacious in a marker group if the posterior probability of the 8-week DCR is sufficiently high as described in the adaptive design methodology. The study schema is shown in Figure 2.
Figure 2. BATTLE trial design schema.

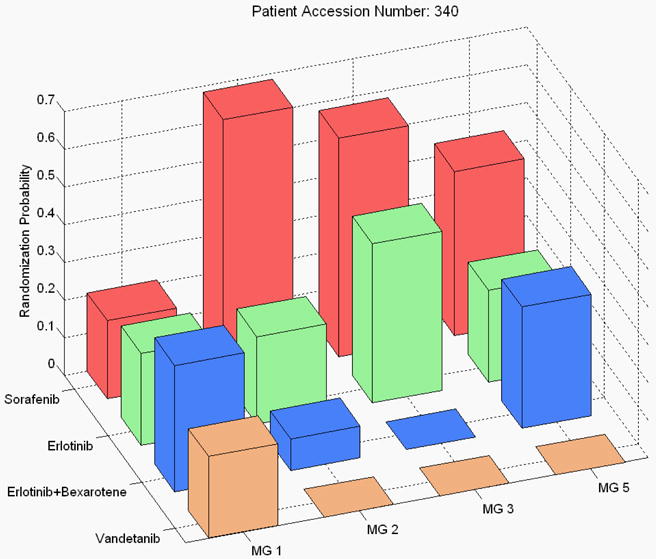
At the conclusion of the study, the overall 8-week DCR was 46%. The study confirmed several pre-specified hypotheses, such as that erlotinib worked well for patients with EGFR mutation, vandetanib worked well for patients with high VEGFR-2 expression, and erlotinib plus bexarotene worked well for patients with high Cyclin D1 expression. The study also generated some intriguing hypotheses, for example, with sorafenib showing efficacy among mutant-KRAS patients, a finding that will be investigated further in future trials. The distribution of the final randomization probability into the four treatments for the biomarker groups 1, 2, 3, and 5 is given in Figure 3. (Note that only 6 patients were in marker group 4, hence, marker group 4 is not shown.)
Figure 3. Probability of adaptive randomization by treatment and marker group in the BATTTLE trial.
A New Paradigm for Personalized Medicine
The BATTLE trial establishes a new paradigm for a personalized approach to treat lung cancer (59, 60). There are many valuable lessons to be learned from this pioneering trial. It has demonstrated that acquiring a fresh biopsy in recurrent lung cancer patients is feasible and important in determining the tumor biomarker profiles at the time of recurrence, to inform the search for the most effective treatment for each individual patient (61). Prospective tissue collection and biomarker analysis provide a wealth of information for future discovery work. The trial had a robust accrual, enrolling an average of 9.5 patients per month, and confirming that the outcome-adaptive randomization design is well received by both the clinical team and patients. Under the Bayesian hierarchical model, the treatment effect and predictive markers are efficiently assessed. This “learn-as-we-go” approach is appealing as it leverages accumulating patient data to improve the treatment outcome as the trial progresses.
Challenges and Learnings
The BATTLE trial faced challenges. To capture the biomarker data, patient eligibility, and efficacy and toxicity outcomes in real time, a web-based data management system had to be developed. This allowed remote data entry and performed data quality control by the built-in data type, value, and range checking. It also had automatic e-mail report generation and data download capability for monitoring the study conduct. For example, when the research nurse entered the patients' baseline information, this data was used for checking the patient's eligibility. The patient's biomarker profile was analyzed and directly entered into the web-based system in the molecular pathology laboratory within two weeks of the biopsy. Upon verifying the patient eligibility and completion of the biomarker profiling, the research nurse could perform the randomization using the randomization button in the web-based application. This called up an adaptive randomization code written in the open-source software, R, via web services. The R code could read the available data, perform Bayesian computation, and randomize patients accordingly. The randomization result was sent to pharmacy for dispensing of drugs. In addition to meeting the general database security requirements, the system also had a role-based security control feature in which each study collaborator had read/write privilege for the relevant data. Such a database system was essential for conducting the BATTLE trial and similar adaptive trials.
In the BATTLE study, 38% of the patients were equally randomized and the remaining 62% were adaptively randomized. In retrospect, the adaptive randomization could possibly have come into effect earlier. The trial design called for enrolling at least one patient in each treatment/marker group pair before adaptive randomization. Due to the fact that there were very few patients in biomarker group 4, the adaptive randomization was delayed. The initial equal randomization could have been replaced by using a fair prior with the proper effective sample size to control the percentage of patients equally randomized. More discussion of the use of outcome adaptive randomization can be found in Lee et al. (62).
Another drawback of the design is that the protocol pre-specified biomarkers and combined markers into groups in designing the BATTLE trial. It turned out that some markers (such as the retinoid X receptors (RXRs)) were not informative in predicting treatment outcome.
Furthermore, grouping markers for dimension reduction is not a good idea because markers in the same group have different predictive strength. For example, for predicting the DCR using erlotinib, EGFR mutation was the strongest predictor followed by the EGFR gene amplification. The EGFR protein expression had little predictive value. Grouping markers dilutes the predictive strength of some of the more important markers. Based on learnings from the BATTLE trial, the BATTLE-2 trial is currently underway using a two-stage design. Predictive markers are being identified in the first stage and applied in the second stage. An important lesson learned is that the outcome-adaptive randomization would benefit patients most if there were effective treatments and associated predictive markers. The study team is working to identify the most appropriate treatments and markers. Adaptive randomization is a sensible way to facilitate this process.
Another adaptive feature in the BATTLE trial was the suspension of randomization in the underperformed treatment/marker pairs. For example, toward the end of the trial, the data suggested that vandetanib did not work for the KRAS/BRAF mutation group and that neither vandetanib nor erlotinib plus bexarotene worked for the VEGF/VEGFR-2 overexpression group. Hence, the randomization probability was set to 0 (Figure 3). The suspension or early stopping rule can avoid assigning patients to ineffective treatments and redirect them to more effective ones. The Bayesian framework allows the information to be updated continuously throughout the trial. The updated posterior distribution can be used for guiding elements of the study conduct, such as the outcome-adaptive randomization, or early stopping due to futility or efficacy.
Although many Bayesian methods have been developed in clinical trials over the years, few have been used in practice. However, the use of Bayesian methods in clinical trials has recently increased substantially (4). Due to the inherent nature of continuously updated information, a Bayesian framework is ideal for adaptive clinical trial designs. The design parameters can be calibrated to control the most frequent type I and type II errors.
In conclusion, the BATTLE study was the first completed, biomarker-based, Bayesian adaptive randomized study in lung cancer. It is inspiring the development of similar adaptive trials (63-70). The real-time biopsy and biomarker profiling, coupled with adaptive randomization, have taken a substantial step toward realizing personalized lung cancer therapy. More such trials should be conducted to refine the design and conduct of adaptive trials in efficiently searching for effective treatments.
Discussion
The use of adaptive designs in the context of early development programs is particularly appealing to biopharmaceutical companies against the current backdrop of increasing competition to get quickly to market and dwindling resources. As discussed in this paper, adaptive designs allow more efficient use of information for decision making, which ultimately translates into improved probability of success and shorter overall time to market for successful products. Adaptive designs, in early and late development, also face additional challenges, from methodological, operational, and regulatory points of view. One of the appeals of early development adaptive designs is their greater acceptance by regulatory agencies. In fact, the FDA draft guidance (8) encourages sponsors to utilize adaptive designs in early development, to improve the efficiency of exploratory studies, as well as to gain experience with the use of adaptive approaches.
The value of adaptive designs as a transformational approach to improve the efficiency of drug development has long been recognized by industry, as well as by regulatory agencies. The 2004 FDA Critical Path Initiative (71) identified adaptive designs as one of the opportunities for solving the pipeline problem then (and still) facing the biopharmaceutical industry. As part of its Pharmaceutical Innovation initiative, the Pharmaceutical Research and Manufacturers of America (PhRMA) created working groups (WGs) in 2005 to evaluate and propose solutions to key drivers of poor performance in drug development. Two of the 10 WGs focused on adaptive designs: the Novel Adaptive Designs WG and the Adaptive Dose Ranging Studies (ADRS) WG. Both have had a significant impact in increasing awareness and acceptance of adaptive designs, across industry and in regulatory agencies around the world.
The ADRS WG focused on evaluating and making recommendations on the use of adaptive designs in the context of dose selection and dose response estimation. The WG conducted several simulation studies to compare different adaptive dose ranging approaches to one another and to conventional fixed designs for dose selection. The conclusions and recommendations from the ADRS WG were published in two impactful white papers (72,73), both of which include discussions from regulators from the FDA, European Medicines Agency (EMA), and Japan's Pharmaceuticals and Medical Devices Agency (PMDA). The key messages in these white papers – and endorsed by regulators – were that (i) poor dose selection and dose response estimation are important drivers of the high attrition rate currently observed in confirmatory studies; (ii) adaptive dose ranging and model-based methods can significantly improve the efficiency and accuracy of dose selection and estimation of dose response; and (iii) without greater investment by sponsors in additional studies (such as dose finding trials), no statistical design or method alone will be able to address the issues leading to today's pipeline shortages.
While some challenges remain with execution of adaptive designs at the operational level, clinical trial teams are learning and improving. Even the conduct of trials with traditional designs is challenging. By improving the systems and structures that allow for execution of adaptive designs, the overall processes of drug development can be improved. The extra effort to design and implement clinical trials using adaptive designs results in significant benefits to patients and improvements in quality, speed, and efficiency of clinical trials and overall drug development.
Contributor Information
Olga Marchenko, Center for Statistics in Drug Development, Innovation, Quintiles, Durham, NC.
Valerii Fedorov, Predictive Analytics, Innovation, Quintiles, Durham, NC.
J. Jack Lee, Department of Biostatistics, Division of Quantitative Sciences, University of Texas MD Anderson Cancer Center, Houston, TX.
Christy Nolan, Innovation, Quintiles, Durham, NC.
José Pinheiro, Quantitative Decision Strategies, Janssen Research & Development, LLC.
References
- 1.Ashby D. Bayesian Statistics in Medicine: A 25 year review. Statistics in Medicine. 2006;25(21):3589–3631. doi: 10.1002/sim.2672. [DOI] [PubMed] [Google Scholar]
- 2.Grieve AP. 25 years of Bayesian methods in the pharmaceutical industry: A personal, statistical bummel. Pharmaceutical Statistics. 2007;6(4):261–281. doi: 10.1002/pst.315. [DOI] [PubMed] [Google Scholar]
- 3.Chevret S. Bayesian adaptive clinical trials: A dream for statisticians only? Statistics in Medicine. 2012;31(11-12):1002–1013. doi: 10.1002/sim.4363. [DOI] [PubMed] [Google Scholar]
- 4.Lee JJ, Chu CT. Bayesian clinical trials in action. Statistics in Medicine. 2012;31(25):2955–2972. doi: 10.1002/sim.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, Liu S, Kim ES, Herbst RS, Lee JJ. Bayesian adaptive design for targeted therapy development in lung cancer – a step toward personalized medicine. Clinical Trials. 2008;5(3):463–467. doi: 10.1177/1740774508091815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim ES, Herbs RS, Wistuba II, et al. The BATTLE trial: Personalizing Therapy for Lung Cancer. Cancer Discovery. 2011;1(1):44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker AD, Sigman CC, Kelloff GJ, et al. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86(1):97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 8.FDA. Guidance for Industry 2010. Adaptive Design Clinical Trials for Drugs and Biologics. http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM201790.pdf.
- 9.Gaydos B, Anderson K, Berry D, et al. Good Practices for Adaptive Clinical Trials in Pharmaceutical Product Development. DIJ. 2009;43:539–556. [Google Scholar]
- 10.He W, Kuznetsova O, Harmer M, et al. Practical Considerations and Strategies for Executing Adaptive Clinical Trials. DIJ. 2012;46:160–174. [Google Scholar]
- 11.Dragalin V. Adaptive designs: terminology and classification. DIJ. 2006;40:425–435. [Google Scholar]
- 12.Rosenberger W, Lachin J. Randomization in Clinical Trials, Theory and Practice. Wiley; 2002. [Google Scholar]
- 13.Rosenberger W, Sverdlov O, Hu F. Adaptive randomization for clinical trials. Journal of Biopharmaceutical Statistics. 2012;22(4):719–736. doi: 10.1080/10543406.2012.676535. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh B, PK S. Handbook of Sequential Analysis. Marcel Dekker Inc; 1991. [Google Scholar]
- 15.Jennison C, Turnbull BW. Group Sequential Methods with Applications to Clinical Trials. CRC Press Inc; 2000. [Google Scholar]
- 16.Proshan MA, Lan KKG, Wittes JT. Statistical Monitoring of Clinical Trials – A Unified Approach. Springer; 2006. [Google Scholar]
- 17.O'Quigley J, Pepe M, Fisher L. Continual reassessment method: A practical design for Phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. [PubMed] [Google Scholar]
- 18.Pronzato L. Adaptive optimization and D-optimum experimental design. The Annals of Statistics. 2000;28(6):1743–1761. [Google Scholar]
- 19.Thall PF, Cook JD. Dose-finding based on efficacy? Toxicity trade-offs. Biometrics. 2004;60(3):684–693. doi: 10.1111/j.0006-341X.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- 20.Chevret S. Statistical Methods for Dose-Finding Experiments. John Wiley & Sons Ltd; 2006. [Google Scholar]
- 21.Berry D, Carlin L, Lee JJ, Muller P. Bayesian Adaptive Methods for Clinical Trials. CRC Press; 2011. [Google Scholar]
- 22.Berry D, Eick S. Adaptive assignment versus balanced randomization in clinical trials: a decision analysis. Statistics in Medicine. 1995;14:231–246. doi: 10.1002/sim.4780140302. [DOI] [PubMed] [Google Scholar]
- 23.Thall P, Wathen J. Practical Bayesian Adaptive Randomization in clinical trials. European Journal of Cancer. 2007;43:859–866. doi: 10.1016/j.ejca.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wathen J, Cook J. Technical report. Department of Biostatistics, M.D Anderson Cancer Center; 2006. Power and bias in adaptively randomized clinical trials. [Google Scholar]
- 25.Azriel D, Mandel M, Rinott Y. The treatment versus experimentation dilemma in dose finding studies. J Statist Plann Inference. 2011;141(8):2759–2768. [Google Scholar]
- 26.Wetherill G. Sequential estimation of quantal response curves. Royal Statistical Society B. 1963;25:1–48. [Google Scholar]
- 27.Lai TL, Robbins H. Adaptive design in regression and control. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:586–587. doi: 10.1073/pnas.75.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Durham SD, Flournoy N. An adaptive design for maximization of a contingent binary response: Adaptive Designs. In: Flournoy N, Rosenberger WF, editors. Institute of Mathematical Statistics. 1995. pp. 179–196. [Google Scholar]
- 29.Azriel D. A note on the robustness of the continual reassessment method. 2011 Preprint. [Google Scholar]
- 30.Bozin A, Zarrop M. Self-tuning extremum optimizer convergence and robustness. ECC. 1991;91:672–677. [Google Scholar]
- 31.Chang HH, Ying Z. Nonlinear sequential designs for logistic item response theory models with applications to computerized adaptive tests. The Annals of Statistics. 2009;37(3):1466–1488. [Google Scholar]
- 32.Ghosh M, Mukhopadhyay N, Sen PK. Sequential Estimation. John Wiley & Sons, Inc.; 1997. [Google Scholar]
- 33.Lai TL, Robbins H. Iterated least squares in multiperiod control. Advances in Applied Mathematics. 1982;3(1):50–73. [Google Scholar]
- 34.Oron AP, Azriel D, Hoff PD. Dose–finding designs: The role of convergence properties. The International Journal of Biostatistics. 7(1) doi: 10.2202/1557-4679.1298. in press. URL http://www.bepress.com/ijb/vol7/iss1/39. [DOI] [PubMed] [Google Scholar]
- 35.Gooley TA, Martin PJ, Lloyd DF, Pettinger M. Simulation as a design tool for phase i/ii clinical trials: An example from bone marrow transplantation. Controlled Clinical Trials. 1994;15:450–460. doi: 10.1016/0197-2456(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 36.Fan SK, Chaloner K. Optimal designs and limiting optimal designs for a trinomial response. Journal of Statistical Planning and Inference. 2004;126(1):347–360. [Google Scholar]
- 37.Rabie H, Flournoy N. In: mODa 7: Advances in Model-Oriented Design and Analysis, chap Optimal designs for contingent responses models. Di Bucchianico A, L, editors. Physical–Verlag; Heidelberg: 2004. pp. 133–142. [Google Scholar]
- 38.Dragalin V, Fedorov V. Adaptive designs for dose-finding based on efficacy-toxicity response. Journal of Statistical Planning and Inference. 2006;136:1800–1823. [Google Scholar]
- 39.Thall PF. Bayesian models and decision algorithms for complex early phase clinical trials. Statistical Science. 2010;25(2):227–244. doi: 10.1214/09-STS315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dragalin V, Fedorov V, Wu Y. Two–stage design for dose-finding that accounts for both efficacy and safety. Statistsics in Medicine. 2008;27:5156–5176. doi: 10.1002/sim.3356. [DOI] [PubMed] [Google Scholar]
- 41.Fedorov WYV, Zhang R. Optimal dose-finding designs with correlated continuous and discrete responses. Statistics in Medicine. 2012;31:217–234. doi: 10.1002/sim.4388. [DOI] [PubMed] [Google Scholar]
- 42.Thall P, Simon R. Practical Bayesian guidelines for Phase IIB clinical trials. Biometrics. 1994;50:337–349. [PubMed] [Google Scholar]
- 43.Thall P, Simon R, Estey E. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Statistics in Medicine. 1995;14:357–379. doi: 10.1002/sim.4780140404. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Liu D. A predictive probability design for phase II cancer clinical trials. Clinical Trials. 2008;5:93–106. doi: 10.1177/1740774508089279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin Y, Shih W. Statistical properties of the traditional algorithm-based designs for phase I cancer clinical trials. Biostatistics. 2001;2(2):203–215. doi: 10.1093/biostatistics/2.2.203. [DOI] [PubMed] [Google Scholar]
- 46.Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Statistics in Medicine. 1998;17:1103–1120. doi: 10.1002/(sici)1097-0258(19980530)17:10<1103::aid-sim793>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 47.Cheung YK. Dose Finding by the Continual Reassessment Method. New York: Chapman & Hall/CRC Press; 2011. [Google Scholar]
- 48.Wang S, O'Neill R, Hung H. Approaches to evaluation of treatment effect in randomized clinical trials with genomic subset. Pharmaceutical Statistics. 2007;6:227–244. doi: 10.1002/pst.300. [DOI] [PubMed] [Google Scholar]
- 49.Lipkovich I, Dmitrienko A, Denne J, Enas G. Subgroup identification based on differential effect search (SIDES): A recursive partitioning method for establishing response to treatment in patient subpopulations. Statistics in Medicine. 2011;30:2601–2621. doi: 10.1002/sim.4289. [DOI] [PubMed] [Google Scholar]
- 50.Lipkovich I, Dmitrienko A. Strategies for identifying predictive biomarkers and subgroupswith enhanced treatment effect in clinical trials using SIDES. Journal of Biopharmaceutical Statistics. doi: 10.1080/10543406.2013.856024. To appear. [DOI] [PubMed] [Google Scholar]
- 51.Stallard N. A confirmatory seamless phase II/III clinical trial design incorporating short-term endpoint information. Statistics in Medicine. 2010;29:959–971. doi: 10.1002/sim.3863. [DOI] [PubMed] [Google Scholar]
- 52.Jenkins M, Stone A, Jennison C. An adaptive seamless phase II/III design for oncology trials with subpopulation selection using correlated survival endpoints. Pharmaceutical Statistics. 2011;10:347–356. doi: 10.1002/pst.472. [DOI] [PubMed] [Google Scholar]
- 53.Friede T, Parsons N, Stallard N. A conditional error function approach for subgroup selection in adaptive clinical trials. Statistics in Medicine. 2012;31(30):4309–20. doi: 10.1002/sim.5541. [DOI] [PubMed] [Google Scholar]
- 54.Hu F, Rosenberger W. The Theory of Response-Adaptive Randomization in Clinical Trials. Wiley Inc; 2006. [DOI] [PubMed] [Google Scholar]
- 55.Berry D. Statistics: A Bayesian perspective. Duxbury; 1996. [Google Scholar]
- 56.Berry D. Bayesian clinical trials. Nature Reviews Drug Discovery. 2006;5:27–36. doi: 10.1038/nrd1927. [DOI] [PubMed] [Google Scholar]
- 57.Albert JH, Chib S. Bayesian analysis of binary and polychotomous response Data. Journal of the American Statistical Association. 1993;88:669–79. [Google Scholar]
- 58.Lara PN, Redman MW, Kelly KM, et al. Disease control rate at 8 weeks predicts clinical benefit in advanced non-small-cell lung cancer: Results from Southwest Oncology Group randomized trials. Journal of Clinical Oncology. 2008;26(3):463–467. doi: 10.1200/JCO.2007.13.0344. [DOI] [PubMed] [Google Scholar]
- 59.Sequist LV, Muzikansky A, Engelman JA. A new BATTLE in the evolving war on cancer. Cancer Discovery. 2011;1(1):14–16. doi: 10.1158/2159-8274.CD-11-0044. [DOI] [PubMed] [Google Scholar]
- 60.Rubin EH, Anderson KM, Gause CK. The BATTLE trial: A bold step toward improving the efficiency of biomarker-based drug development. Cancer Discovery. 2011;1(1):17–20. doi: 10.1158/2159-8274.CD-11-0036. [DOI] [PubMed] [Google Scholar]
- 61.Tam AL, Kim ES, Lee JJ, et al. Feasibility of image-guided transthoracic core-needle biopsy in the BATTLE lung trial. Journal of Thoracic Oncology. 2013 doi: 10.1097/JTO.0b013e318287c91e. Article in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JJ, Chen N, Yin G. Worth adapting? Revisiting the usefulness of outcome-adaptive randomization. Clinical Cancer Research. 2012;18(17):4498–4507. doi: 10.1158/1078-0432.CCR-11-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JJ, Gu X, Liu S. Bayesian adaptive randomization designs for targeted agent development. Clinical Trials. 2010;7(5):584–596. doi: 10.1177/1740774510373120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Printz C. BATTLE to personalize lung cancer treatment. Novel clinical trial design and tissue gathering procedures drive biomarker discovery. Cancer. 2010;116(14):3307–3308. doi: 10.1002/cncr.25493. [DOI] [PubMed] [Google Scholar]
- 65.Gold KA, Kim ES, Lee JJ, Wistuba II, Farhangfar CJ, Hong WK. The BATTLE to personalize lung cancer prevention through reverse migration. Cancer Prevention Research. 2011;4(7):962–972. doi: 10.1158/1940-6207.CAPR-11-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai TL, Lavori PW, Shih MCI, Sikic BI. Clinical trial designs for testing biomarker-based personalized therapies. Clinical Trials. 2012;9(2):141–154. doi: 10.1177/1740774512437252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rubin EH, Gilliland DG. Drug development and clinical trials - The path to an approved cancer drug. Nature Reviews Clinical Oncology. 2012;9(4):215–222. doi: 10.1038/nrclinonc.2012.22. [DOI] [PubMed] [Google Scholar]
- 68.Kelloff GJ, Sigman CC. Cancer biomarkers: Selecting the right drug for the right patient. Nature Reviews Drug Discovery. 2012;11(3):201–214. doi: 10.1038/nrd3651. [DOI] [PubMed] [Google Scholar]
- 69.Berry DA, Herbst RS, Rubin EH. Reports from the 2010 clinical and translational cancer research think tank meeting: Design strategies for personalized therapy trials. Clinical Cancer Research. 2012;18(3):638–644. doi: 10.1158/1078-0432.CCR-11-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allison M. Reinventing clinical trials. Nature Biotechnology. 2012;30(1):41–49. doi: 10.1038/nbt.2083. [DOI] [PubMed] [Google Scholar]
- 71.U.S.A Food and Drug Administration. Innovation/Stagnation: Challenge and opportunity on the critical path to new medical products. 2004 http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm.
- 72.Bornkamp B, Bretz F, Dmitrienko A, Enas G, Gaydos B, Hsu CH, Koenig F, Krams M, Liu Q, Neuenschwander B, Parke T, Pinheiro J, Roy A, Sax R, Shen F. Innovative approaches for designing and analyzing adaptive dose-ranging trials (with Discussion) Journal of Biopharmaceutical Statistics. 2007;17:965–995. doi: 10.1080/10543400701643848. [DOI] [PubMed] [Google Scholar]
- 73.Pinheiro J, Sax R, Antonijevic Z, Bornkamp B, Bretz F, Chuang-Stein C, Dragalin V, Fardipour P, Gallo P, Gillespie W, Hsu CH, Miller F, Padmanabhan SK, Patel N, Perevozskaya I, Roy A, Sanil A, Smith JR. Adaptive and Model-based Dose Ranging Trials: Quantitative Evaluation and Recommendations (with discussion) Statistics in Biopharmaceutical Research. 2010;2(4):435–454. [Google Scholar]