

Abstract
The molecular mechanisms of short-term plasticity observed during synaptic transmission are unknown. To determine whether the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins play a role in short-term plasticity, Botulinum toxins A, E, and F, were used to disrupt SNARE protein function in cultured hippocampal neurons. Although low concentrations of all of the toxins significantly reduced evoked release, they differentially affected short-term plasticity as assessed by the paired-pulse ratio, regardless of the initial release probability and size of the readily releasable pool of the synapse. The toxin effects on the paired-pulse ratio resulted in different phenotypes dependent on the toxin cleavage site. Together, these data indicate proteolysis of SNARE proteins alters facilitation and depression in a specific way.
Keywords: botulinum toxins, readily releasable pool, release probability, short-term plasticity
The molecular mechanisms of short-term plasticity and its molecular components are important to understanding information processing in the brain. Currently, three parameters provide useful information when studying synaptic modulation and are used to model synaptic modulation: (i) the initial release probability of the synapse (1–3), (ii) the size of the readily releasable pool (RRP) of the synapse (3), and (iii) the individual release probability of each individual vesicle (the α-factor) (4, 5). Because the initial release probability of a synapse can indicate the likelihood of a successful release in the second pulse of a paired stimulation protocol (1, 3), an indirect way to determine the release probability of a neuron is to measure the paired-pulse ratio (PPR) (6). Synapses that facilitate with a PPR greater than one exhibit a low initial release probability, whereas synapses that depress with a PPR of less than one represent cells with high initial release probability (6). Although these parameters have been used to predict facilitation and depression for model systems, initial release probability and the α-factor ultimately depend on the calcium concentration within the terminal (6). However, the action of calcium on these parameters has not provided insight about the molecular mechanisms of facilitation and depression.
This study sought to answer the questions: What is the molecular mechanism that controls the modulation of facilitation and depression? Does the N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex play a role in the molecular mechanism of modulation of facilitation and depression, independent of the parameters of initial release probability, size of the RRP, and α-factor? To address these issues about the mechanisms of facilitation and depression, SNARE proteins were modified. To determine how modification of these complexes alters paired-pulse facilitation (PPF) and paired-pulse depression (PPD), the present study used clostridial toxins [Botulinum toxins (Botox) A, E, and F] that have known specific protease activity on SNARE proteins (7).
Despite reductions in individual synapse's initial release probability and size of the RRP, with no change in the α-factor, each toxin treatment differentially affected the PPRs, independent of external calcium concentration. These results suggest that, in the presence of clostridial toxins, synaptic plasticity is not solely determined by initial release probability, RRP, or calcium levels, but rather by the relative contribution of various SNARE complexes.
Materials and Methods
Neuronal Cell Cultures. Primary cultures of hippocampal neurons from P0 rat pups were prepared as described (8) with the following changes: Neurobasal A medium with B27 and glutamine were used according to the manufacturer's instructions (Invitrogen). Botox A, (Sigma), Botox F (Calbiochem), and Botox E (Wako) were added to cultures in varying concentrations for 20–28 h before electrophysiology or imaging experiments were carried out. Botox E was activated by trypsin nicking according to the manufacturer's instructions.
Electrophysiology. Whole-cell patch clamp recordings were made from 11- to 14-day-old monosynaptic excitatory CA1 autapses (8) by using an Axopatch 200A amplifier. Borosilicate patch pipettes of 4–6 MΩ contained 130 mM K gluconate, 10 mM KCl, 4 mM NaCl, 1 mM EGTA, 2 mM MgCl2, 10 mM Hepes, 4 mM MgATP, 0.3 mM Tris-GTP, and 14 mM phosphocreatine. Final pH was 7.35, and the osmolarity was 295 mOsm. Unless noted, all electrophysiology experiments were carried out in an external medium containing 137 mM NaCl, 5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, and 10 mM glucose. Final pH was 7.35 and the osmolarity was adjusted to 315 mOsm by adding sorbitol. All chemicals were from Sigma.
Cells were first current-clamped to determine their passive membrane properties and action potential firing properties. Cells that gave voltage depolarization to +55 mV and had an access resistance of <20 mOhm were used for experiments. The membrane potential of the cells were then voltage clamped to -70 mV. Synaptic responses were elicited by giving a 1-ms step pulse to depolarize the cells. For experiments involving paired pulses, two 1-ms depolarizing step pulses were given 50 ms apart, with a 15-s interval between paired pulses. To ensure that the contamination of the action current were not the major source of signal, cells that had been toxin treated that gave no excitatory postsynaptic current (EPSC) response were averaged to determine the charge integral of the autaptic contamination. This average number was then checked against all of the EPSCs, and the EPSCs were corrected for this autaptic action current contamination.
During experiments aimed at determining the total amount of asynchronous release, a 1-ms step depolarizing pulse was given, and data were collected for 1.5 s after the pulse. The pulse was then given every 15 s, and data were collected again.
Data were acquired by using a custom-made program written in labview by Lee Campbell (The Salk Institute). The data were acquired at 10 kHz and filtered by using a low-pass Bessel filter set at 2 kHz. EPSC charge integrals were analyzed by using a custom written program in matlab (Mathworks, Natick, MA).
Values of the experiments are given as the mean ± SEM. Cumulative distributions were compared with the Kolmogorov–Smirnoff test, and the means were compared with a paired Student's t test.
To compare the percentage change in the PPR response between the different toxin treatments, each individual toxin-treated cell's PPR was divided by its respective average control response of its sister cultures. A one-way ANOVA and a Tukey test were used to compare the different means of the PPRs between the different toxin treatments. To determine the PPRs, the EPSC charge integral of the second pulse was divided by the EPSC charge integral of the first pulse: PPR = EPSC P2/EPSC P1.
To determine whether toxin treatments affected synchronous and asynchronous release in a similar manner, the number of individual asynchronous release between control cells and various treatments were counted and EPSC charge integrals were measured. Individual asynchronous events were counted in a manner similar to previous studies (9, 10) using the mini analysis program (version 5.5.5; Synaptosoft, Decatur, GA) for 1 s after the 1-ms depolarizing pulse. The EPSC charge integrals were measured by integrating for 150 ms after the onset of the EPSC for each condition and were plotted as means. The ratio of the mean EPSC charge integral was determined and is defined as the EPSC charge integral (experimental)/EPSC charge integral (control). This ratio of the synchronous treatments was used to test the assumption that the various treatments affected the synchronous release and the asynchronous release in the same manner. To determine whether they were affected similarly, the ratios of the EPSC charge integrals were multiplied by the total amount of asynchronous events individually counted in the control to give a predicted number of asynchronous events. If the asynchronous release and the synchronous release were affected in the same manner by the toxin treatment, than the predicted number of events should equal the number of experimentally determined events: Predicted asynchronous events = EPSC charge integral mean (experimental)/EPSC charge integral mean (control) × asynchronous total (control).
This predicted number was then compared against the actual number of asynchronous events found in the experimental conditions by using a Student's t test.
FM1-43 Loading and Destaining. For the imaging experiments measuring the RRP, the cells were bathed in the extracellular medium containing 136 mM NaCl, 2.5 mM KCl, 10 mM glucose, 10 mM Hepes, 2 mM CaCl2, and 1.3 mM MgCl2. For imaging experiments measuring the release probability, cells were bathed in medium containing 137 mM NaCl, 5 mM KCl, 10 mM Hepes, 10 mM glucose, 5 mM CaCl2, and 1 mM MgCl2. All solutions were 315 mOsm and had a pH of 7.4 and contained NMDA and non-NMDA receptor antagonists (10 μM NBQX and 50 μM DL-APV) to block recurrent activity. All chemicals were from Sigma.
Release probability and the size of the RRP were estimated as described (11, 12) and conventional neuronal cultures that were 21–24 days in vitro were used. For detailed methods, see supporting information, which is published on the PNAS web site.
To determine the α-factor, the experimentally derived initial release probabilities were divided by the experimentally derived RRP: α-factor = RP/RRP
Values of the experiments are given as the mean ± SEM. Cumulative distributions are compared with the Kolmogorov-Smirnoff test, and the means are compared with a paired two-tailed Student's t test.
Results
Submaximal Concentrations of Botox A Causes PPF. Although at saturating doses of toxin, synaptic transmission is blocked by Botox A and F (Fig. 8, which is published as supporting information on the PNAS web site), it is known that submaximal concentrations of Botox A can cause activity-dependent facilitation (13). To test how submaximal doses of Botox A (60 pM) affect short-term plasticity, paired-pulses were applied to toxin-treated autaptic cells that had ≈30% of evoked transmission remaining (Fig. 8). In control recordings, paired EPSCs at 50-ms interstimulus intervals exhibited no facilitation (0.95 ± 0.03 SEM; n = 5). However, EPSCs recorded from Botox A-treated cultures showed robust PPF (1.4 ± 0.12 SEM; n = 11) (Fig. 1 A and B) that was an ≈45% increase in the PPR (see Fig. 6). This result confirms previous observations (13) that partial cleavage of SNAP 25 at amino acid 197 by submaximal doses of Botox A causes synaptic transmission to undergo PPF.
Fig. 1.

Botox A treatment causes PPF, whereas Botox F treatment does not. (A) Representative traces of untreated and Botox A-treated autaptic cultures in response to paired-pulse stimulation. (B) Paired-pulse data plotted as a ratio of the paired-pulse EPSC charge integrals. n = 5 for control; n = 9 for 60 pM Botox A. The mean PPR for the control was 0.97 ± 0.03 SEM and the mean PPR for the Botox A-treated culture was 1.4 ± 0.12 SEM. (C) Representative traces of autaptic cultures untreated and Botox F-treated in response to paired-pulse stimulation. (D) Paired-pulse data plotted as a ratio of the paired-pulse EPSC charge integrals. The mean PPR for the control was 0.90 ± 0.07 SEM, and the mean PPR for the Botox F-treated cultures was 0.80 ± 0.08 SEM. n = 10 for control and 60 pM Botox F.
Fig. 6.

Botox A, E, and F result in a different percentage change in the PPR. PPRs were expressed as a percentage change over the control cells. Each individual toxin-treated response was divided by the average control response of their sister cultures. Botox A-treated neurons had a percentage change of 49 ± 10% SEM, whereas Botox E-treated neurons were 28 ± 4.7% SEM, and Botox F-treated neurons were 11 ± 8.6% SEM. A one-way ANOVA test demonstrated a significant difference in the mean of the three conditions (P < 0.0001). A Tukey test revealed that Botox A vs. Botox E was significantly different (P < 0.05), Botox A vs. Botox F was significantly different (P < 0.001), and Botox E vs. Botox F was significantly different (P < 0.01).
Submaximal Concentrations of Botox F Do Not Cause PPF. It was important to determine whether the PPF observed was caused by the specific cleavage of SNAP-25 or of other members of the SNARE complex resulted in a similar PPF phenotype. Cultures were treated overnight with submaximal concentrations of Botox F (60 pM) that left ≈40% of evoked transmission remaining (Fig. 8). Recordings of paired EPSCs consistently demonstrated PPD (0.80 ± 0.08 SEM; n = 10) (Fig. 1 C and D) in Botox F-treated cells; however, this result was not significantly different from parallel recordings in nontreated sister cultures (0.90 ± 0.07 SEM; n = 10; P > 0.42) (Fig. 1D). Therefore, removal of the last 58 aa of VAMP by Botox F has a qualitatively different result on the PPR than removal of the last 9 aa of SNAP-25 by Botox A and demonstrates that VAMP is important for synaptic transmission to undergo PPF.
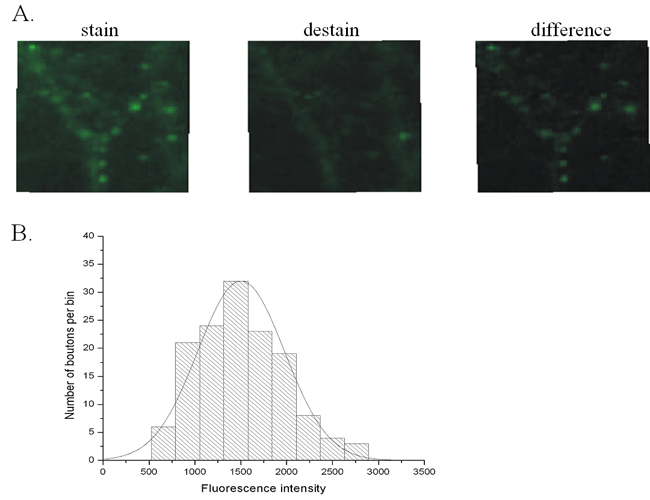
Botox A and F Lower the Release Probability and Size of the RRP. Both Botox A and F caused a reduction in synaptic transmission; Botox F showed no effect on the PPR compare to its control, whereas Botox A caused an increase in the PPR compared to its control. It is possible that this difference may be due to differential effects of the toxins on a heterogeneous population of synapses in the neurons, where the effects of submaximal concentrations of toxin differentially affected individual synapses; synapses unaffected by the toxin might therefore account for the effects of the individual toxins on the PPR. To test this idea, the release probability and the RRP of toxin-treated cultures were measured by using imaging of FM1-43 dye loading and release in individual synapses (11, 12, 14) (see Materials and Methods for details and Supporting Text and Fig. 9, which are published as supporting information on the PNAS web site).
Because the synaptic currents of Botox A- and F-treated autaptic cultures were recorded in 5 mM Ca2+, the FM1-43 release probability experiments were also carried out in 5 mM Ca2+. By using the same external Ca2+ concentration in the FM1-43 imaging experiments and the autaptic recording, it allowed for correlation between the PPRs found in the toxin-treated cells and the individual release probabilities of the toxin-treated terminals. However, for the measurement of the size of the RRP, 2 mM Ca2+ was used in the external solution because it has been shown that 2 mM Ca2+ results in an accurate estimation of the RRP using FM1-43 (12).
Briefly, the release probability was measured by delivering 20 action potentials at 0.5 Hz to conventionally plated neuronal cultures in the presence of the FM1-43 dye. Release probability is determined by dividing the average number of vesicles stained by the number of stimulus delivered (11). Cumulative frequency histograms of individual synapses from both Botox A- and F-treated cultures demonstrated a mean release probability 40% less than synapses in untreated sister cultures (Fig. 2 A and B). Importantly, the frequency distribution histograms were unimodal and by this analysis indicate there was not a population of synapses that were untouched by toxin, (15) suggesting that all synapses had reduced amounts of noncleaved SNARE proteins (SNAP-25 or VAMP) and had effected release probability in the same manner (Fig. 2C) To measure the size of the RRP, a stimulus of 40 action potentials at 20 Hz was used (12) and an average number of vesicles was determined by an the experimentally measured image intensity of one vesicle. Cumulative frequency histograms of individual synapses from toxin-treated cultures was 40% lower than control recordings (Fig. 2 D and E). Frequency distribution histogram were unimodal, and by this measurement suggest that the RRP of synapses were equally affected by the toxins (Fig. 2F). These results demonstrate that despite the same 40% reduction in the initial release probability and the size of the RRP by Botox A and F, only Botox A treatment results in PPF, whereas Botox F did not.
Fig. 2.

FM1-43 imaging experiments of Botox A- and F-treated cultures show a decrease in initial release probability and RRP of individual synapses. (A) Cumulative frequency graph showing the release probability of each individual synapse of untreated neurons or neurons treated with 60 pM Botox F for 20–28 h measured by FM1-43. The mean release probability of the Botox F-treated cultures was 0.287 ± 0.009 SEM, and the mean release probability of the untreated cultures was 0.507 ± 0.013 SEM. Five separate experiments were done for the toxin-treated cultures, whereas four separate experiments were performed on untreated cultures. A total of 283 synapses were counted for Botox F-treated cultures, whereas 308 synapses were counted for untreated synapses. Coverslips were all used from sister cultures and were 21–24 days old in vitro.(B) Cumulative frequency graph showing the release probability of each individual synapse of untreated neurons or neurons treated with 60 pM Botox A for 20–28 h measured by FM1-43. The mean release probability of the Botox A-treated cultures was 0.220 ± 0.005 SEM, whereas the mean release probability of the untreated cultures was 0.387 ± 0.009 SEM. Five separate experiments were done for the toxin-treated cultures, and four separate experiments were performed on untreated cultures. A total of 375 synapses were counted for Botox A-treated cultures, whereas 349 synapses were counted for untreated synapses. Coverslips were all used from sister cultures and were 21–24 days old in vitro. (C) Cumulative frequency graph showing that the distributions of the Botox A- and F-treated cultures are similar. Each individual experimental group from the release probability experiments was divided by its median, and then each was plotted as a cumulative frequency graph to determine whether there was any difference in the shapes of the distributions. (D) Cumulative frequency graph showing the RRP of each individual synapse of untreated neurons or neurons treated with 60 pM Botox F for 20–28 h measured by FM1-43. The mean vesicle number for Botox F-treated cultures was 5.18 ± 0.176 SEM, whereas the mean vesicle number for untreated cultures was 8.55 ± 0.247 SEM. Four separate experiments were carried out with the control and Botox F-treated cultures. A total of 307 synapses were counted for Botox F-treated terminals, whereas 316 synapses were counted for untreated terminals. (E) Cumulative frequency graph showing that the RRP of each individual synapse of untreated neurons or neurons treated with 60 pM Botox A for 20–28 h measured by FM1-43. The mean vesicle number for Botox A-treated cultures was 6.1 ± 0.163 SEM, whereas the mean vesicle number for untreated cultures was 9.1 ± 0.204 SEM. Four separate experiments were carried out with the control and Botox A-treated cultures. A total of 297 synapses were counted for Botox A-treated terminals, whereas 349 synapses were counted for untreated terminals. (F) Cumulative frequency graph showing that the distributions of the Botox A- and F-treated cultures are similar. Each individual experimental group from the RRP experiments was divided by its median, and then each was plotted as a cumulative frequency graph to determine whether there was any difference in the shapes of the distributions.
The Individual Release Probabilities of the Vesicles Remain Unchanged. Although these results revealed no correlation between the RRP, initial release probability, and PPR in the toxin-treated cells, it was still possible that the toxins changed the individual release probabilities of the synaptic vesicles to become higher in the Botox F treatment and lower in the Botox A treatment. To test this possibility, the distribution of the α-factors, release probability of a single vesicle, was analyzed. The α-factor is the ratio of the initial release probability that was experimentally determined in Fig. 2 A and B divided by their respective the experimentally derived RRP in Fig. 2 D and E (see Materials and Methods for more detail). Cumulative frequency histograms of the α-factors revealed no difference between the α-factors of the toxin-treated cultures and their respective controls (Fig. 3C). This analysis confirmed that Botox A and F did not affect the release probability of individual vesicles (Fig. 3C).
Fig. 3.

The α-factors are unchanged by Botox A and F. (A) Cumulative frequency graph showing that the α-factors of each individual synapse of untreated neurons or neurons treated with 60 pM Botox F for 20–28 h. (B) Cumulative frequency graph showing that the alpha of each individual synapse of untreated neurons or neurons treated with 60 pM Botox A for 20–28 h. (C) Cumulative frequency graph showing the distributions of the Botox A- and F-treated cultures are similar. Each individual group was divided by its median and then plotted as a cumulative frequency graph.
Botox F-Treated Cultures Exhibit PPF in Low Calcium. A previous report has demonstrated that facilitation is occluded by the effects of PPD (9). Although there are no other known effects of Botox F, it was possible that Botox F was cleaving another molecule responsible for facilitation. To test this hypothesis, recordings were made from 60 pM Botox F-treated cultures by using 1.5 mM Ca2+ and 2.5 mM Mg2+ in the external solution, which is known to cause PPF in autapses (9). Under these conditions, PPF was observed in untreated neurons (1.2 ± 0.06 SEM; n = 5) and in Botox F-treated cultures (1.4 ± 0.8 SEM; n = 5; Fig. 4). These results indicate that, in Botox F-treated terminals, facilitation is still intact. Furthermore, Botox F had no effect on the synaptic plasticity relative to control in high or low Ca2+ because Botox F gave PPD in high Ca2+ (Figs. 1 C and D and 4)
Fig. 4.

Botox F-treated cultures show PPF in low calcium/high magnesium. Paired-pulse data plotted as a ratio of the paired-pulse EPSC charge integrals. n = 5 for control; n = 5 for 60 pM Botox F. The mean PPR for the control was 1.233 ± 0.055 SEM, and the mean PPR for the Botox F-treated samples was 1.424 ± 0.085 SEM. Recordings were done in 1.5 mM Ca2+/2.5 mM Mg+2.
Submaximal Doses of Botox E Does Not Cause PPF or PPD. Because only Botox A, and not Botox F, affected PPRs, it was important to determine whether this was due to their difference in respective targets, VAMP vs. SNAP 25. It is known that Botox E also cleaves SNAP 25, but at a site 17 aa upstream from the Botox A-cleavage site (16). A dose-dependent reduction in the amplitude of the EPSC was found in response to increasing amounts of Botox E, with a complete blockade of synaptic transmission found at two of the highest doses of Botox E used (60 and 600 pM, Fig. 10, which is published as supporting information on the PNAS web site).
However, at submaximal concentrations of Botox E (Fig. 5), which left ≈20% of evoked transmission, Botox E treatment produced no PPD or PPF on its own (PPR = 1.0 ± 0.04 SEM; n = 14) but removed the normal PPD observed in untreated cultures (PPR = 0.81 ± 0.04 SEM; n = 12; P < 0.01) because the Botox E cells had a 20% increase in PPR (Fig. 6). Therefore, cleavage of SNAP 25 at amino acid 180 by Botox E gives a different phenotype than that seen with cleavage of SNAP-25 at amino acid 197 by Botox A (Fig. 6).
Fig. 5.

Botox E blocks evoked release, but submaximal doses do not cause PPF or PPD. Paired-pulse data plotted as a ratio of the paired-pulse EPSC charge integrals. The mean PPR for the control was 0.806 ± 0.043 SEM, and the mean PPR for the Botox E-treated cultures was 1.04 ± 0.039 SEM. n = 12 for control; n = 14 for 3 pM Botox E.
The SNARE Complex Is Involved in Regulating Asynchronous Release. Previous experiments have shown that there are two components of release, a synchronous and asynchronous component, and has lead to the hypothesis that one calcium sensor controls synchronous release while the other sensor controls asynchronous release and facilitation (6). It has been demonstrated that asynchronous release decouples with the same time constant as facilitation and has been used as evidence that the mechanism of asynchronous release and PPF maybe similar (9). Because the toxin cleavage would differentially affect SNARE complex formation (7) and based on the fact that three different toxin treatments lead to three different percentage changes in the PPR (Fig. 6), determining how asynchronous release is affected by the toxins different effects on the SNARE complex could provide direct evidence as to whether a single sensor or second sensor regulates asynchronous release and facilitation.
To test this hypothesis, the total number of asynchronous events in control cells and toxin-treated cells were counted, and the charge integrals of synchronous release of the toxin-treated and control cells were measured. The charge integrals of the synchronous release of the toxin-treated cells and control cells were then divided by each other to obtain a ratio of the synchronous release between the toxin-treated and the control recordings. This ratio was multiplied by the total number of asynchronous events for the control cells to obtain a predicted value of asynchronous events for the toxin-treated cells. If the toxins affected the synchronous release and the asynchronous release in the same manner, then there should be no significant difference between the predicted amount of asynchronous release for the experimental treatment and the actual amount obtained for the experimental treatment (see Materials and Methods for further details). Under the conditions of lower calcium or treatment with Botox A or F, no difference in the predicted amount of asynchronous release and the measured asynchronous release was observed (Fig. 7). In contrast, Botox E-treated cells showed an amount of asynchronous release higher than predicted (7.8 ± 1.2 SEM, n = 5 vs. 2.8 ± 0.49 SEM, n = 5; P < 0.01 Student's t test). The higher amount of asynchronous release in the Botox E-treated cells demonstrates that differences in SNARE complex stability and formation differentially affect asynchronous release and leads to the conclusion that the SNARE complex also regulates asynchronous release.
Fig. 7.

The SNARE complex regulates asynchronous release. (A) Comparison of EPSC charge integral in 5 mM Ca2+ vs. 1 mM Ca2+. The ratio of the EPSC charge integral in 1 mM Ca2+ to 5 mM Ca2+ was 0.35. (B) Total number of asynchronous events in 5 mM Ca2+ vs. 1 mM Ca2+. The predicted number of events was 3.8 ± 0.82 SEM, whereas the 1 mM Ca2+ was 3.8 ± 0.62 SEM. Student's t test did not reveal a difference between the predicted total and the 1 mM total. n = 5 for 5 mM Ca2+ and 1 mM Ca2+. (C) Comparison of EPSC charge integral in 5 mM Ca2+ vs. Botox A cultures. The ratio of the EPSC charge integral in 1 mM Ca2+ to 5 mM Ca2+ was 0.26. (D) Total number of asynchronous events in 5 mM Ca2+ vs. 1 mM Ca2+. The predicted number of events was 5.6 ± 1.5 SEM, whereas the Botox A amount was 5.8 ± 2.9 SEM. Student's t test did not reveal a difference between the predicted total and the 1 mM total. n = 5 for 5 mM Ca2+- and Botox A-treated cultures. (E) Comparison of EPSC charge integral in 5 mM Cal2+ vs. Botox F. The ratio of the EPSC charge integral in 1 mM Ca2+ to 5 mM Ca2+ was 0.26. (F) Total number of asynchronous events in 5 mM Ca2+ vs. Botox F. The predicted number of events was 4.1 ± 1.2 SEM, whereas the Botox F amount was 4.4 ± 0.9 SEM. Student's t test did not reveal a difference between the predicted total and the 1 mM total. n = 5 for 5 mM Ca2+- and Botox F-treated cultures. (G) Comparison of EPSC charge integral in 5 mM Ca2+ vs. Botox E. The ratio of the EPSC charge integral in 1 mM Ca2+ to 5 mM Ca2+ was 0.16. (H) Total number of asynchronous events in 5 mM Ca2+ and Botox E. The predicted number of events was determined as described in Materials and Methods. Student's t test revealed a difference between the predicted total and the Botox E total. (P < 0.01). The predicted amount was 2.848 ± 0.49 SEM, whereas the Botox E amount was 7.8 ± 1.15 SEM. n = 5 for 5 mM Ca2+- and Botox E-treated cultures.
Discussion
Release Probability Uncoupled. A dominant view in the field of synaptic physiology states that lowering the initial release probability or the size of the RRP always results in facilitation (3, 6, 11). According to this theory, disruption of SNARE function with Botox treatments should induce PPF because both Botox A and F lower initial release probability and the size of the RRP by 40%. Contrary to this prediction, Botox F treatment did not result in facilitation.
In the absence of modifications to the molecular components of the synaptic release machinery, the initial release probability and the RRP normally predict whether a synapse will facilitate or depress (2, 3). This study describes how cleavage of SNARE proteins by Botox affects the RRP and initial release probability parameters and how these changes relate to the molecular events leading to facilitation and depression. Cleavages of SNARE proteins by Botox have uncovered an intermediate step or steps important for the regulation of facilitation and depression. Evidence supporting this idea comes from other studies in the brain showing that the size of the RRP in the climbing fiber or parallel fiber of Purkinje cells or the CA1–CA3 synapse cannot predict facilitation or depression (17, 18). Furthermore, a recent study (19) shows that overexpression of the neuronal calcium sensor-1 (NCS-1) protein causes neurons that normally depress, to facilitate, without affecting the basal release probability of the individual cells.
Facilitation. Despite the same external calcium concentration, toxin cleavage of SNAREs affected PPRs differently (Figs. 1, 5, and 6) and suggests that calcium buffering alone cannot account for changes in short-term plasticity. Calcium buffering has been proposed as a dominant mechanism involved in controlling facilitation and depression (for a review, see ref. 6). Studies on the Calyx of Held (20) and in mice that lacked either Calbindin 28K (21) or parvalbuminum (22) provide support for the calcium buffering hypothesis.
It is possible that changes in calcium dynamics within the presynaptic terminal can explain some of the effects of the toxins on the PPR, because SNAP-25 can interact with calcium channels. However, currently no evidence exists that VAMP interacts with calcium channels (23). Although earlier reports have demonstrated that there is no difference in calcium dynamics with the Botox treatments (24, 25) a recent study showed that the Botox A cleaved form of SNAP-25, but not the Botox E cleaved form of SNAP-25 alters the ability of neurons to respond to calcium (26) and may explain the differences in PPR between the Botox A and E toxin treatments. Further studies are needed to determine how the molecular release machinery and calcium buffering in the terminal cooperate to modulate short-term plasticity.
Vesicular Release Probability. As reported, the vesicular release probability between the control cells and the toxin treatments was similar by FM1-43 data. Because it is not known whether 40 AP at 20 Hz completely labels the RRP of the toxin-treated terminals, it is possible that the α-factor is misreported. However, if only a fraction of the RRP was stained in the toxin-treated cells, then the reported data would have overestimated the α-factor because α is determined by dividing initial release probability by the size of the RRP. Regardless, if there are inaccuracies with RRP measurements and the α-factor measurement in the toxin-treated cells, one would expect that the control cells should have had completely different distributions for the α-factor. Again, this difference was not observed. Because the α-factor is only a ratio of the initial release probability divided by the RRP and is not indicative of the vesicular release probability on the second pulse, it is possible that the α-factors of the toxin-treated vesicles are different on the second pulse of the paired-pulse protocol and would explain the differences in the PPR.
Potential Evidence for Two Calcium Sensors. Alternative hypotheses have been proposed that state that either one calcium sensor (6, 27, 28) or two calcium sensors (6, 29, 30) play a role in the mechanisms of facilitation or depression. Several lines of evidence suggest the existence of a second calcium sensor: (i) synaptotagmin knockout animals have asynchronous release intact, which is responsive to calcium (31–33); (ii) evoked release displays biphasic decay, suggestive of two sensors (29, 30); and (iii) the observed differential effects of Br2+ and Sr2+ on synaptic transmission (34).
There are a host of molecular candidates as a putative second calcium sensor involved in facilitation (6, 35), and a possible interpretation of the data is that a separate second calcium sensor protein independently interacts with the distal part of the SNARE complex in addition to the synaptotagmin I-binding site between the Botox A- and E-cleavage sites (36). Botox E treatment caused a larger increase in the amount of asynchronous release observed than predicted if toxin similarly affected synchronous release and asynchronous release (Fig. 7). This increase of asynchronous release may be explained by assuming that a second sensor is involved in the facilitation of normal synaptic terminals, because the asynchronous component of release maybe related to the facilitation mechanism (9). Because Botox E cleavage leaves the distal part of the SNARE complex intact, whereas Botox F cleavage does not, it is likely that the distal part of the SNARE complex is involved with regulating facilitation/depression. Botox E- and F-cleaved complexes are deficient in binding synaptotagmin I, whereas Botox A complexes are not (36), but have different effects on the change in the PPR (Fig. 6).
Although the above arguments focus on the relationship between that synaptic release probability and a separate PPF sensor, it is possible that the toxin cleavage somehow has minimized PPD but does not involve a second PPF sensor. Previous results have shown that, if the initial release probability is low, then the release probability of the synapse will increase on the second pulse (2, 3). Although the release probability of the synapse after the second pulse was not measured, it is possible that the toxins were differentially affecting the synapses ability to respond to the second pulse. It can be argued that the release probability is a product of the actual release probability of an available vesicle po and the probability of availability pa (37). Based on this scenario, one could explain the Botox A affect as a graded change in pa, lowering the product and thereby avoiding depletion on the second pulse. Evidence supporting this comes from Xu et al. (38) who showed that Botox A slows down the exocytotic burst of Botox A-cleaved complexes, whereas Botox E would lower the graded response even further, and the Botox F effect might be seen as an all-or-nothing knockout of docked vesicles, not changing the depletion of the remaining docked vesicles. Moreover, it has also been shown that Botox E and A differentially effect relative sensitivity of evoked release to calcium (39) and mutations in VAMP (40, 41) differentially affect the calcium cooperativity of evoked release. Because the vesicles' ability to respond to calcium has been postulated to result in difference types of short-term plasticity (42), the ability of these toxin-cleaved complex vesicles to respond to the calcium may result in the different PPR phenotypes. Interestingly, tetanus toxin and Botox F cleave VAMP at different cleavage sites and demonstrate PPD similar to their parallel control cells (ref. 13 and Fig. 1) rather than the predicted PPF. These results, in addition to genetic evidence from Drosophila, (43) emphasize the importance of the VAMP molecule in the regulation of PPF/PPD.
Supplementary Material
Acknowledgments
I thank Dr. Jeremy Seamans for teaching me electrophysiology and for advice, Dr. Charles F. Stevens for providing laboratory space to carry out this study, Dr. Yongling Zhu for help with the FM1-43 experiments, and Drs. Mara Darsow, Juan Pina-Crespo, and Steve Heinemann for critical reading of the manuscript and advice. This work was supported by the Howard Hughes Medical Institute.
Author contributions: S.M.Y. designed research, performed research, analyzed data, and wrote the paper.
Abbreviations: RRP, readily releasable pool; PPR, paired-pulse ration; SNARE, N-ethylmaleimide-sensitive factor attachment protein receptor; PPF, paired-pulse facilitation; PPD, paired-pulse depression; EPSC, excitatory postsynaptic current.
References
- 1.Debanne, D., Guerineau, N. C., Gahwiler, B. H. & Thompson, S. M. (1996) J. Physiol. 491, 163-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens, C. F. & Wang, Y. (1995) Neuron 14, 795-802. [DOI] [PubMed] [Google Scholar]
- 3.Dobrunz, L. E. & Stevens, C. F. (1997) Neuron 18, 995-1008. [DOI] [PubMed] [Google Scholar]
- 4.Schikorski, T. & Stevens, C. F. (1997) J. Neurosci. 17, 5858-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanse, E. & Gustafsson, B. (2001) J. Physiol. 531, 481-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zucker, R. S. & Regehr, W. G. (2002) Annu. Rev. Physiol. 64, 355-405. [DOI] [PubMed] [Google Scholar]
- 7.Schiavo, G., Matteoli, M. & Montecucco, C. (2000) Physiol. Rev. 80, 717-766. [DOI] [PubMed] [Google Scholar]
- 8.Bekkers, J. M. & Stevens, C. F. (1991) Proc. Natl. Acad. Sci. USA 88, 7834-7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mennerick, S. & Zorumski, C. F. (1995) J. Physiol. 488, 85-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahamimoff, R. & Yaari, Y. (1973) J. Physiol. 228, 241-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murthy, V. N., Sejnowski, T. J. & Stevens, C. F. (1997) Neuron 18, 599-612. [DOI] [PubMed] [Google Scholar]
- 12.Murthy, V. N. & Stevens, C. F. (1999) Nat. Neurosci. 2, 503-507. [DOI] [PubMed] [Google Scholar]
- 13.Owe-Larsson, B., Kristensson, K., Hill, R. H. & Brodin, L. (1997) Eur. J. Neurosci. 9, 1773-1777. [DOI] [PubMed] [Google Scholar]
- 14.Ryan, T. A., Reuter, H. & Smith, S. J. (1997) Nature 388, 478-482. [DOI] [PubMed] [Google Scholar]
- 15.Motulsky, H. (1995) Intuitive Biostatistics (Oxford Univ. Press, New York).
- 16.Schiavo, G., Santucci, A., Dasgupta, B. R., Mehta, P. P., Jontes, J., Benfenati, F., Wilson, M. C. & Montecucco, C. (1993) FEBS Lett. 335, 99-103. [DOI] [PubMed] [Google Scholar]
- 17.Xu-Friedman, M. A., Harris, K. M. & Regehr, W. G. (2001) J. Neurosci. 21, 6666-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanse, E. & Gustafsson, B. (2001) J. Physiol. 537, 141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sippy, T., Cruz-Martin, A., Jeromin, A. & Schweizer, F. E. (2003) Nat. Neurosci. 6, 1031-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felmy, F., Neher, E. & Schneggenburger, R. (2003) Neuron 37, 801-811. [DOI] [PubMed] [Google Scholar]
- 21.Blatow, M., Caputi, A., Burnashev, N., Monyer, H. & Rozov, A. (2003) Neuron 38, 79-88. [DOI] [PubMed] [Google Scholar]
- 22.Caillard, O., Moreno, H., Schwaller, B., Llano, I., Celio, M. R. & Marty, A. (2000) Proc. Natl. Acad. Sci. USA 97, 13372-13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atlas, D. (2001) J. Neurochem. 77, 972-985. [DOI] [PubMed] [Google Scholar]
- 24.Gundersen, C. B., Katz, B. & Miledi, R. (1982) Proc. R. Soc. London Ser. B 216, 369-376. [DOI] [PubMed] [Google Scholar]
- 25.Molgo, J., Comella, J. X., Angaut-Petit, D., Pecot-Dechavassine, M., Tabti, N., Faille, L., Mallart, A. & Thesleff, S. (1990) J. Physiol. (Paris) 84, 152-166. [PubMed] [Google Scholar]
- 26.Verderio, C., Pozzi, D., Pravettoni, E., Inverardi, F., Schenk, U., Coco, S., Proux-Gillardeaux, V., Galli, T., Rossetto, O., Frassoni, C. & Matteoli, M. (2004) Neuron 41, 599-610. [DOI] [PubMed] [Google Scholar]
- 27.Katz, B. & Miledi, R. (1968) J. Physiol. 195, 481-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertram, R., Sherman, A. & Stanley, E. F. (1996) J. Neurophysiol. 75, 1919-1931. [DOI] [PubMed] [Google Scholar]
- 29.Barrett, E. F. & Stevens, C. F. (1972) J. Physiol. 227, 691-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goda, Y. & Stevens, C. F. (1994) Proc. Natl. Acad. Sci. USA 91, 12942-12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshihara, M. & Littleton, J. T. (2002) Neuron 36, 897-908. [DOI] [PubMed] [Google Scholar]
- 32.Geppert, M., Goda, Y., Hammer, R. E., Li, C., Rosahl, T. W., Stevens, C. F. & Sudhof, T. C. (1994) Cell 79, 717-727. [DOI] [PubMed] [Google Scholar]
- 33.Littleton, J. T., Stern, M., Schulze, K., Perin, M. & Bellen, H. J. (1993) Cell 74, 1125-1134. [DOI] [PubMed] [Google Scholar]
- 34.Zengel, J. E. & Magleby, K. L. (1980) J. Gen. Physiol. 76, 175-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atwood, H. L. & Karunanithi, S. (2002) Nat. Rev. Neurosci. 3, 497-516. [DOI] [PubMed] [Google Scholar]
- 36.Zhang, X., Kim-Miller, M. J., Fukuda, M., Kowalchyk, J. A. & Martin, T. F. (2002) Neuron 34, 599-611. [DOI] [PubMed] [Google Scholar]
- 37.Quastel, D. M. (1997) Biophys. J. 72, 728-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu, T., Binz, T., Niemann, H. & Neher, E. (1998) Nat. Neurosci. 1, 192-200. [DOI] [PubMed] [Google Scholar]
- 39.Keller, J. E. & Neale, E. A. (2001) J. Biol. Chem. 276, 13476-13482. [DOI] [PubMed] [Google Scholar]
- 40.Deitcher, D. L., Ueda, A., Stewart, B. A., Burgess, R. W., Kidokoro, Y. & Schwarz, T. L. (1998) J. Neurosci. 18, 2028-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart, B. A., Mohtashami, M., Trimble, W. S. & Boulianne, G. L. (2000) Proc. Natl. Acad. Sci. USA 97, 13955-13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trommershauser, J., Schneggenburger, R., Zippelius, A. & Neher, E. (2003) Biophys. J. 84, 1563-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshihara, M., Ueda, A., Zhang, D., Deitcher, D. L., Schwarz, T. L. & Kidokoro, Y. (1999) J. Neurosci. 19, 2432-2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}