

Abstract
Hearing loss leads to a host of cellular and synaptic changes in auditory brain areas that are thought to give rise to auditory perception deficits such as temporal processing impairments, hyperacusis, and tinnitus. However, little is known about possible changes in synaptic circuit connectivity that may underlie these hearing deficits. Here, we show that mild hearing loss as a result of brief noise exposure leads to a pronounced reorganization of local excitatory and inhibitory circuits in the mouse inferior colliculus. The exact nature of these reorganizations correlated with the presence or absence of the animals' impairments in detecting brief sound gaps, a commonly used behavioral sign for tinnitus in animal models. Mice with gap detection deficits (GDDs) showed a shift in the balance of synaptic excitation and inhibition that was present in both glutamatergic and GABAergic neurons, whereas mice without GDDs showed stable excitation–inhibition balances. Acoustic enrichment (AE) with moderate intensity, pulsed white noise immediately after noise trauma prevented both circuit reorganization and GDDs, raising the possibility of using AE immediately after cochlear damage to prevent or alleviate the emergence of central auditory processing deficits.
SIGNIFICANCE STATEMENT Noise overexposure is a major cause of central auditory processing disorders, including tinnitus, yet the changes in synaptic connectivity underlying these disorders remain poorly understood. Here, we find that brief noise overexposure leads to distinct reorganizations of excitatory and inhibitory synaptic inputs onto glutamatergic and GABAergic neurons and that the nature of these reorganizations correlates with animals' impairments in detecting brief sound gaps, which is often considered a sign of tinnitus. Acoustic enrichment immediately after noise trauma prevents circuit reorganizations and gap detection deficits, highlighting the potential for using sound therapy soon after cochlear damage to prevent the development of central processing deficits.
Keywords: midbrain, noise trauma, tinnitus, uncaging
Introduction
Cochlear injury by loud noise, ototoxic drugs, or traumatic brain injury induces changes in neuronal, synaptic, and circuit properties in the central auditory system and often leads to perceptual deficits including temporal processing impairments (Phillips et al., 1994), hyperacusis (Knipper et al., 2013), and tinnitus (Baguley et al., 2013). In both humans and animals, hyperacusis and tinnitus are associated with a hyperactive state of central auditory areas (Melcher et al., 2000; Brozoski et al., 2002; Seki et al., 2003; Ma et al., 2006; Bauer et al., 2008; Middleton et al., 2011; Vogler et al., 2011; Llano et al., 2012; Manzoor et al., 2012; Li et al., 2013; Kalappa et al., 2014) caused by increased neuronal membrane excitability (Li et al., 2013; Li et al., 2015) or decreased synaptic inhibition (Wang et al., 2009; Middleton et al., 2011; Yang et al., 2011; Pilati et al., 2012).
Dysfunction of the inferior colliculus (IC), a prominent auditory midbrain structure, has been implicated in temporal processing deficits (Walton, 2010), hyperacusis (Melcher et al., 2000), audiogenic seizures (Millan et al., 1986; Garcia-Cairasco, 2002), and tinnitus (Berger et al., 2015). The IC is a major subcortical integration center in the vertebrate brain, receiving ascending inputs from almost all auditory brainstem nuclei (Adams, 1979; Malmierca et al., 2005), commissural inputs from the contralateral IC (Saldaña et al., 1992, Orton and Rees, 2014), and descending inputs from the auditory cortex (Saldaña et al., 1996; Gao et al., 1998; Bajo et al., 2012). In addition, the IC contains an extensive network of intrinsic excitatory and inhibitory connections that are thought to form the majority of IC synapses (Saldaña et al., 1992; Saldaña and Merchán, 2005) and to provide gain control (Grimsley et al., 2013). Hearing-loss-induced changes in intrinsic IC circuits may thus contribute to IC hyperactivity, which is a hallmark of tinnitus in both humans and animal models (Melcher et al., 2000; Ma et al., 2006; Bauer et al., 2008; Dong et al., 2010; Manzoor et al., 2012; Ropp et al., 2014).
Here, we used laser-scanning photostimulation (LSPS) with caged glutamate to map local synaptic inputs to glutamatergic and GABAergic neurons in the central nucleus of the IC (CNIC) of control and noise-traumatized mice. We found that noise trauma leads to a reorganization of both excitatory and inhibitory local CNIC circuits that was cell-type and input specific and correlated with the presence or absence of sound gap detection deficits (GDDs), which is commonly interpreted as a sign of tinnitus in animal models (Turner et al., 2006; Dehmel et al., 2012; Li et al., 2013; Li et al., 2015; Wu et al., 2016). Acoustic enrichment (AE) with mild-intensity pulsed white noise immediately after noise exposure prevented the reorganization of CNIC circuits and GDDs. Our results link patterns of circuit reorganization in the CNIC with a temporal auditory processing deficit and raise the possibility of using early AE after cochlear trauma to prevent the emergence of temporal processing deficits and tinnitus.
Materials and Methods
Animals and preparation.
Experimental procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Vgat-ires-cre and dT-loxP mice were purchased from The Jackson Laboratories. Vesicular glutamate transporter 2 (Vglut2)-cre mice were a gift from Dr. Rebecca Seal. The background strain of all of the mice was C57BL/6J. Vglut2-cre and vgat-ires-cre mice were crossed with dT-loxP mice to generate vglut2-cre-dT-loxP and vgat-ires-cre-dT-loxP strains. Behavioral experiments, slice preparation, and recordings were carried out between the hours of 7:00 A.M. and 7:00 P.M.
Noise exposure.
Mice of either sex [postnatal day (P) 20–23] were randomly assigned to the noise-exposed (NE) or the control group. For unilateral noise exposure, mice were deeply anesthetized with isoflurane. A pipette tip connected to a 2.5 cm piece of plastic tubing was inserted into the left ear canal and the other end of the tubing was attached to a close-field speaker (MF1-M; Tucker-Davis Technologies). Noise exposure consisted of band-pass noise with a 1 kHz bandwidth that was centered at 16 kHz and presented at a 116 dB sound pressure level (SPL) for 45 min. Mice in the control group were not given noise exposure, but, similar to experimental animals, were exposed to isoflurane anesthesia during auditory brainstem response (ABR) measurements.
Gap inhibition of the acoustic startle response.
Behavioral evidence for central hearing deficits was tested using the gap detection method (Turner et al., 2006). Gap detection was tested in all mice before noise exposure (P19–P22) and 7 d after noise exposure (P26–P30) using a custom-built system (Clause et al., 2011). Gap detection testing was carried out in a sound-attenuating chamber (Medical Associates). A narrow band-pass background sound (1 kHz bandwidth centered at 10, 12, 16, 20, 24, and 32 kHz) was presented at 70 dB SPL for 8–25 s (randomly varied) before presentation of an acoustic startle stimulus (white noise, 115 dB SPL, 20 ms). In 50% of trials, a 50 ms gap was introduced into the background sound 130 ms before the presentation of the startle stimulus. Startle response magnitude was measured (in arbitrary units) as the maximum peak-to-peak amplitude of the downward force exerted by mice onto the piezoelectric platform. Trials were combined and played back digitally from a single computer file to avoid unwanted sound artifacts generated by initiating new stimulation protocols. Gap detection ability was determined for each sound frequency using the gap startle ratio, which is the ratio of the startle response amplitude in trials with gaps in the background sound at a given sound frequency over the startle response magnitude in trials without gaps at the same sound frequency. Gap startle ratios closer to 0 indicate a greater gap inhibition of the acoustic startle reflex (ASR) and gap startle ratios closer to 1 indicate less gap inhibition.
Behavioral classification criteria.
The behavioral threshold for a deficit in gap detection was defined as an increase in the gap detection ratio that was at least 2 SDs above the average change in gap detection ratios observed for age-matched control mice over a 7 d period (Li et al., 2013). To control for potential strain differences in gap detection ability, behavioral threshold scores were determined independently for vglut2-cre-dTloxP and vgat-ires-cre-dT-loxP mice (Fig. 1). The distribution of changes in gap detection in control mice from each strain were fitted with a normal distribution (vglut2-cre-dT-loxP; μ = 0.02, SD σ = 0.141, n = 12 animals: vgat-ires-cre-dT-loxP; μ = 0.02, SD σ = 0.145, n = 12 animals) and threshold scores were determined to be 0.30 (vglut2-cre-dT-loxP) and 0.31 (vgat-ires-cre-dT-loxP), similar to the values reported previously for other mouse strains (Li et al., 2013). NE mice that exhibited an increase in the gap startle ratio that exceeded these thresholds for at least one sound frequency were categorized as mice with GDDs (GDD mice). To control for GDDs unrelated to noise exposure, control mice exhibiting an increase in the gap startle ratio that exceeded the threshold in the initial testing (one animal for vglut2-cre-dT-loxP and one animal for vgat-ires-cre-dT-loxP) were excluded from further analysis. To ensure that all mice were able to detect the gap in the background sounds before noise exposure, gap startle ratios for individual sound frequencies were required to be <0.9 before noise exposure to be included in postnoise exposure analysis (Li et al., 2013). One mouse had no gap startle ratios below 0.9 for any tested frequency and was therefore excluded. Using these criteria, nine of 18 (50%) NE vglut2-cre-dT-loxP mice and 10 of 19 (53%) NE vgat-ires-cre-dT-loxP mice showed behavioral GDDs.
Figure 1.

Effects of noise exposure on gap detection and PPI. A, Distribution of changes in gap startle ratios (response to startle stimulus with gap present divided by response to startle stimulus alone) over a 1 week period in control mice. Data fitted with normal distribution (gray curve, μ = 0.02, σ = 0.145, n = 67 sound frequencies). Gap ratio changes >2σ above the distribution mean (0.31) are considered a GDD. B, Cumulative probability distribution of gap ratio changes after noise exposure. C, Summary graphs of gap startle ratios before (open bars) and 7 d after noise exposure (closed bars). Gap detection ratios remain stable in no-GDD mice, but are increased for higher sound frequencies in GDD mice. See Figure 2 source data 1 for raw data and exact p values. D, Summary graphs of PPI startle ratio before and after noise exposure. Error bars indicate SEM. Asterisks indicate statistical significance. *p < 0.05, **p < 0.01. E, Summary graphs of ABR thresholds before and 7 d after noise exposure in vgat-ires-cre-dT mice. Thresholds for higher sound frequencies were elevated after noise exposure in both no-GDD and GDD mice. Error bars indicate SEM. *p < 0.05, **p < 0.01. A–E are from vgat-ires-cre-dT-loxP mice; similar results were obtained from vglut2-cre-dT-loxP mice (data not shown).
Prepulse inhibition (PPI) of the acoustic startle response.
For PPI testing, a 50 ms prepulse sound (1 kHz bandwidth centered at 10, 12, 16, 20, 24, and 32 kHz) was presented 130 ms before the presentation of the startle stimulus (white noise, 115 dB SPL, 20 ms). The sound intensity of the prepulse was similar to the intensity of the background sound used in gap detection trials (70 dB SPL). PPI was evaluated for each sound frequency with the PPI startle ratio, which is the ratio of the startle response amplitude in trials with prepulse over the startle response amplitude in trials without prepulse. PPI startle ratios closer to 0 indicate stronger PPI of the startle response and PPI startle ratios closer to 1 indicate weaker PPI of the startle response.
ABR.
ABR thresholds were measured before noise exposure (P19–P22) and 7 d after noise exposure (P26–P30). Measurements were conducted in a sound-attenuating chamber (Medical Associates) using a Z-Series 3-DSP Bioacoustic System (Tucker Davis Technologies) with subdermal electrodes placed at the vertex, a ground electrode placed ventral to the right pinna, and the reference electrode placed ventral to the left pinna. Stimuli were produced using the System 3 software package (Tucker Davis Technologies). During ABR measurements, animals were anesthetized with isoflurane and their body temperatures were maintained around 36.5–38.5°C with a heating pad. ABR thresholds were obtained with 1 ms clicks as well as 3 ms tone bursts of 10, 12, 16, 20, 24, and 32 kHz presented at various sound intensities at a rate of 18.56/s. Evoked potentials were averaged 1024 times and filtered using a 300–3000 Hz band-pass filter.
AE.
AE consisted of pulsed white noise (75 dB SPL) delivered in a sound-attenuating chamber as described previously (Clause et al., 2014). Pulse length was set to 138 ms and was presented pseudorandomly with an interpulse length of 0–450 ms and a duty cycle of 47%. AE was started immediately after noise exposure and continued for 7 d until animals were used for ABR measurements, behavioral testing, and preparation of brain slices. Age-matched mice in the AE control group (AE-only) were placed in the AE chamber without noise stimulation, after which they were treated identically to NE mice that received AE (NE-AE).
Slice preparation.
Coronal brainstem slices were prepared from mice of either sex at P26–P30. For slice preparation, animals were deeply anesthetized with isoflurane, decapitated, and their brains were removed. Coronal midbrain slices (300 μm thick) were then prepared as described previously (Sturm et al., 2014) and incubated at 34°C in artificial CSF (ACSF) containing the following (in mm): 0.25 7H2O × MgSO4, 124 NaCl, 5 KCl, 10 dextrose, 1.25 KH2PO4, 26 NaHCO3, 2 CaCl2. Brain slices were kept in an interface chamber for 1 h (30 m incubation at 34°C followed by 30 m rest at 22–25°C) before beginning recordings.
Electrophysiological recordings.
Recordings were performed in a submersion-type chamber (3–4 ml/min perfusion with oxygenated ACSF at 22–25°C) mounted on an upright microscope (AxioExaminer A1; Zeiss) and were targeted at dTomato-expressing neurons under epifluorescent illumination. Borosilicate glass recording pipettes (3–6 MΩ) contained a potassium-based internal solution containing the following (in mm): 115 K-gluconic acid (C6H11O7K), 5 KCl, 11 EGTA, 1 MgCl2 × 6H2O, 1 CaCl2 × 2H2O, 10 HEPES, 0.3 GTP disodium salt, 2.0 ATP disodium salt, and 0.5% biocytin, pH 7.2, 314 mOsm/L. Whole-cell currents or membrane voltages were acquired (Multiclamp 700B) at a sampling rate of 4 kHz. Current–voltage (IV) plots were generated in current-clamp mode with 15 pA current steps. Spontaneous excitatory (sEPSC) and inhibitory (sIPSC) currents were recorded in voltage-clamp mode at −65 and 0 mV, respectively.
Blinding procedure.
Slice recordings and analysis of mapping and electrophysiological data, as well as gathering and analyzing ABRs, was performed blind to the behavioral classification. Analysis of ABRs was performed blind to the behavioral classification and startle data were analyzed blind to the mapping data. Synaptic input mapping was performed without information about the ABR and startle behavior data and analysis of electrophysiological data and ABR thresholds was performed blind to gap detection behavioral status. For analysis, data from all animals were combined into a single software folder, with data from each cell being identified only by the animal code and recording date. After completion of all analysis, cells were matched with gap detection behavioral status.
Synaptic input mapping.
The spatial distribution of presynaptic inputs to IC neurons was determined using focal photolysis of 4-methoxy-7-nitroindolinyl-caged-L-glutamate (MNI glutamate, 0.2 mm; Tocris Bioscience) using a custom-built system to control the location and duration (1 ms) of uncaging light spots (Sturm et al., 2014). In short, a 355 nm UV laser light (3510-30; DPSS Laser) was steered using galvo-driven mirrors (6210H; Cambridge Technology) and focused to a 20 μm, 2 mW spot at the position of the slice. Uncaging sites, electrophysiological data acquisition, and analysis were under the control of custom-written LabView software linked to pClamp software. Uncaging occurred once per second at sites chosen in a random sequence. Input maps were only collected if cells had a holding current <−100 pA and access resistance <50 MΩ. Excitatory and inhibitory synaptic responses were isolated by holding cells at −65 and 0 mV, respectively. For each condition, one to three input maps were obtained and the average of these maps was used for analysis.
Mapping analysis.
Responses due to direct action of uncaged glutamate on the recorded neuron (direct responses) were distinguished from synaptic responses based upon their latencies of <7 ms (Sturm et al., 2014). Stimulation sites that elicited direct glutamate responses from the recoded neuron were excluded from input maps. Spontaneous and uncaging-evoked synaptic events were distinguished using the following procedures. First, to be considered as an evoked synaptic response, an event's absolute amplitude had to exceed a threshold of 4σ of the baseline activity (the 100 ms period before glutamate uncaging). Thresholds were calculated for each stimulation trial to account for variations in baseline activity. Second, to be considered as an evoked synaptic event, its onset latency had to be between 10 and 60 ms. Finally, candidate presynaptic sites had to fulfill at least one of three additional criteria to be considered a valid synaptic response: (1) suprathreshold events were elicited in at least two stimulation iterations at that site; (2) a suprathreshold event was detected in a single stimulation iteration at that site, but the event consisted of several components rather than single-peaked, single component event, which is characteristic of spontaneous synaptic currents; or (3) if a suprathreshold event was detected in a single stimulation iteration at that site, then an event with similar onset latency (<10 ms difference) had to be elicited from an immediately adjacent stimulation site.
Excitatory and inhibitory synaptic input map areas were calculated as the sum of individual excitatory and inhibitory synaptic response sites, respectively. The synaptic charge (in picocoulombs) elicited from a valid synaptic response site was calculated over a 150 ms window, from 10 to 160 ms poststimulus, and averaged across all available mapping iterations. For each neuron, the total amounts of excitatory and inhibitory synaptic charge were determined as the sum of all excitatory and inhibitory synaptic charges. Synaptic charge per stimulation site was calculated as the total synaptic charge (either excitatory or inhibitory) for a given neuron divided by the number of synaptic response sites (either excitatory or inhibitory) for that neuron.
Excitation: inhibition (E:I) indices for individual cells were calculated in terms of input area and input charge according to the following equation:
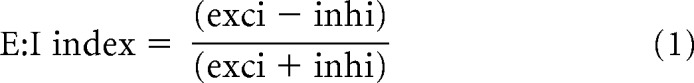 |
where the terms “exci” and “inhi” refer to the corresponding input areas or input charges for each cell, respectively.
Spontaneous synaptic event analysis.
sEPSCs and sIPSCs were analyzed using MiniAnalysis software (Synaptosoft). The frequencies and amplitudes of sEPSCs and sIPSCs were determined for each recorded neuron. E:I indices of total synaptic charge were calculated according to the following equation:
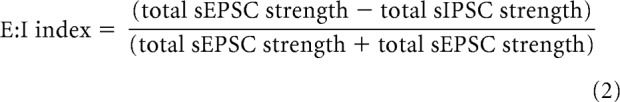 |
where total sEPSC strength is the sum of sEPSC amplitudes (in picoamperes) recorded over a 60 s period and total sIPSC strength is the sum of sIPSC amplitudes (also in picoamperes) recorded over 60 s.
Distinguishing type 1 and type 2 vgat+ neurons.
To distinguish between type 1 and type 2 GABAergic neurons, an inhibitory input map threshold was set. Neurons that exhibited inhibitory input map areas ≤0.25 × 105 μm2 and total inhibitory input charges ≤5 pC were considered type 2 neurons. Neurons that exhibited either inhibitory input map area >0.25 × 105 μm2 or total inhibitory input charge >5 pC were considered type 1 neurons.
Intrinsic properties of type 1 and type 2 vgat+ neurons.
Intrinsic membrane properties were derived from IV plots collected in current-clamp mode using 15 pA steps. Input resistance was measured with −15 pA injections from rest. Depolarization and repolarization slopes of action potentials were measured as the maximum slope during the depolarization phase and the minimum slope during repolarization phase of a spike, respectively. Action potential half-height width was measured as the width of a spike when voltage = spike threshold + (spike amplitude/2). Spike threshold was measured as the membrane potential at which the depolarization slope shows the first abrupt change.
Gap detection behavior analysis.
Gap detection ratios were determined for each sound frequency as the average startle amplitude in the presence of a background sound gap divided by the average startle magnitude in the absence of a sound gap. Gap ratio changes were defined as the gap detection ratio before noise exposure (or control) subtracted from the gap detection ratio obtained 7 d later.
PPI behavior analysis.
PPI ratios were determined for each sound frequency as the average startle amplitude in the presence of a prepulse divided by the average startle amplitude in the absence of that prepulse. PPI ratio changes were defined as the PPI ratio obtained before noise exposure (or control) subtracted from the PPI ratio obtained 7 d later.
ABR threshold analysis.
ABR thresholds were analyzed with Biosig software and were defined as the minimum sound intensity (in decibels SPL) that a given click or tone burst was able to elicit at least two waves in the ABR waveform.
Statistical analysis.
Data are presented as box-and-whisker plots, with median, interquartile range, and total range values. Data were tested for normal distribution using Bartlett's test. For independent, two-group comparisons, Student's t tests (two-tailed) and Mann–Whitney tests were used to determine statistical significance (GraphPad Prism). For two-group comparisons that involved repeated measures (gap behavior, PPI behavior, and ABR thresholds), paired t tests (two-tailed) were used. In the case of two-group comparisons involving repeated measures in which data were non-normally distributed, Wilcoxon tests were used. For three- and four-group comparisons, One-way ANOVAs (for normally distributed data) and Kruskal–Wallis tests (for non-normally distributed data) were used to determine statistical significance (GraphPad Prism). Multiple comparisons were corrected for with Tukey's method (ANOVAs) and Dunn's pairwise method (Kruskal–Wallis). For cumulative probably data, Kolmogorov–Smirnov tests were used. Statistical significance was set to p < 0.05.
Results
A subset of noise-traumatized mice developed temporal processing deficits
In both humans and rodent models, noise-induced hearing loss leads to central processing deficits or behavioral signs of tinnitus in only a fraction of the population (for human data, see Yankaskas, 2013; for rodent data, see Dehmel et al., 2012; Li et al., 2013; Li et al., 2015; Wu et al., 2016). We elicited noise-induced hearing loss by unilaterally exposing 20- to 23-d-old (P20–P23) mice for 45 min to continuous narrow-band noise (116 dB, 1 kHz band-width centered at 16 kHz). One week later, mice were assessed for hearing threshold shifts using ABRs and tested for behavioral GDDs using the acoustic startle gap detection method (Turner et al., 2006; Dehmel et al., 2012; Li et al., 2013). This method is based upon inhibition of the ASR by a silent gap embedded in a background sound preceding the startle stimulus by 130 ms. Despite some limitations (Eggermont, 2013; Brozoski et al., 2016), a decrease in gap-mediated inhibition of the ASR is widely regarded as a behavioral sign of tinnitus in animal models (Turner et al., 2006; Li et al., 2013; Wu et al., 2016).
Consistent with previous reports (Li et al., 2013; Li et al., 2015; Wu et al., 2016), noise exposure induced a significant reduction in gap-mediated inhibition of the ASR in ∼50% of mice (19 of 37; Figs. 1,2). Impaired gap detection was found only for sound frequencies that were at or above the hearing-loss frequency range (16–32 kHz; Fig. 1A–C), similar to the situation in human patients, in whom the frequency of tinnitus is usually at or above the frequency of hearing loss. The degree of hearing loss after noise exposure was mild (10–20 dB threshold shift at 16–32 kHz) and was similar between NE mice with and without GDDs (ABR threshold shift 16–32 kHz: no GDD: 17.9 ± 2.9 dB, GDD: 18.2 ± 2.1, n = 33 animals, p = 0.93, two-tailed Student's t test; Fig. 1E). We also found no significant differences between GDD and no-GDD mice with respect to either the amplitudes or the latencies of click- or tone-evoked ABR waves I or II or in the ratios of wave II/wave I amplitudes (data not shown).
Figure 2.

Noise-induced reorganization of synaptic input maps onto glutamatergic IC neurons. A, Mice were noise exposed at P20–P23 and tested 7 d later for GDDs. Nineteen of 37 NE mice (51%) developed a GDD. B, Schematic of mapping excitatory (red E) and inhibitory (red I) synaptic inputs to glutamatergic (vglut2+) neurons (black encircled E). Excitatory and inhibitory synaptic input maps are obtained at a membrane holding voltage (Vm) of −65 mV (left) and 0 mV (right), respectively. Example maps are overlaid over a photograph of the corresponding IC slice. C, Examples of input maps from control, NE with GDD, and noise exposed without GDD (no-GDD) mice. Traces illustrate excitatory (¢) and inhibitory (Δ) synaptic responses to glutamate uncaging (*) at the locations indicated by symbols. Uncaging sites that elicited direct responses in the recorded neuron are marked as black. Examples maps and recordings traces are from single stimulation iterations. D, Noise trauma decreased inhibitory input area (top) and total inhibitory charge (bottom) in GDD mice, but not in no-GDD mice (median inhi area: control = 2.95 × 105 μm2, n = 12 neurons, n = 6 animals, GDD = 0.60 × 105 μm2, n = 10 neurons, n = 6 animals, no-GDD = 1.0 × 105 μm2, n = 12 neurons, n = 7 animals, F(2,31) = 6.13, p = 0.006, one-way ANOVA; median inhi charge: control = 88.3 pC, n = 12 neurons, n = 6 animals, GDD = 9.6 pC, n = 10 neurons, n = 6 animals, no-GDD = 32.6, n = 11 neurons, n = 7 animals, H = 14.13, p = 0.0009, Kruskal–Wallis test). Total excitatory charge (bottom) was decreased in no-GDD mice compared with GDD mice (median exci charge: control = 17.3 pC, n = 14 neurons, n = 6 animals, GDD = 39.4 pC, n = 10 neurons, n = 6 animals, no-GDD = 7.1 pC, n = 12 neurons, n = 7 animals, H = 6.43, p = 0.04, Kruskal–Wallis test). Data are shown as box-and-whisker plots; midline depicts median, box encompasses interquartile range and error bars represent total range. E, Excitatory and inhibitory synaptic inputs for individual neuron. Lines connect data from individual neurons. F, Mean of E:I indices of individual neurons for input area (left) and input charge (right; median E:IArea: control = −0.59, n = 12 neurons, n = 6 animals, GDD = 0.40, n = 10 neurons, n = 6 animals, no-GDD = −0.24, n = 12 neurons, n = 7 animals, F(2,31) = 17.72, p < 0.0001, one-way ANOVA; median E:ICharge: control = −0.81, n = 12 neurons, n = 6 animals, GDD = 0.83, n = 10 neurons, n = 6 animals, no-GDD = −0.55, n = 11 neurons, n = 7 animals, F(2,30) = 21.62, p < 0.0001, one-way ANOVA). *p < 0.05, **p < 0.01 in post hoc, pairwise assessments corrected for multiple comparisons.
Impaired gap detection was not due to an impairment in the detection of background sounds in which the silent gaps were embedded because PPI of the ASR with prepulses of similar intensities and frequencies as the background sounds used in gap detection testing was unaffected (PPI ratio change: control: −0.12 ± 0.02, n = 90 sound frequencies, no GDD: −0.10 ± 0.03, n = 100 sound frequencies, GDD: 0.00 ± 0.03, n = 65 sound frequencies, H = 5.11 p = 0.08, Kruskal–Wallis test; Fig. 1D). Together, these results indicate that, whereas all NE mice exhibited a similar degree of hearing loss, only ∼50% of exposed mice exhibited GDDs.
Reorganization of synaptic inputs onto excitatory IC neurons
We next investigated whether the presence or absence of GDDs correlates with the degree or the type of noise-induced reorganization of intrinsic CNIC circuits. To this end, we obtained whole-cell patch-clamp recordings from CNIC neurons in brain slices contralateral to the NE ear and used LSPS with caged glutamate to map the spatial distribution and strengths of their synaptic inputs (Fig. 2). Because excitatory and inhibitory neurons in the IC cannot be reliably distinguished on the basis of morphological or physiological criteria (Malmierca et al., 1993; Sivaramakrishnan et al., 2001), we first recorded only from identified glutamatergic neurons using a mouse line in which the expression of the fluorescent protein dtTomato is restricted to glutamatergic neurons that express the Cre protein under the promoter for the vesicular glutamate transporter 2 (vglut2).
Glutamatergic CNIC neurons received both excitatory and inhibitory local inputs, which were recorded while voltage clamping the neurons at −65 or 0 mV, respectively (Fig. 2B,C). In unexposed control mice, local input maps were dominated by synaptic inhibition because inhibitory map areas were significantly larger than excitatory areas (n = 12 neurons, n = 6 animals, t(11) = 4.56, p = 0.0008, two-tailed paired t test) and because total inhibitory charges were significantly larger than total excitatory charges (n = 12 neurons, n = 6 animals, W = 58, p = 0.021, two-tailed Wilcoxon matched-pairs signed-rank test; Fig. 2D,E). This resulted in a negative E:I index for input area and input charge (Fig. 2F). The dominance of inhibition at P26–P30 was similar to the dominance of inhibition at P19–P22 (Sturm et al., 2014), consistent with a mature state of IC circuitry by the end of the third postnatal week (Shnerson and Willott, 1979, Romand and Ehret, 1990, 1992; Yu et al., 2005).
All NE mice showed a reorganization of synaptic inputs to glutamatergic CNIC neurons. However, the degree and type of this reorganization was different in mice with and without GDDs. In mice with GDDs, excitatory input maps were similar as in the control animals (Fig. 2C,D), but inhibitory input maps were significantly smaller [80% area reduction, F(2,31) = 6.13, p = 0.006, one-way ANOVA, 95% confidence interval (CI) of control vs GDD = 0.52–3.26 × 105 μm2, corrected pairwise comparison) and also weaker (90% charge reduction, H = 14.13, p = 0.0009, Kruskal–Wallis test, mean rank difference for control vs GDD = 15.53, Dunn's test; Fig. 2C,D). Therefore, the mean E:I index of glutamatergic neurons in GDD mice was shifted from the negative value in control mice to a positive value (E:I Area: F(2,31) = 17.72, p < 0.0001, one-way ANOVA, 95% CI of control vs GDD = −1.28 to −0.51, corrected pairwise comparison; E:I Charge: F(2,30) = 21.62, p < 0.0001, one-way ANOVA, 95% CI of control vs GDD = −1.63 to −0.63, corrected pairwise comparison; Fig. 2E). Therefore, in contrast to the dominance of inhibition of local inputs in control animals, in GDD mice, local inputs to glutamatergic CNIC neurons are dominated by excitation.
NE mice without GDDs (no-GDD mice) also showed a reorganization of local inputs to glutamatergic CNIC neurons, but this reorganization was different from that of GDD mice. First, the total excitatory synaptic charge in no-GDD mice was larger than in GDD mice (H = 6.43, p = 0.042, Kruskal–Wallis test, mean rank difference of GDD vs no-GDD = 11.55, Dunn's test; Fig. 2D). Second, synaptic inhibition was not significantly reduced in no-GDD mice (Fig. 2C,D). Third, in no-GDD mice, there was no significant change of the mean E:I index, indicating that local inputs to glutamatergic CNIC neurons remained dominated by inhibition (Fig. 2D,F).
Next, we investigated whether noise exposure changes spontaneous synaptic events, which, in addition to local IC connections, also reflect changes in ascending or descending extrinsic inputs. All noise-traumatized mice regardless of their gap detection performance exhibited an increase in the frequency, but not in the amplitude, of sEPSCs (H = 16.89, p = 0.0002, Kruskal–Wallis test, mean rank difference of control vs GDD = −16.69, mean rank difference of control vs no-GDD = −9.86, Dunn's tests; Fig. 3B,C). The frequencies and amplitudes of sIPSCs were unaffected by noise exposure (Fig. 3B,C). To determine the E:I balance of spontaneous synaptic currents, we calculated the E:I index for the summed synaptic charges received by individual excitatory neurons over a 60 s period (see Materials and Methods; Fig. 3D). Similar to the E:I indices for local inputs, the E:I index of spontaneous synaptic events in GDD mice shifted to positive values (F(2,26) = 7.72 p = 0.002, one-way ANOVA, 95% CI of control vs GDD = −1.23 to −0.28, corrected pairwise comparison), whereas they were unchanged in no-GDD mice (95% CI of control vs no-GDD = −0.78–0.15, corrected pairwise comparison; Fig. 3D).
Figure 3.

Noise-trauma-induced changes in spontaneous synaptic events in glutamatergic CNIC neurons. A, Example traces of sEPSCs from control, GDD, and no-GDD mice. Individual events are averages of 30–100 individual events from single neurons. B, Same as A but for sIPSCs. C, Summary graphs for sPSC frequency (left) and amplitude (right). sEPSC frequency was increased in both GDD and no-GDD mice compared with control mice (median exci frequency: control = 0.45 Hz, n = 12 neurons, n = 6 animals, GDD = 1.48 Hz, n = 9 neurons, n = 6 animals, no-GDD = 1.0 Hz, n = 11 neurons, n = 6 animals, H = 16.89, p = 0.0002, Kruskal–Wallis test). sIPSC frequency was indistinguishable between groups (H = 1.85, p = 0.40, Kruskal–Wallis test). Amplitudes of sEPSC and sIPSC did not differ between control and NE groups (sEPSC amplitude, F(2,29) = 0.135, p = 0.88, one-way ANOVA; sIPSC amplitude, H = 3.42, p = 0.18, Kruskal–Wallis test). D, The E:I index calculated for the sum of PSC amplitudes over 60 s was increased in GDD mice relative to control mice (median E:I index: control = −0.45, n = 10 neurons, n = 6 animals, GDD = 0.58, n = 9 neurons, n = 6 animals, no-GDD= −0.15, n = 10 neurons, n = 6 animals, F(2,26) = 7.72, p = 0.002, one-way ANOVA). Data are shown as box-and-whisker plots. *p < 0.05, **p < 0.01 in post hoc, pairwise assessments corrected for multiple comparisons.
In summary, these results demonstrate that traumatic noise exposure leads to a substantial reorganization of synaptic inputs received by glutamatergic CNIC neurons and that the magnitude and nature of this reorganization are different in mice with and without behavioral GDDs. In no-GDD mice, synaptic reorganization maintains the overall E:I balance, whereas in GDD mice, the pattern of reorganization leads to a profound disturbance of the E:I balance, resulting in a dominance of excitation.
Two types of GABAergic IC neurons
We next examined the effects of noise trauma on the synaptic inputs to inhibitory GABAergic neurons, which constitute 20–25% of the neuronal population in the IC (Oliver et al., 1994). GABAergic neurons were identified using mice in which dtTomato expression is restricted to neurons that express the vesicular GABA transporter (vgat-ires-cre-dT-loxP mice), a marker for all GABAergic IC neurons (Ito et al., 2009). In our mapping experiments, we could distinguish two types of GABAergic neurons based on their synaptic input patterns and their responses to direct glutamate stimulation (Fig. 4). Type 1 neurons (21/30) received both excitatory and inhibitory local inputs, with a dominance of inhibition, whereas type 2 GABAergic neurons (9/30 neurons) received predominantly excitatory inputs with very few or no inhibitory inputs (inhi input area ≤0.25 × 105 μm2 and inhi input charge ≤5 pC, see Materials and Methods; Fig. 4A). Compared with type 1 neurons, type 2 GABAeric neurons also had 2-fold larger responses to glutamate uncaging at the soma (direct responses; peak direct amplitude: U = 23.5, p = 0.0007, Mann–Whitney test; peak direct charge: U = 34, p = 0.0048, Mann–Whitney test; Fig. 4A,B) and type 2 neurons received more and larger sEPSCs (frequency: U = 13, p = 0.006, Mann–Whitney Test, amplitude: U = 14.5, p = 0.006, Mann–Whitney Test; Fig. 4C). The basic electrical membrane properties were similar for type 1 and type 2 neurons (Fig. 4, Table 1). Type 1 and type 2 neurons likely correspond to the two major classes of GABAergic neurons described previously in the IC (Ito et al., 2009; Ito et al., 2012; Ito et al., 2014). Type 2 neurons likely correspond to collico-thalamic GABAergic projection neurons, which receive dense, axosomatic glutamatergic synapses, whereas type 1 neurons likely correspond to local GABAergic interneurons, which receive fewer glutamatergic inputs that are located predominantly on dendrites (Ito et al., 2012; Ito et al., 2014).
Figure 4.
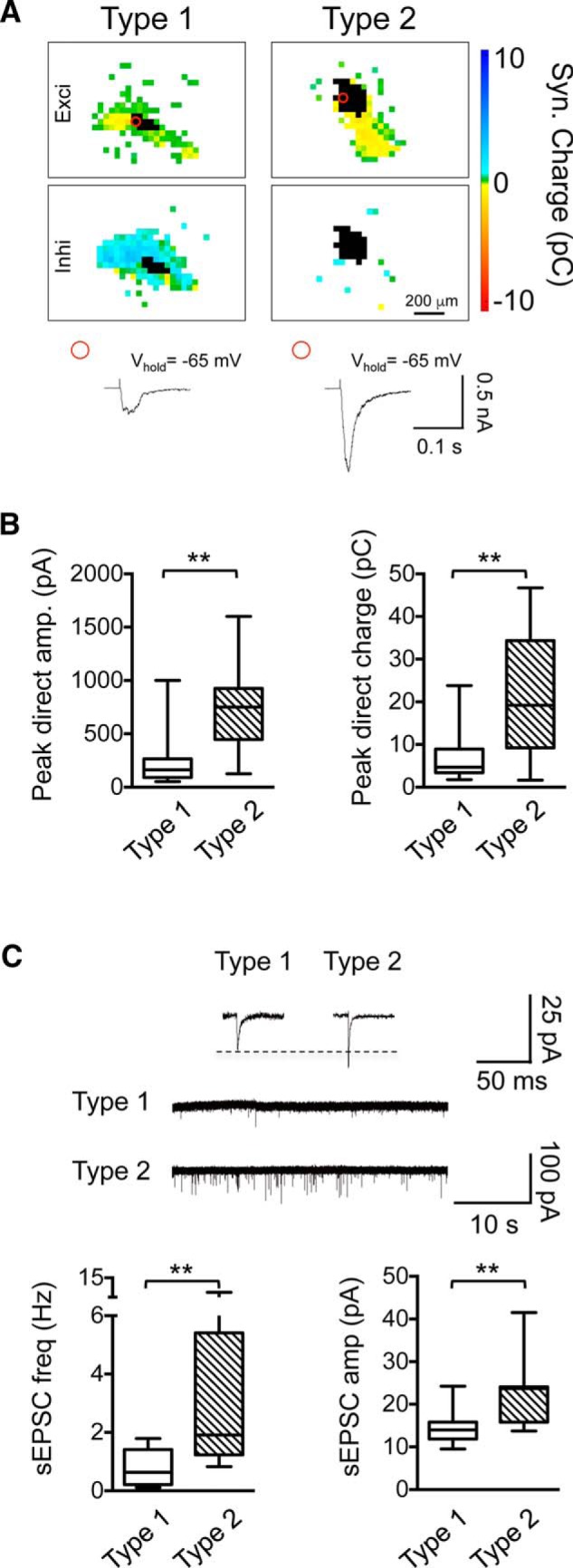
Two types of GABAergic CNIC neurons distinguished by local inputs. A, Example input maps of a type 1 and a type 2 GABAergic neuron. Type 1 neurons receive both excitatory and inhibitory inputs, whereas type 2 neurons receive predominantly excitatory inputs. Stimulation sites eliciting direct glutamate responses are in black. Example traces represent membrane currents elicited by glutamate uncaging over the soma. Circle indicates location of stimulation site. B, Direct response amplitude (left) and charge (right) are significantly larger in type 2 than in type 1 neurons (median peak amplitude, type 1 = 162.0 pA, n = 21 neurons, n = 10 animals, type 2 = 750.0 pA, n = 9 neurons, n = 8 animals, U = 23.50, p = 0.0007, two-tailed Mann–Whitney test; median peak charge, type 1 = 4.7 pC, n = 21 neurons, n = 10 animals, type 2 = 19.18 pC, n = 9 neurons, n = 8 animals, U = 34.0, p = 0.005, two-tailed Mann–Whitney test). C, Frequency and amplitudes of sEPSCs are significantly greater for type 2 neurons than for type 1 neurons (median frequency, type 1 = 0.63 Hz, n = 14 neurons, n = 8 animals, type 2 = 1.91 Hz, n = 7 neurons, n = 6 animals, U = 13.0, p = 0.006, two-tailed Mann–Whitney test; median amplitude, type 1 = 14.0 pA, n = 14 neurons, n = 8 animals, type 2 = 23.7 pA, n = 7 neurons, n = 6 animals, U = 14.5, p = 0.008, two-tailed Mann–Whitney test). Data are shown as box-and-whisker plots. Asterisks indicate statistical significance. *p < 0.05, **p < 0.01.
Table 1.
Intrinsic properties of type 1 and type 2 GABAergic IC neurons
| GABAergic neuron | Input resistance (MΩ) | Depolarization slope (V/S) | Repolarization slope (V/s) | Half-height width (ms) | Spike threshold (mV) |
|---|---|---|---|---|---|
| Type 1 | 262.8 ± 42.9 | 38.7 ± 4.4 | −38.0 ± 3.4 | 1.4 ± 0.1 | −33.1 ± 1.8 |
| Type 2 | 245.7 ± 53.9 | 40.4 ± 3.5 | −41.2 ± 3.3 | 1.4 ± 0.1 | −32.5 ± 1.7 |
All recordings were performed in vgat + IC neurons from non-noise-exposed animals (input resistance, type 1: n = 10 neurons, n = 5 animals, type 2: n = 6 neurons, n = 4 animals, p = 0.82; depolarization slope, type 1: n = 9 neurons, n = 5 animals, type 2: n = 6 neurons, n = 5 animals, p = 0.79; repolarization slope, type 1: n = 9 neurons, n = 5 animals, type 2: n = 6 neurons, n = 4 animals, p = 0.53; half-height width, type 1: n = 9 neurons, n = 5 animals, type 2: n = 6 neurons, n = 4 animals, p = 0.55; spike threshold, type 1: n = 10 neurons, n = 5 animals, type 2: n = 6 neurons, n = 4 animals, p = 0.82). Depolarization slope: maximum depolarizing slope, repolarization slope: minimum repolarizing slope, spike threshold: minimum voltage of first spike generation.
The excitatory input maps of type 1 and type 2 GABAergic CNIC neurons were similar to each other and were also similar to the excitatory input maps of glutamatergic neurons (exci input area: F(2,41) = 0.35, p = 0.78, one-way ANOVA; exci postsynaptic charge: H = 0.05, p = 0.98, Kruskal–Wallis test). The inhibitory input maps of type 1 GABAergic neurons were not significantly different from the inhibitory input maps of glutamatergic neurons (inhi input area: t(31) = 1.11, p = 0.28, two-tailed Student's t test; inhi postsynaptic charge: U = 101, p = 0.36, Mann–Whitney test). However, the mean E:I index calculated for individual type 1 GABAergic neurons was less negative than for glutamatergic neurons (Table 2), indicating that local inputs to type 1 GABAergic neurons are less dominated by inhibition than local inputs to glutamatergic neurons.
Table 2.
E:I balance for excitatory and inhibitory IC neurons
| vglut2+ | Type 1 vgat+ | p value | |
|---|---|---|---|
| E:I index input area | −0.50 ± 0.09 | −0.23 ± 0.07 | 0.030 |
| E:I index input charge | −0.58 ± 0.15 | −0.28 ± 0.1 | 0.048 |
| E:I index spontaneous events | −0.46 ± 0.09 | −0.27 ± 0.12 | 0.25 |
All recordings were performed in either vgat + or vglut2 + IC neurons from non-noise-exposed animals (E:I index input area, vglut2+: n = 12 neurons, n = 6 animals, vgat+: n = 18 neurons, n = 10 animals, p = 0.030, Student's t test; E:I index input charge, vglut2+: n = 12 neurons, n = 6 animals, vgat+: n = 20 neurons, n = 10 animals, p = 0.048, Mann–Whitney test; E:I index spontaneous events, vglut2+: n = 10 neurons, n = 6 animals, vgat+: n = 14 neurons, n = 10 animals, p = 0.25, Student's t test).
Reorganization of synaptic inputs onto GABAergic CNIC neurons
Noise trauma affected the local inputs to type 1 GABAergic neurons, but not to type 2 neurons. In GDD mice, excitatory input maps to type 1 neurons were smaller and weaker (exci input area: F(2,37) = 7.16, p = 0.002, one-way ANOVA, 95% CI of control vs GDD = 0.36–1.75 × 105 μm2, corrected pairwise comparison; exci input charge: H = 10.44, p = 0.005, Kruskal–Wallis test, mean rank difference of control vs GDD = 13.57, Dunn's test; Fig. 5A–C), whereas inhibitory input maps were not changed significantly (Fig. 5B). Therefore, in GDD mice, the E:I indices of type 1 neurons shifted to more negative values, indicating a shift toward increased inhibition (input area: H = 13.01, p = 0.0015, Kruskal–Wallis test, mean rank difference of control vs GDD = 14.8, Dunn's test; postsynaptic charge: H = 13.35, p = 0.0013, Kruskal–Wallis test, mean rank difference of control vs GDD = 16.2, Dunn's test; Fig. 5D,E). Spontaneous synaptic events in type 1 from GDD mice neurons were not different from controls (Fig. 6).
Figure 5.

Noise-induced reorganization of synaptic input maps onto type 1 GABAergic neurons. A, Schematic of synaptic input mapping of type 1 GABAergic neurons (black triangle). Excitatory (circle) and inhibitory (triangle) inputs are shown in red. B, Examples of input maps for type 1 GABAergic neurons in control, GDD, and no-GDD mice. Current traces illustrate excitatory (circle) and inhibitory (triangle) synaptic responses to glutamate uncaging (asterisk) at the locations indicated by symbols. Uncaging sites that elicited direct responses are in black. C, Excitatory input area and total excitatory charge were decreased in GDD mice, relative to control mice (median exci area: control = 1.33 × 105 μm2, n = 20 neurons, n = 10 animals, GDD = 0.25 × 105 μm2, n = 12 neurons, n = 5 animals, no-GDD = 0.65 × 105, n = 9 neurons, n = 6 animals, F(2,37) = 7.16, p = 0.002, one-way ANOVA; median exci charge, control = 23.7 pC, n = 20 neurons, n = 10 animals, GDD = 3.5 pC, n = 11 neurons, n = 5 animals, no-GDD = 7.9 pC, n = 9 neurons, n = 6 animals, H = 10.44, p = 0.005, Kruskal–Wallis test). Inhibitory input area and total inhibitory charge were decreased in no-GDD mice compared with control mice (median inhi area: control = 2.25 × 105 μm2, n = 21 neurons, n = 10 animals, GDD = 1.55 × 105 μm2, n = 13 neurons, n = 5 animals, no-GDD = 0.93 × 105, n = 9 neurons, n = 6 animals, H = 10.97, p = 0.004, Kruskal–Wallis test; median inhi charge, control = 86.3 pC, n = 21 neurons, n = 10 animals, GDD = 38.3 pC, n = 13 neurons, n = 5 animals, no-GDD = 29.4 pC, n = 9 neurons, n = 6 animals, H = 6.02, p = 0.049, Kruskal–Wallis test). D, Relationship between excitatory and inhibitory synaptic inputs for individual neuron. Lines connect measurements from individual cells. E, E:I indices of control, GDD, and no-GDD mice. In GDD mice, the E:I indices for input area and input charge were shifted to more negative values relative to both control and no-GDD mice (median E:IArea: control = −0.28, n = 20 neurons, n = 10 animals, GDD = −0.65, n = 12 neurons, n = 5 animals, no-GDD = −0.21, n = 9 neurons, n = 6 animals, H = 13.01, p = 0.0015, Kruskal–Wallis test, median E:ICharge: control = −0.36, n = 20 neurons, n = 10 animals, GDD = −0.84, n = 12 neurons, n = 5 animals, no-GDD = −0.69, n = 9 neurons, n = 6 animals, H = 13.35, p = 0.0013, Kruskal–Wallis test). Data are shown as box-and-whisker plots. *p < 0.05, **p < 0.01 in post hoc, pairwise assessments corrected for multiple comparisons.
Figure 6.

Spontaneous synaptic events onto type 1 GABAergic neurons in noise-traumatized mice. A, Example traces of sEPSCs from control, GDD, and no-GDD mice. Individual event traces are the average of 30–100 events from a single cell. B, Same as A but for sIPSCs. C, Frequencies and amplitudes of sEPSCs were increased in no-GDD mice (median sEPSC frequency: control = 0.63 Hz, n = 14 neurons, n = 8 animals, GDD = 0.33 Hz, n = 15 neurons, n = 6 animals, no-GDD = 1.50 Hz, n = 7 neurons, n = 5 animals, F(2,33) = 7.39, p = 0.0022, one-way ANOVA; median sEPSC amplitude: control = 14.0 pA, n = 14 neurons, n = 8 animals, GDD = 13.0 pA, n = 15 neurons, n = 6 animals, no-GDD = 24.1 pA, n = 7 neurons, n = 5 animals, H = 8.80, p = 0.0123, Kruskal–Wallis test). The frequencies and amplitudes of sIPSCs were indistinguishable between groups (sIPSC frequency, F(2,33) = 1.141, p = 0.33, one-way ANOVA; sIPSC amplitude, F(2,33) = 0.236, p = 0.79, one-way ANOVA). D, E:I index calculated for the sum of PSC amplitudes over 60 s was increased in no-GDD mice relative to both GDD and control mice (median E:I index: control = −0.32, n = 14 neurons, n = 8 animals, GDD = −0.62, n = 15 neurons, n = 6 animals, no-GDD = 0.67, n = 7 neurons, n = 5 animals, F(2,33) = 19.74, p < 0.0001, one-way ANOVA). Data are shown as box-and-whisker plots. *p < 0.05, **p < 0.01 in post hoc, pairwise assessments corrected for multiple comparisons.
In no-GDD mice, the excitatory input maps of type 1 GABAergic neurons were not statistically different from control mice (Fig. 5C), but the inhibitory input maps were smaller and weaker (inhi input area: H = 10.97, p = 0.004, Kruskal–Wallis test, mean rank difference of control vs no-GDD = 15.8, Dunn's test; inhi postsynaptic charge: H = 6.022, p = 0.049, Kruskal–Wallis test, mean rank difference of control vs no-GDD = 12.0, Dunn's test; Fig. 5C). However, these changes did not shift the median E:I index, which remained similar to controls (Fig. 5D,E). In no-GDD mice, type 1 GABAergic neurons had increased spontaneous excitatory activity involving both EPSC amplitudes (H = 8.8, p = 0.012, Kruskal-Wallis test, mean rank difference of GDD vs no-GDD = 12.0, Dunn's test) and frequencies (F(2,33) = 7.39, p = 0.0022, one-way ANOVA, 95% CI of control vs no-GDD = −2.06 to −0.22 Hz, corrected pairwise comparison; Fig. 6A,C), whereas sIPSCs remained unchanged (Fig. 6B,C). As a result, the mean E:I index of spontaneous synaptic events for type 1 neurons shifted from the negative values present in control and GDD mice to a positive value (F(2,33) = 19.74, p < 0.0001, one-way ANOVA, 95% CI of control vs no-GDD = −1.23 to −0.39, 95% CI of GDD vs no-GDD = −1.48 to −0.644, corrected pairwise comparisons; Fig. 6D), indicating increased synaptic excitation of type 1 GABAergic neurons in no-GDD mice.
In contrast to type 1 neurons, noise trauma had no effect on the synaptic inputs of type 2 neurons (Fig. 7). In both GDD and no-GDD mice, the excitatory input maps of type 2 GABAergic neurons were indistinguishable from control mice with respect to size and total postsynaptic charge (Fig. 7B). In addition, sEPSC amplitudes and frequencies were also unaffected by noise trauma (Fig. 7C). Together, these results demonstrate a cell-type- and input-specific reorganization of synaptic connections onto GABAergic CNIC neurons. In GDD mice, synaptic inputs to type 1 neurons became more dominated by inhibition, leading to a disinhibition of the CNIC in mice with behavioral GDDs.
Figure 7.

Synaptic inputs onto type 2 GABAergic neurons are unchanged in noise-traumatized mice. A, Examples of excitatory input maps of type 2 GABAergic neurons from control, GDD, and no-GDD mice. Uncaging sites that elicited direct responses in the recorded neuron are marked in black. B, Noise exposure had no effect on excitatory input area (left) or on total excitatory postsynaptic charge (right; area: control, n = 9 neurons, n = 8 animals, GDD, n = 7 neurons, n = 4 animals, no-GDD, n = 10 neurons, n = 6 animals, H = 0.067, p = 0.97, Kruskal–Wallis test; charge: control, n = 9 neurons, n = 8 animals, GDD, n = 7 neurons, n = 4 animals, no-GDD, n = 10 neurons, n = 6 animals, F(2,23) = 0.49, p = 0.62, one-way ANOVA). C, Amplitudes (top) and frequency of spontaneous EPSCs are unchanged by noise trauma (control, n = 7 neurons, n = 6 animals, GDD, n = 6 neurons, n = 3 animals, no-GDD, n = 10 neurons, n = 6 animals, amplitudes: F(2,20) = 1.13, p = 0.34, one-way ANOVA; frequency: F(2,20) = 1.00, p = 0.38, one-way ANOVA). Data are shown as box-and-whisker plots. *p < 0.05, **p < 0.01 in post hoc, pairwise assessments corrected for multiple comparisons.
Posttraumatic AE prevents circuit reorganization and GDDs
In the primary visual and auditory cortices, sensory deprivation can open periods of enhanced synaptic plasticity during which sensory experience can reshape neuronal circuits (He et al., 2006; Zhou et al., 2011; Zhu et al., 2014). Noise-induced hearing loss may open a similar sensitive period in the CNIC that enables local circuit reorganization, which may be influenced by posttraumatic auditory experience. To test this idea, we exposed mice immediately after noise trauma for 7 d to moderate intensity (75 dB), pulsed white noise. We chose pulsed white noise because of its strong effect in preventing map refinement in the developing primary auditory cortex (Zhang et al., 2002) and in interfering with developmental sharpening of sound frequency tuning of IC neurons (Sanes et al., 1983) without, however, affecting tonotopic refinement in lower brainstem nuclei (Clause et al., 2014).
Posttraumatic AE with pulsed white noise prevented the noise-induced reorganizations of both excitatory and inhibitory input maps to glutamatergic and GABAergic CNIC neurons (Fig. 8). In NE-AE mice, excitatory and inhibitory input maps and E:I indices of glutamatergic CNIC neurons remained indistinguishable from control mice (Fig. 8A–C). Likewise, input maps and E:I indices of type 1 GABAergic neurons were also indistinguishable between NE-AE and control mice (Fig. 8D–F). AE itself had no effect on local CNIC circuits. Age-matched control mice, which did not receive noise trauma but did receive AE, showed no changes in intrinsic CNIC circuitry, as indicated by stable excitatory and inhibitory input maps and E:I indices for glutamatergic neurons (Fig. 8G,I) and for type 1 GABAergic neurons (Fig. 8H,I). Therefore, posttraumatic AE with moderate intensity, pulsed white noise prevented noise-induced circuit reorganization without having an effect on CNIC circuits in nontraumatized control mice.
Figure 8.

AE with pulsed white noise inhibits noise-trauma-induced circuit reorganization. A, Excitatory input maps of glutamatergic neurons (Vglut2+) in control, GDD, no-GDD (same data as in Figs. 2, 5), and NE-AE mice (median exci area: control = 1.15 × 105 μm2, n = 15 neurons, n = 6 animals, NE-AE = 1.43 × 105 μm2, n = 9 neurons, n = 4 animals, F(3,42) = 1.78, p = 0.180, one-way ANOVA: 95% CI of difference between control vs NE-AE = −1.45–0.92 × 105 μm2, corrected pairwise comparison after one-way ANOVA). B, Same as A but for inhibitory input maps (median inhi area: control = 2.95 × 105 μm2, n = 12 neurons, n = 6 animals, NE-AE = 3.88 × 105 μm2, n = 9 neurons, n = 4 animals, F(3,39) = 6.008, p = 0.0018, one-way ANOVA: 95% CI of difference between control vs NE-AE = −2.44–1.11 × 105 μm2, corrected pairwise comparison after one-way ANOVA). C, E:I indices of glutamatergic neurons. The E:I indices from NE-AE animals are not significantly different from those of control animals (median E:I index, control = −0.59, n = 12 neurons, n = 6 animals, NE-AE = −0.44, n = 9 neurons, n = 4 animals, F(3,39) = 11.18, p < 0.0001, one-way ANOVA: 95% CI of difference between control vs NE-AE = −0.65–0.24, corrected pairwise comparison after one-way ANOVA). D–F, Same as in A–C but for type 1 GABAergic neurons (vgat+). D, Excitatory input maps (median exci area: control = 1.33 × 105 μm2, n = 20 neurons, n = 10 animals, NE-AE = 2.01 × 105 μm2, n = 8 neurons, n = 4 animals, H = 17.50, p = 0.0006, Kruskal–Wallis test, mean rank difference for control vs NE-AE = −5.36, p > 0.05, Dunn's test after Kruskal–Wallis test). E, Inhibitory input maps (median inhi area, control = 2.25 × 105 μm2, n = 21 neurons, n = 10 animals, NE-AE = 1.38 × 105 μm2, n = 9 neurons, n = 4 animals, H = 10.60, p = 0.014, Kruskal–Wallis test, mean rank difference for control vs NE-AE = 4.93, p > 0.05, Dunn's test after Kruskal–Wallis test). F, E:I indices (median E:I index, control = −0.28, n = 20 neurons, n = 10 animals, NE-AE = −0.14, n = 8 neurons, n = 4 animals, H = 15.17, p = 0.0017, Kruskal–Wallis test, mean rank difference of between control vs NE-AE = −5.50, p > 0.05, Dunn's test after Kruskal–Wallis test). G, AE applied to animals without noise trauma had no effect on excitatory or inhibitory input maps of glutamatergic neurons (vglut2+; exci area: control, n = 15 neurons, n = 6 animals, AE-only, n = 7 neurons, n = 4 animals, t(20) = 0.850, p = 0.41, two-tailed Student's t test: inhi area: control, n = 12 neurons, n = 6 animals, AE-only, n = 7 neurons, n = 4 animals, t(17) = p = 0.12, two-tailed Student's t test). H, Same as G but for type 1 GABAergic neurons (vgat+; exci area: control, n = 20 neurons, n = 10 animals, AE-alone, n = 9 neurons, n = 3 animals, t(27) = 0.99, p = 0.33, two-tailed Student's t test; inhi area: control, n = 21 neurons, n = 10 animals, AE-alone, n = 9 neurons, n = 3 animals, t(28) = 1.07, p = 0.29, two-tailed Student's t test). I, AE in nontraumatized animals did not change E:I indices for glutamatergic or for type 1 GABAergic neurons (Vglut2+ neurons: control, n = 12 neurons, n = 6 animals, AE-only, n = 7 neurons, n = 4 animals, t(17) = 1.08, p = 0.30, two-tailed Student's t test: Vgat+ neurons: control, n = 20 neurons, n = 10 animals, AE-only, n = 9 neurons, n = 3 animals, t(27) = 1.75, p = 0.09, two-tailed Student's t test). Data are shown as box-and-whisker plots. *p < 0.05, **p < 0.01.
AE also prevented the emergence of GDDs. Noise-traumatized mice with AE had stable gap detection ratios before and after noise exposure (gap ratio change, NE-AE = 0.02 ± 0.02, n = 84 sound frequencies; Fig. 9A). AE significantly reduced the fraction of NE animals showing GDDs from 51% (19/37 animals) to 12% (3/25 animals) (p = 0.0025, two-tailed Fisher's exact test; Fig. 9C). In contrast, AE did not change PPI of the ASR (Fig. 9B), indicating that the effect of AE was specific for gap detection and that AE did not affect hearing sensitivity or the neuronal circuits that mediate acoustic startle behavior. In addition, the protective effects of AE against noise-induced circuit reorganization and GDDs were not due to a maintenance or restoration of hearing (Zhu et al., 2014) because AE did not reverse ABR threshold shifts for either low (10–16 kHz: t(28) = 0.725, p = 0.472, two-tailed Student's t test) or high (20–32 kHz: t(28) = 0.185, p = 0.854, two-tailed Student's t test) sound frequencies (Fig. 9D). Finally, the effects of AE were only present in noise-traumatized mice. Control mice (no noise trauma) receiving identical AE showed no changes in gap inhibition of the ASR (p = 0.59, n = 16 animals, Kolmogorov–Smirnov test; Fig. 9E), prepulse inhibition of the ASR (p = 0.83, n = 16 animals, Kolmogorov–Smirnov test; Fig. 9F), or hearing thresholds (10–16 kHz: p = 0.44, n = 6 animals, Wilcoxon test; 20–32 kHz: p = 0.60, n = 6 animals, two-tailed paired t test; Fig. 9G).
Figure 9.

AE prevents the development of GDDs. A, AE prevented the noise-induced increase in gap detection ratios observed in GDD mice (gap ratio change: control = 0.03 ± 0.02, n = 89 sound frequencies, GDD = 0.18 ± 0.02, n = 93 sound frequencies, no-GDD = 0.00 ± 0.01, n = 110 sound frequencies, NE-AE = 0.02 ± 0.02, n = 84 sound frequencies, p < 0.0001, Kruskal–Wallis test). B, PPI of the startle response was not significantly different between groups (PPI ratio change, control = −0.12 ± 0.03, n = 90 sound frequencies; GDD = 0.00 ± 0.03, n = 65 sound frequencies, no-GDD = −0.10 ± 0.04, n = 100 sound frequencies, NE-AE = −0.15 ± 0.04, n = 89 sound frequencies). C, Percentage of NE animals that developed GDDs (black) was decreased by AE (green; NE without AE, n = 37 animals, NE with AE, n = 25, p = 0.0025, two-tailed Fisher's exact test). D, AE did not affect noise-induced ABR threshold shifts at low or high frequencies (10–16 kHz: NE = 13.6 ± 2.8 dB, n = 17 animals, NE+AE = 16.2 ± 2.0 dB, n = 13 animals, t(28) = 0.73, p = 0.47, two-tailed Student's t test; 20–32 kHz: NE = 16.5 ± 3.0 dB, n = 17 animals, NE+AE = 15.8 ± 2.7 dB, n = 13 animals, t(28) = 0.185, p = 0.85, two-tailed Student's t test). E, AE delivered to control mice without noise exposure had no effect on gap detection ratios (gap ratio change, control = 0.03 ± 0.02, n = 89 sound frequencies, AE-only = 0.04 ± 0.02, n = 75 sound frequencies, p = 0.59, Kolmogorov–Smirnov test). F, Same as E but for PPI inhibition of the ASR (PPI ratio change, control = −0.12 ± 0.03, n = 90 sound frequencies, AE-only = −0.11 ± 0.04, n = 77 sound frequencies, p = 0.83, Kolmogorov–Smirnov test). G, AE for 7 d delivered to control mice without noise exposure did not change ABR thresholds (10–16 kHz: median threshold before AE = 27.5 dB, after AE = 34.0 dB, n = 6 animals, p = 0.43, Wilcoxon test; 20–32 kHz: median threshold before AE = 29.2 dB, after AE = 30.8, n = 6 animals, p = 0.60, two-tailed paired t test). Data in D and G are shown as box-and-whisker plots. *p < 0.05, **p < 0.01.
Discussion
In this study, we demonstrate that mild noise-induced hearing loss induced a substantial reorganization of excitatory and inhibitory local circuits in the CNIC and that the nature of these reorganizations correlated with the animals' ability to detect silent gaps. In GDD mice, but not in no-GDD mice, glutamatergic CNIC neurons received less synaptic inhibition, which shifted the E:I balance toward excitation. Type 1 GABAergic CNIC neurons showed the reverse, receiving less excitation but maintaining inhibition, thus shifting the E:I balance toward inhibition. The decreased inhibition of excitatory neurons and the increased inhibition of inhibitory neurons indicate an overall functional “disinhibition” of the CNIC in GDD mice. Noise-induced IC plasticity of intrinsic CNIC circuits and its associated hearing deficits are sensitive to auditory experience because posttraumatic exposure to moderate-intensity patterned white noise prevented both noise-induced circuit reorganization and GDDs.
Gap detection impairment after noise trauma
The deficit in gap detection we observed in ∼50% of NE mice is consistent with impaired temporal processing after hearing loss (Giraudi-Perry et al., 1982; Rybalko et al., 2005). Deficits in the detection of silent gaps are also commonly interpreted as a behavioral sign of tinnitus (Wang et al., 2009; Engineer et al., 2011; Middleton et al., 2011; Dehmel et al., 2012; Li et al., 2013; Kalappa et al., 2014; Kalappa et al., 2015; Li et al., 2015; Wu et al., 2016) based on the idea that the tinnitus masks silent gaps, making them less perceivable (Turner et al., 2006). More recently, the idea that tinnitus masks sound gaps has been challenged by studies of tinnitus patients who show gap-induced inhibition of the ASR at frequencies well outside of the tinnitus frequency (Fournier et al., 2013), as well as tinnitus patients showing no deficits if silent gaps are identified consciously in psychoacoustic tasks (Boyen et al., 2015). Therefore, whereas it is at least plausible that GDD mice may experience tinnitus, further support from additional behavioral paradigms will be necessary to clarify this issue.
Cell-type-specific organization of synaptic input maps in the CNIC
Our mapping of local synaptic inputs to IC neurons revealed three cell-type-specific input map configurations in the CNIC, providing new insight into the organization of intrinsic IC connectivity. First, glutamatergic neurons receive local excitatory and inhibitory inputs with an E:I balance that is strongly biased toward inhibition. This is similar to the input patterns described previously for a population of unidentified, but presumed glutamatergic, IC neurons in 3-week-old mice (Sturm et al., 2014), supporting the notion that the developmental refinement of IC circuits is largely completed by the third postnatal week (Ehret et al., 1992; Yu et al., 2005). Second, type 1 GABAergic neurons, which likely correspond to local interneurons (Ito et al., 2009), also receive excitatory and inhibitory local inputs, but the E:I index for type 1 neurons is only slightly biased toward inhibition. Together, these results suggest that the dominant effect of the intrinsic synaptic network in the CNIC is inhibitory. Finally, type 2 GABAergic neurons receive almost exclusively excitatory intrinsic inputs. Although the projection patterns of type 1 and type 2 GABAergic neurons remain to be identified, many properties of type 2 neurons resemble those of thalamic-projecting GABAergic neurons (Ito et al., 2009). The lack of local inhibitory inputs to type 2 neurons is consistent with their role in providing fast feedforward inhibition from the colliculus to the thalamic medial geniculate nucleus (Ito et al., 2012).
Reorganization of intrinsic CNIC circuits after noise trauma
Noise-induced hearing loss consistently triggered a reorganization of the intrinsic CNIC network. However, the magnitude and patterns of synaptic reorganization correlated with behaviorally measured gap detection, such that only GDD mice exhibited a functional disinhibition of local IC circuitry. In GDD mice, glutamatergic IC neurons lost local inhibition, which shifted their E:I balance toward excitation (Figs. 2, 3). Type 1 GABAergic IC neurons lost synaptic excitation, which shifted their E:I balance toward inhibition (Figs. 5, 6). This disinhibition of intrinsic CNIC circuits in GDD mice likely contributes to the IC hyperactivity that is a neurological hallmark of tinnitus and hyperacusis in rodent models (Bauer et al., 2008), as well as in human tinnitus patients (Melcher et al., 2000). A disinhibition of the IC may provide increased excitatory drive to the medial geniculate nucleus, contributing to the thalamocortical dysrhythmia that is observed in tinnitus patients (van Gendt et al., 2012, De Ridder et al., 2015).
It remains to be shown whether functional reorganization is accompanied or followed by a structural reorganization such as axonal pruning/growth or remodeling of dendritic branches or spines. Few studies have investigated the effects of hearing-loss on structural IC connectivity (Nordeen et al., 1983; Moore et al., 1988), intrinsic connections or neuronal morphology. Considering the close relationship of functional and structural reorganization in many brain areas (Hensch, 2005; Holtmaat et al., 2009; Clause et al., 2014), structural remodeling of intrinsic IC connections after cochlear trauma is highly plausible. Compared with the dynamic regulation of synaptic strengths and neuronal membrane excitability, structural changes are usually long lasting and can permanently impair sensory processing (Wiesel et al., 1963; Hofer et al., 2009; Sengpiel, 2014) and thus could underlie the persistence of temporal processing deficits, hyperacusis, and tinnitus.
Increased activity in the IC is evident shortly after noise trauma (Willott et al., 1982) and likely reflects an increase in afferent activity from the dorsal cochlear nucleus (DCN) because early noise-induced IC hyperactivity is abolished by ablation of the DCN (Brozoski et al., 2012; Manzoor et al., 2012). The shift in the E:I balance in the CNIC of GDD animals 7 d after noise trauma suggests that changes in IC circuits itself can contribute to hyperactivity, which in turn may trigger cortical reorganizations (Noreña et al., 2003; Engineer et al., 2011; Llano et al., 2012). The IC also receives efferent cortical projections, which play an important role in mediating plasticity in the IC (Bajo et al., 2010) and thus could also play a role in the initiation of or determination of the nature of IC circuit reorganization after hearing loss.
It should be noted that, in our experiments, mice underwent traumatic noise exposure at 3 weeks of age, which raises the possibility that some of the effects that we observed reflect a disturbance of development. However, we find this possibility to be unlikely because of a number of studies showing adult-like properties in the IC of three week-old mice, including tonotopic organization (Romand and Ehret, 1990, Yu et al., 2005), thresholds and latencies of tone-evoked responses (Ehret and Romand, 1992), stimulus following (Sanes and Constantine-Paton, 1985), and dynamic intensity ranges and intensity functions (Shnerson and Willott, 1979). Another question that remains to be addressed is whether the GDDs that we observed 1 week after noise trauma are permanent or temporary. In our ongoing preliminary results (data not shown), we found no spontaneous recovery from these deficits over a 2 month period, which suggests that these behavior deficits are permanent, although more extensive studies need to be done to address this issue.
Prevention of IC circuit reorganization by AE
We found that AE with pulsed white noise immediately after noise exposure could prevent the reorganization of synaptic circuitry in the CNIC. Previous studies have shown that stimulation with pulsed white noise during development prevents the refinement of cortical tonotopic maps and frequency tuning of IC neurons (Sanes et al., 1983; Zhang et al., 2002). Although the 3- to 4-week-old mice used in this study are not fully mature, it is unlikely that the effects of white noise on inhibiting CNIC reorganization reflect a disturbance of normal developmental refinement because pulse white noise had no effect in control mice without acoustic trauma. Instead, we propose that noise trauma and the resulting hearing loss opened a “sensitive period” in the IC similar to the reopening of a “critical period” in mature visual cortex by dark rearing (He et al., 2006) or in auditory cortex by long-term noise exposure (Zhou et al., 2011). Previous studies have shown that AE after noise exposure can prevent reorganization of tonotopic maps in auditory cortex (Noreña et al., 2005), although the conservation of map organization in these experiments may have resulted from a recovery of ABR thresholds (Noreña et al., 2005), which we did not observe in our conditions. In the cerebral cortex, reopening of critical period plasticity involves a decrease in GABAergic inhibition (Huang et al., 2010; Zhou et al., 2011; Gu et al., 2016), which also occurs in the IC after noise trauma (this study; Milbrandt et al., 2000; Dong et al., 2010). Because local CNIC circuits are dominated by inhibition (i.e., negative E:I ratios), it is conceivable that AE prevented a reorganization of CNIC circuits by increasing net inhibitory activity via the dominance of intrinsic inhibition.
Prevention of GDDs by AE
Posttraumatic AE not only prevented circuit reorganization in the CNIC, but also reduced the occurrence of GDDs significantly (from 51% to 12% of mice). These results encourage further studies to test immediate posttraumatic acoustic stimulation as a prophylactic measure in humans to prevent the emergence of central hearing deficits, including tinnitus, after acoustic trauma. Although acoustic stimulation (e.g., sound therapy) to treat tinnitus has been tried for many years, it has been largely unsuccessful (Vanneste et al., 2013), perhaps because sound therapy is usually started months or years after tinnitus has been present (i.e., after the closure of a trauma-induced sensitive period). It will be interesting to determine what types of acoustic stimulation are the most effective in preventing synaptic reorganization and GDDs. A better understanding of the mechanisms by which auditory experience influences posttraumatic refinement may eventually lead to new clinical interventions that can alleviate central hearing deficits after noise trauma, traumatic brain injury, or treatment with ototoxic drugs.
Footnotes
This work was supported by the National Institutes of Health (National Institute on Deafness and Communication Disorders Grant 04399 to K.K. and Grant 1F30DCO14177 to J.J.S.) and the University of Pittsburgh Physicians Academic Foundation (K.K.). We thank A. Barth, A. Magnusson, S. Ross, S. Sadagopan, R. Seal, and P. Strick for comments on previous versions of the manuscript; Naama Balass for editorial support; and the members of the Auditory Research Group at the University of Pittsburgh for discussions.
The authors declare no competing financial interests.
References
- Adams JC. (1979) Ascending projections to the inferior colliculus. J Comp Neurol 183:519–538. 10.1002/cne.901830305 [DOI] [PubMed] [Google Scholar]
- Baguley D, McFerran D, Hall D (2013) Tinnitus. Lancet 382:1600–1607. 10.1016/S0140-6736(13)60142-7 [DOI] [PubMed] [Google Scholar]
- Bajo VM, King AJ (2012) Cortical modulation of auditory processing in the midbrain. Front Neural Circuits 6:114. 10.3389/fncir.2012.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo VM, Nodal FR, Moore DR, King AJ (2010) The descending corticocollicular pathway mediates learning-induced auditory plasticity. Nat Neurosci 13:253–260. 10.1038/nn.2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CA, Turner JG, Caspary DM, Myers KS, Brozoski TJ (2008) Tinnitus and inferior colliculus activity in chinchillas related to three distinct patterns of cochlear trauma. J Neurosci Res 86:2564–2578. 10.1002/jnr.21699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JI, Coomber B (2015) Tinnitus-related changes in the inferior colliculus. Front Neurol 6:61. 10.3389/fneur.2015.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyen K, Başkent D, van Dijk P (2015) The gap detection test: can it be used to diagnose tinnitus? Ear Hear 36:e138–145. 10.1097/AUD.0000000000000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozoski TJ, Bauer CA (2016) Animal models of tinnitus. Hear Res 338:88–97. 10.1016/j.heares.2015.10.011 [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Bauer CA, Caspary DM (2002) Elevated fusiform cell activity in the dorsal cochlear nucleus of chinchillas with psychophysical evidence of tinnitus. J Neurosci 22:2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozoski TJ, Wisner KW, Sybert LT, Bauer CA (2012) Bilateral dorsal cochlear nucleus lesions prevent acoustic-trauma induced tinnitus in an animal model. J Assoc Res Otolaryngol 13:55–66. 10.1007/s10162-011-0290-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rűbsamen R, Kandler K (2014) The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82:822–835. 10.1016/j.neuron.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Nguyen T, Kandler K (2011) An acoustic startle-based method of assessing frequency discrimination in mice. J Neurosci Methods 200:63–67. 10.1016/j.jneumeth.2011.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ridder D, Vanneste S, Langguth B, Llinas R (2015) Thalamocortical dysrhythmia: a theoretical update in tinnitus. Front Neurol 6:124–137. 10.3389/fneur.2015.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S, Mulders WH, Rodger J, Woo S, Robertson D (2010) Acoustic trauma evokes hyperactivity and changes in gene expression in guinea-pig auditory brainstem. Eur J Neurosci 31:1616–1628. 10.1111/j.1460-9568.2010.07183.x [DOI] [PubMed] [Google Scholar]
- Dehmel S, Eisinger D, Shore SE (2012) Gap prepulse inhibition and auditory brainstem-evoked potentials as objective measures for tinnitus in guinea pigs. Front Syst Neurosci 6:42. 10.3389/fnsys.2012.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont JJ. (2013) Hearing loss, hyperacusis, or tinnitus: what is modeled in animal research? Hear Res 295:140–149. 10.1016/j.heares.2012.01.005 [DOI] [PubMed] [Google Scholar]
- Ehret G, Romand R (1992) Development of tone response thresholds, latencies and tuning in the mouse inferior colliculus. Brain Res Dev Brain Res 67:317–326. 10.1016/0165-3806(92)90233-M [DOI] [PubMed] [Google Scholar]
- Engineer ND, Riley JR, Seale JD, Vrana WA, Shetake JA, Sudanagunta SP, Borland MS, Kilgard MP (2011) Reversing pathological neural activity using targeted plasticity. Nature 470:101–104. 10.1038/nature09656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier P, Hébert S (2013) Gap detection deficits in humans with tinnitus as assessed with the acoustic startle paradigm: does tinnitus fill in the gap? Hear Res 295:16–23. 10.1016/j.heares.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Gao E, Suga N (1998) Experience-dependent corticofugal adjustment of midbrain frequency map in bat auditory system. Proc Natl Acad Sci U S A 95:12663–12670. 10.1073/pnas.95.21.12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cairasco N. (2002) A critical review on the participation of inferior colliculus in acoustic-motor and acoustic-limbic networks involved in the expression of acute and kindled audiogenic seizures. Hear Res 168:208–222. 10.1016/S0378-5955(02)00371-4 [DOI] [PubMed] [Google Scholar]
- Giraudi-Perry DM, Salvi RJ, Henderson D (1982) Gap detection in hearing-impaired chinchillas. J Acoust Soc Am 72:1387–1393. 10.1121/1.388444 [DOI] [PubMed] [Google Scholar]
- Grimsley CA, Sanchez JT, Sivaramakrishnan S (2013) Midbrain local circuits shape sound intensity codes. Front Neural Circuits 7:174. 10.3389/fncir.2013.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Tran T, Murase S, Borrell A, Kirkwood A, Quinlan EM (2016) Neuregulin-dependent regulation of fast-spiking interneuron excitability controls the timing of the critical period. J Neurosci 36:10285–10295. 10.1523/jneurosci.4242-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HY, Hodos W, Quinlan EM (2006) Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. J Neurosci 26:2951–2955. 10.1523/JNEUROSCI.5554-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. (2005) Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6:877–888. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M (2009) Experience leaves a lasting structural trace in cortical circuits. Nature 457:313–317. 10.1038/nature07487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. 10.1038/nrn2699 [DOI] [PubMed] [Google Scholar]
- Huang S, Gu Y, Quinlan EM, Kirkwood A (2010) A refractory period for rejuvenating GABAergic synaptic transmission and ocular dominance plasticity with dark exposure. J Neurosci 30:16636–16642. 10.1523/jneurosci.4384-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Oliver DL (2012) The basic circuit of the IC: tectothalamic neurons with different patterns of synaptic organization send different messages to the thalamus. Front Neural Circuits 6:48. 10.3389/fncir.2012.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Oliver DL (2014) Local and commissural IC neurons make axosomatic inputs on large GABAergic tectothalamic neurons. J Comp Neurol 522:3539–3554. 10.1002/cne.23623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bishop DC, Oliver DL (2009) Two classes of GABAergic neurons in the inferior colliculus. J Neurosci 29:13860–13869. 10.1523/JNEUROSCI.3454-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Brozoski TJ, Turner JG, Caspary DM (2014) Single unit hyperactivity and bursting in the auditory thalamus of awake rats directly correlates with behavioural evidence of tinnitus. J Physiol 592:5065–5078. 10.1113/jphysiol.2014.278572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV, Tzounopoulos T (2015) Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci 35:8829–8842. 10.1523/JNEUROSCI.5176-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipper M, Van Dijk P, Nunes I, Rüttiger L, Zimmermann U (2013) Advances in the neurobiology of hearing disorders: recent developments regarding the basis of tinnitus and hyperacusis. Prog Neurobiol 111:17–33. 10.1016/j.pneurobio.2013.08.002 [DOI] [PubMed] [Google Scholar]
- Li S, Choi V, Tzounopoulos T (2013) Pathogenic plasticity of Kv7.2/3 channel activity is essential for the induction of tinnitus. Proc Natl Acad Sci U S A 110:9980–9985. 10.1073/pnas.1302770110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kalappa BI, Tzounopoulos T (2015) Noise-induced plasticity of KCNQ2/3 and HCN channels underlies vulnerability and resilience to tinnitus. Elife 4. 10.7554/eLife.07242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano DA, Turner J, Caspary DM (2012) Diminished cortical inhibition in an aging mouse model of chronic tinnitus. J Neurosci 32:16141–16148. 10.1523/JNEUROSCI.2499-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WL, Hidaka H, May BJ (2006) Spontaneous activity in the inferior colliculus of CBA/J mice after manipulations that induce tinnitus. Hear Res 212:9–21. 10.1016/j.heares.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Malmierca MS, Blackstad TW, Osen KK, Karagülle T, Molowny RL (1993) The central nucleus of the inferior colliculus in rat–a Golgi and computer reconstruction study of neuronal and laminar structure. J Comp Neurol 333:1–27. 10.1002/cne.903330102 [DOI] [PubMed] [Google Scholar]
- Malmierca MS, Saint Marie RL, Merchan MA, Oliver DL (2005) Laminar inputs from dorsal cochlear nucleus and ventral cochlear nucleus to the central nucleus of the inferior colliculus: two patterns of convergence. Neuroscience 136:883–894. 10.1016/j.neuroscience.2005.04.040 [DOI] [PubMed] [Google Scholar]
- Manzoor NF, Licari FG, Klapchar M, Elkin RL, Gao Y, Chen G, Kaltenbach JA (2012) Noise-induced hyperactivity in the inferior colliculus: its relationship with hyperactivity in the dorsal cochlear nucleus. J Neurophysiol 108:976–988. 10.1152/jn.00833.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher JR, Sigalovsky IS, Guinan JJ Jr, Levine RA (2000) Lateralized tinnitus studied with functional magnetic resonance imaging: abnormal inferior colliculus activation. J Neurophysiol 83:1058–1072. [DOI] [PubMed] [Google Scholar]
- Middleton JW, Kiritani T, Pedersen C, Turner JG, Shepherd GM, Tzounopoulos T (2011) Mice with behavioral evidence of tinnitus exhibit dorsal cochlear nucleus hyperactivity because of decreased GABAergic inhibition. Proc Natl Acad Sci U S A 108:7601–7606. 10.1073/pnas.1100223108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbrandt JC, Holder TM, Wilson MC, Salvi RJ, Caspary DM (2000) GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hear Res 147:251–260. 10.1016/S0378-5955(00)00135-0 [DOI] [PubMed] [Google Scholar]
- Millan MH, Meldrum BS, Faingold CL (1986) Induction of audiogenic seizure susceptibility by focal infusion of excitant amino acid or bicuculline into the inferior colliculus of normal rats. Exp Neurol 91:634–639. 10.1016/0014-4886(86)90059-2 [DOI] [PubMed] [Google Scholar]
- Moore DR, Kowalchuk NE (1988) Auditory brainstem of the ferret: effects of unilateral cochlear lesions on cochlear nucleus volume and projections to the inferior colliculus. J Comp Neurol 272:503–515. 10.1002/cne.902720405 [DOI] [PubMed] [Google Scholar]
- Nordeen KW, Killackey HP, Kitzes LM (1983) Ascending projections to the inferior colliculus following unilateral cochlear ablation in the neonatal cochlear ablation in the neonatal gerbil, Meriones unguiculatus. J Comp Neurol 214:144–153. 10.1002/cne.902140204 [DOI] [PubMed] [Google Scholar]
- Noreña AJ, Eggermont JJ (2003) Changes in spontaneous neural activity immediately after an acoustic trauma: implications for neural correlates of tinnitus. Hear Res 183:137–153. 10.1016/S0378-5955(03)00225-9 [DOI] [PubMed] [Google Scholar]
- Noreña AJ, Eggermont JJ (2005) Enriched acoustic environment after noise trauma reduces hearing loss and prevents cortical map reorganization. J Neurosci 25:699–705. 10.1523/JNEUROSCI.2226-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver DL, Winer JA, Beckius GE, Saint Marie RL (1994) Morphology of GABAergic neurons in the inferior colliculus of the cat. J Comp Neurol 340:27–42. 10.1002/cne.903400104 [DOI] [PubMed] [Google Scholar]
- Orton LD, Rees A (2014) Intercollicular commissural connections refine the representation of sound frequency and level in the auditory midbrain. eLife 3. 10.7554/eLife.03764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DP, Rappaport JM, Gulliver JM (1994) Impaired word recognition in noise by patients with noise-induced cochlear hearing loss: contribution of temporal resolution defect. Am J Otol 15:679–686. [PubMed] [Google Scholar]
- Pilati N, Large C, Forsythe ID, Hamann M (2012) Acoustic over-exposure triggers burst firing in dorsal cochlear nucleus fusiform cells. Hear Res 283:98–106. 10.1016/j.heares.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romand R, Ehret G (1990) Development of tonotopy in the inferior colliculus. I. Electrophysiological mapping in house mice. Brain Res Dev Brain Res 54:221–234. 10.1016/0165-3806(90)90145-O [DOI] [PubMed] [Google Scholar]
- Ropp TJ, Tiedemann KL, Young ED, May BJ (2014) Effects of unilateral acoustic trauma on tinnitus-related spontaneous activity in the inferior colliculus. J Assoc Res Otolaryngol 15:1007–1022. 10.1007/s10162-014-0488-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalko N, Syka J (2005) Effect of noise exposure on gap detection in rats. Hear Res 200:63–72. 10.1016/j.heares.2004.08.014 [DOI] [PubMed] [Google Scholar]
- Saldaña E, Merchán MA (1992) Intrinsic and commissural connections of the rat inferior colliculus. J Comp Neurol 319:417–437. 10.1002/cne.903190308 [DOI] [PubMed] [Google Scholar]
- Saldaña E, Merchán MA (2005) Intrinsic and commissural connections of the inferior colliculus. In: Winer JA, Schreiner CE (eds), pp 155–181. The inferior colliculus. New York: Springer. [DOI] [PubMed] [Google Scholar]
- Saldaña EM, Feliciano M, Mugnaini E (1996) Distribution of descending projections from primary auditory neocortex to inferior colliculus mimics the topography of intracollicular projections. J Comp Neurol 371:15–40. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Constantine-Paton M (1983) Altered activity patterns during development reduce neural tuning. Science 221:1183–1185. 10.1126/science.6612332 [DOI] [PubMed] [Google Scholar]
- Sanes DH, Constantine-Paton M (1985) The development of stimulus following in the cochlear nerve and inferior colliculus of the mouse. Brain Res 354:255–267. 10.1016/0165-3806(85)90177-4 [DOI] [PubMed] [Google Scholar]
- Seki S, Eggermont JJ (2003) Changes in spontaneous firing rate and neural synchrony in cat primary auditory cortex after localized tone-induced hearing loss. Hear Res 180:28–38. 10.1016/S0378-5955(03)00074-1 [DOI] [PubMed] [Google Scholar]
- Sengpiel F. (2014) Plasticity of the visual cortex and treatment of amblyopia. Curr Biol 24:R936–R940. 10.1016/j.cub.2014.05.063 [DOI] [PubMed] [Google Scholar]
- Shnerson A, Willott JF (1979) Development of inferior colliculus response properties in C57BL/6J mouse pups. Exp Brain Res 37:373–385. [DOI] [PubMed] [Google Scholar]
- Sivaramakrishnan S, Oliver DL (2001) Distinct K currents result in physiologically distinct cell types in the inferior colliculus of the rat. J Neurosci 21:2861–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm J, Nguyen T, Kandler K (2014) Development of intrinsic connectivity in the central nucleus of the mouse inferior colliculus. J Neurosci 34:15032–15046. 10.1523/JNEUROSCI.2276-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JG, Brozoski TJ, Bauer CA, Parrish JL, Myers K, Hughes LF, Caspary DM (2006) Gap detection deficits in rats with tinnitus: a potential novel screening tool. Behav Neurosci 120:188–195. 10.1037/0735-7044.120.1.188 [DOI] [PubMed] [Google Scholar]
- van Gendt MJ, Boyen K, de Kleine E, Langers DR, van Dijk P (2012) The relation between perception and brain activity in gaze-evoked tinnitus. J Neurosci 32:17528–17539. 10.1523/JNEUROSCI.2791-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste S, van Dongen M, De Vree B, Hiseni S, van der Velden E, Strydis C, Joos K, Noreña A, Serdijn W, De Ridder D (2013) Does enriched acoustic environment in humans abolish chronic tinnitus clinically and electrophysiologically? A double blind placebo controlled study. Hear Res 296:141–148. 10.1016/j.heares.2012.10.003 [DOI] [PubMed] [Google Scholar]
- Vogler DP, Robertson D, Mulders WH (2011) Hyperactivity in the ventral cochlear nucleus after cochlear trauma. J Neurosci 31:6639–6645. 10.1523/JNEUROSCI.6538-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton JP. (2010) Timing is everything: temporal processing deficits in the aged auditory brainstem. Hear Res 264:63–69. 10.1016/j.heares.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Brozoski TJ, Turner JG, Ling L, Parrish JL, Hughes LF, Caspary DM (2009) Plasticity at glycinergic synapses in dorsal cochlear nucleus of rats with behavioral evidence of tinnitus. Neuroscience 164:747–759. 10.1016/j.neuroscience.2009.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH (1963) Single-cell responses in striate cortex of kittens deprived of vision in one eye. J Neurophysiol 26:1003–1017. [DOI] [PubMed] [Google Scholar]
- Willott JF, Lu SM (1982) Noise-induced hearing loss can alter neural coding and increase excitability in the central nervous system. Science 216:1331–1334. 10.1126/science.7079767 [DOI] [PubMed] [Google Scholar]
- Wu C, Martel DT, Shore SE (2016) Increased synchrony and bursting of dorsal cochlear nucleus fusiform cells correlate with tinnitus. J Neurosci 36:2068–2073. 10.1523/JNEUROSCI.3960-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Weiner BD, Zhang LS, Cho SJ, Bao S (2011) Homeostatic plasticity drives tinnitus perception in an animal model. Proc Natl Acad Sci U S A 108:14974–14979. 10.1073/pnas.1107998108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankaskas K. (2013) Prelude: noise-induced tinnitus and hearing loss in the military. Hear Res 295:3–8. 10.1016/j.heares.2012.04.016 [DOI] [PubMed] [Google Scholar]
- Yu X, Wadghiri YZ, Sanes DH, Turnbull DH (2005) In vivo auditory brain mapping in mice with Mn-enhanced MRI. Nat Neurosci 8:961–968. 10.1038/nn1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LI, Bao S, Merzenich MM (2002) Disruption of primary auditory cortex by synchronous auditory inputs during a critical period. Proc Natl Acad Sci U S A 99:2309–2314. 10.1073/pnas.261707398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Panizzutti R, de Villers-Sidani E, Madeira C, Merzenich MM (2011) Natural restoration of critical period plasticity in the juvenile and adult primary auditory cortex. J Neurosci 31:5625–5634. 10.1523/JNEUROSCI.6470-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wang F, Hu H, Sun X, Kilgard MP, Merzenich MM, Zhou X (2014) Environmental acoustic enrichment promotes recovery from developmentally degraded auditory cortical processing. J Neurosci 34:5406–5415. 10.1523/JNEUROSCI.5310-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]