

Abstract
Chemical bonds are a key determinant of the structure and properties of a material. Thus, rationally designing arbitrary materials requires complete control over the bond. While atomic bonding is dictated by the identity of the atoms, nanoparticle superlattice engineering, where nano-particle “atoms” are held together by DNA “bonds”, offers a route to design crystal lattices in a way that nature cannot: through altering the oligonucleotide bond. Herein, the use of RNA, as opposed to DNA, is explored by synthesizing superlattices in which nanoparticles are bonded by DNA/DNA, RNA/RNA, and DNA/RNA duplexes. By moving beyond nanoparticle superlattices assembled only with DNA, a new degree of freedom is introduced, providing programmed responsiveness to enzymes and greater bond versatility. Therefore, the oligonucleotide bond can have programmable function beyond dictating the structure of the material and moves nanoparticle superlattices closer to naturally occurring biomaterials, where the line between structural and functional elements is blurred.
Graphical abstract
Introduction
DNA is a powerful ligand for programming the assembly of nanoparticles into superlattices with a vast number of crystallographic symmetries.1a–g This can be achieved by using a programmable atom equivalent (PAE), which consists of a nanoparticle core densely functionalized with geometrically defined oligonucleotides, where DNA mediates interactions between nanoparticles. The oligonucleotide density and rigid nanoparticle core impose a radial orientation of the DNA and valency to the nanoparticles. Initially, spherical gold nano-particles (AuNPs) were studied as PAE cores, but subsequent work has found that this approach is core generalizable, as other inorganic2a,b and organic1f cores, anisotropic cores,3a,b as well as biological materials, such as proteins,4 can be assembled using the same design rules.1d The unifying element of all these studies is the DNA “bond” that programs nanoparticle interactions and drives their assembly into ordered crystalline structures. While recent work has been dedicated to understanding the function of these materials including emergent plasmonic2a,5 and catalytic properties,4,6 these properties are predominantly derived from the nanoparticle core. Studies of how the bond contributes to the functional properties of the crystalline superlattice are absent.
When considering materials that could in principle be used as a programmable ligand to assemble nanoparticles, DNA is not the only candidate. Specifically, the incorporation of RNA into nanoparticle superlattices would enable new classes of functional and stimuli-responsive superstructures that are not achievable with DNA or solely by engineering the PAE building block core. Though RNA is chemically similar to DNA (the primary difference is the presence of a 2′-hydroxyl (2′-OH) group in RNA), it has a vast chemical, structural, and functional design space that exceeds that of DNA.7 For example, in cells, while DNA is often found in the form of long double helices,8 RNA is generally composed of short helices surrounded by loops and bulges.9 Notable forms of biofunctional RNA include small interfering RNA (siRNA) that can regulate gene expression,10 ribozymes (ribonucleic acid enzymes) which are catalytic RNA molecules,11 and riboswitches which are structures formed in mRNA that can regulate gene expression in bacteria12 and even act as stimuli responsive sensors.13 While the vast chemical and biological space that RNA occupies may appear to make it an ideal ligand for endowing PAEs with additional functionalities, its instability and vulnerability to nuclease-catalyzed hydrolysis14 provides a substantial barrier to realizing biomaterials based upon RNA.
Research on synthesizing RNA biomaterials has focused on the analogy to DNA hybridization, where rigidity is imposed by the DNA hybridization events, which leads to rigid structures and therefore valency.15 This approach, based purely on DNA hybridization, has been extended to RNA for the synthesis of micrometer scale RNA filaments, molecular jigsaw puzzles,16 and square-shaped RNA particles.17a,b In order for these syntheses to work for RNA, however, a hierarchical multistep process is required, whereas DNA structures can typically be made in a “one pot” synthesis.18 In addition, the DNA and RNA-based hybridization approaches require the use of both simulation and experiment to rationally design the 3D RNA architectures through initial computer modeling.19 This strategy is conceptually related but different from the method discussed herein for forming nanoparticle-based templated bonds, where the rigid nanoparticle core leads to a radial upright orientation of the densely packed DNA, leading to valency imposed by the core.
Therefore, the well-understood nature of DNA programmable assembly, through the established design rules for the rational construction of DNA nanoparticle superlattices,1d provides the perfect platform for exploring the degree to which non-DNA oligonucleotides can serve as programmable “bonds”. Here, the conventional design space for DNA-programmable assembly is transformed by introducing oligonucleotide identity (i.e., PAEs held together by DNA/DNA, RNA/RNA, or DNA/RNA duplexes) as an important design parameter. Similar to conventional DNA-based assembly, the programmable nature of the oligonucleotide bond is the driving force, and is independent of the oligonucleotide identity such that DNA/DNA, RNA/RNA, and DNA/RNA duplexes are all suitable programmable ligands. However, the ability to tune the bond identity enables the rational design of responsive materials, whereby the oligonucleotide bond identity and interparticle distance dictate the response to enzymes.
Results and Discussion
Design of Nanoparticle Superlattices with Different Oligonucleotide Bonds
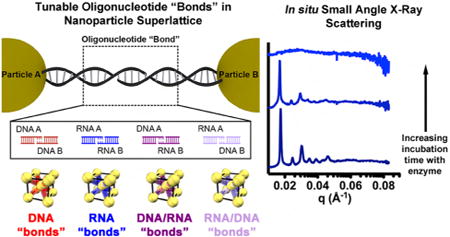
Design rules for the synthesis of nanoparticle superlattices with a variety of crystallographic symmetries have been established, which allow one to independently adjust each of the relevant crystallographic parameters, including particle size, periodicity, and interparticle distance.1d Because these design rules are based upon explorations of DNA as the programmable ligand, one must first explore how the oligonucleotide bond identity affects the programmable assembly of nanoparticle superlattices. We hypothesize that since RNA/RNA and RNA/DNA binding proceeds in a similar fashion to DNA/DNA binding, the use of DNA, RNA, or a DNA/RNA heteroduplex will not significantly change the resulting nanoparticle superlattice crystal structure. As an initial proof-of-concept study, a two-component system which is expected to yield superlattices with a body-centered cubic (bcc) crystallographic symmetry, was evaluated. Four binary sets of particles were functionalized with DNA or RNA with non-self-complementary sticky ends, such that particle A can only bind to particle B and vice versa (Figure 1a). The DNA and RNA design (Table S1 and Figure S1) contains short overhang regions on the 3′ end of the linkers, which facilitate the interactions between nanoparticles. For each sample, particle A and particle B were mixed in a 1:1 ratio so that each sample was allowed to form aggregates. The possible permutations of oligonucleotide bonds are A-DNA/B-DNA (red), A-RNA/B-RNA (blue), A-DNA/B-RNA (dark purple), and A-RNA/B-DNA (light purple).
Figure 1.
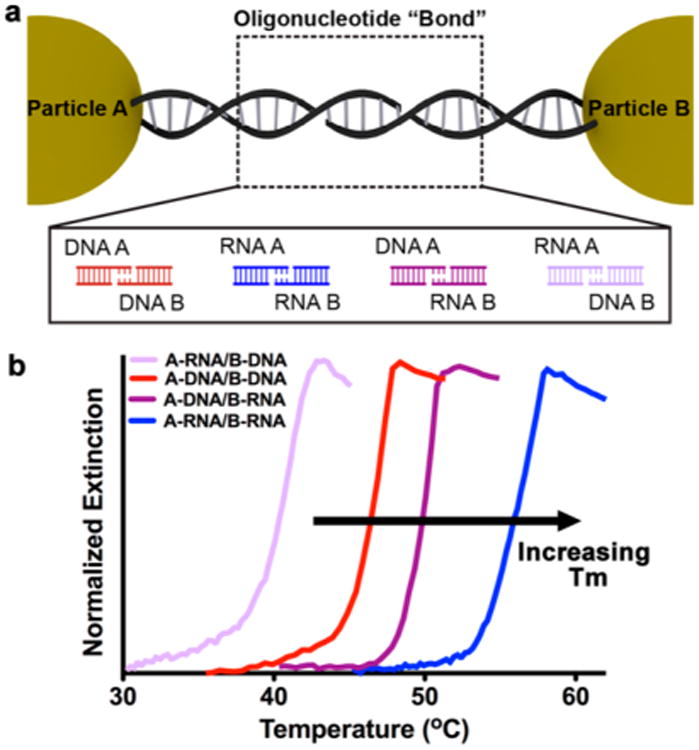
Nanoparticle superlattices synthesized with modular oligonucleotide bonds. (a) Two-component system where particles A and B are linked by non-self-complementary sticky ends. Four types of oligonucleotide bonds are explored: DNA/DNA (red), RNA/RNA (blue), DNA/RNA (dark purple), and RNA/DNA (light purple). (b) UV–vis melts of aggregates linked with four different oligonucleotide bond compositions at short linker lengths (color scheme is the same as in (a)). Tunable melting transitions (Tm) emerge based on oligonucleotide bond composition.
Synthesis and Characterization of DNA and RNA Nanoparticle Superlattices
It is well-known that the density of DNA affects the cooperative melting transition and crystallization of PAEs.20,21 In order to explore new oligonucleotide identities as bonding ligands, a method to functionalize particles with RNA at a density similar to that attainable with DNA must be developed (Figure S2).21 In previous reports, RNA immobilized on gold nanoparticles (AuNPs) has been exclusively in the form of double stranded RNA,22a,b where a backfill molecule was added to passivate the remaining gold surface to account for the lower loading of RNA compared to what is observed with DNA.21 But herein, RNA particles A and B need to be synthesized with single- stranded RNA without backfill molecules. Therefore, RNA particles A and B were synthesized using methods analogous to their DNA counterparts (Figure S2). This was found to significantly increase the density of thiolated RNA on particles A and B from 25–60 pmol/cm2 to 50–75 pmol/cm2, thus allowing RNA particle A and particle B to be analogous to their DNA counterparts in terms of oligonucleotide density. Once this has been accomplished, the only parameters that need to be optimized to synthesize nanoparticle superlattices with different bond compositions are the strength and length of the sticky end, the spacer unit between the nanoparticle surface, and the oligonucleotide recognition sequence (Figure S1).
It has been widely observed that PAEs exhibit cooperative and sharp melting transitions (transition widths of 2–8 °C) compared to linear nucleic acids, which exhibit broad melting transitions (transition width ~20 °C).20,23a,b A typical melting experiment involves monitoring the optical extinction at 260 and 520 nm, which is dampened for room temperature assembled PAE aggregates, but increases as the temperature is raised and the nanoparticles begin to dissociate. All aforementioned PAE-oligonucleotide combinations (Figure 1a) exhibited sharp and cooperative melting transitions (Figure 1b). Notably, the characteristic melting transition (Tm) for the RNA-PAE aggregates occurred about 13 °C higher than analogous DNA-PAE aggregates (full width at half-maximum (fwhm) = 2.7 °C for both), where the only difference is the identity of the oligonucleotide. A similar stabilization effect exists in molecular duplexes of RNA, which exhibit greater thermal stability and higher melting temperatures than their DNA counterparts.7 Additionally, the two PAE aggregates held together by heteroduplexes exhibited two distinct melting transitions (Tm = 39 °C, fwhm = 3 °C for aggregates with RNA-PAE A; Tm = 50 °C, fwhm = 1.3 °C for aggregates with DNA-PAE A). While this result may appear surprising given the similarity of RNA and DNA, the position of the melting transitions for the two heteroduplexes can be understood by examining the molecular counterparts for their sticky ends, where the same trend in melting temperatures was observed for molecular duplexes of similar sequences as the sticky ends.24a,b More specifically, the stability of homopurine-homopyrimidine oligomer duplexes mirrors the trend in superlattice melting temperatures when looking at sticky end identity,25a-c thus demonstrating that the characteristics of hybrid molecular duplexes are maintained when they are used as programmable ligands. Additionally, adenine DNA/uracil RNA heteroduplexes are known to be exceptionally unstable.24b,26a,b This hetero-duplex is analogous to the sticky end interaction in A-RNA/B-DNA and thus explains the lower melting temperature. Finally, this trend in melting temperatures was found to persist as the sticky end was increasingly moved away from the particle surface by utilizing longer linker oligonucleotides (Figure S3).
Recent work has demonstrated that slowly cooling PAEs through their melting transition is an effective method for synthesizing micron-scale single crystals.27 To test whether RNA-programmable assembly could also be used to form such large scale crystals, PAE aggregates were slowly cooled (0.01 °C/min) from 5 to 10 °C above their melting temperature down to room temperature. Small-angle X-ray scattering (SAXS) was used to confirm the bcc crystallographic symmetry1d (Figure 2a) across three different oligonucleotide linker length scales and four different oligonucleotide bond compositions (Figure 2b). Despite differences at the molecular level, both DNA and RNA can be used interchangeably with the same crystal design principles, as evidenced by SAXS and SEM. For example, the macroscopic crystallites formed by this slow cooling process were examined by SEM and determined to be rhombic dodecahedra (Figure 2c), as observed in pure DNA systems.27 Though the translated sequences used for assembly are identical (i.e., the DNA and RNA used on all A type particles had the same sequence just a different oligonucleotide identity), the A-DNA/B-DNA superlattices exhibited the largest interparticle distance, while the A-RNA/B-RNA superlattices consistently exhibited the shortest inter-particle distances (Table 1; SI eq 1). These data can be understood by looking at the typical characteristics of the molecular duplexes and specifically the 0.275 nm rise per base pair for RNA (A-form) as compared with 0.34 nm for DNA (B-form).7,28 DNA/RNA heteroduplexes are typically intermediate in pitch, however, it is difficult to predict the properties of a DNA-RNA heteroduplex compared to its homoduplex counterpart, as they are known to be highly sequence specific.24a
Figure 2.

Body-centered cubic nanoparticle superlattices. (a) Depictions of body-centered cubic (bcc) nanoparticle superlattices of four different oligonucleotide bond compositions (to-scale). Gold nanoparticles are shown in yellow and the oligonucleotide bond in red, blue, or purple. (b) Small-angle X-ray scattering (SAXS) of nanoparticle superlattices with four different “bond” compositions and three different interparticle distances (from bottom to top: short (46-base pair (-bp)), medium (67-bp), and long (128-bp) linkers). (c) Scanning electron microscopy (SEM) images of nanoparticle super-lattices in the solid state. In all cases, single crystal rhombic dodecahedra are observed. Scale bars = 100 nm.
Table 1. Interparticle Distance and Crystal Domain Size Calculated from SAXS Dataa.
| Linker | Interparticle Distance (nm) | Crystal Domain Size (nm) |
|---|---|---|
| Short Linker (46-bp) | 26.70 | 700 |
| 26.60 | 725 | |
| 25.39 | 690 | |
| 25.35 | 680 | |
|
| ||
| Medium Linker (67-bp) | 38.82 | 985 |
| 36.36 | 1010 | |
| 35.71 | 980 | |
| 34.55 | 925 | |
|
| ||
| Long Linker (128-bp) | 48.06 | 1315 |
| 47.73 | 1280 | |
| 47.12 | 1305 | |
| 44.05 | 1205 | |
The interparticle distance is defined as the distance from the center-to-center of each nanoparticle on the bcc diagonal and is therefore the length of the oligonucleotide between the nanoparticles plus the sum of the nanoparticle radii (N = 2). Color scheme is as follows: A-DNA/B-DNA (red), A-RNA/B-DNA (light purple), A-DNA/B-RNA (dark purple), A-RNA/B-RNA (blue).
Use of the Scherrer equation (SI eq 2) allows one to calculate the mean crystallite size for a given sample, defined as the average diameter of a single crystalline domain. These calculations show that the grain sizes are all very similar regardless of bond type (Table 1), thus demonstrating the power of programmable assembly for generating crystals of similar size but with different oligonucleotide constituents. Together, these data demonstrate that tuning the oligonucleotide bond is an important new handle for on-demand materials properties including melting temperature and interparticle distance in crystalline nanoparticle materials. Finally, SAXS patterns of analogous DNA and RNA superlattices stored at 25 °C were obtained throughout the course of 100 days (Figure S4). These data revealed that the superlattices remain well ordered, with the interparticle distance changing <1 nm and crystalline domain size changing <20 nm over this time period (Table S2). This demonstrates that the RNA stability is adequate for its use as a programmable ligand in nanoparticle superlattices.
Isostructural Nanoparticle Superlattices Exhibit Tunable Responsiveness to Enzymes
Having shown that DNA/DNA, RNA/RNA, and DNA/RNA duplexes can all serve as programmable ligands to synthesize nanoparticle superlattices, it was explored whether the oligonucleotide bond could play a significant role in dictating the properties of the material, rather than acting as a passive “glue”. To this end, time-dependent SAXS measurements were performed to probe the interaction of nanoparticle superlattices with ribonuclease (RNase) A (Figure 3), an enzyme that is known to recognize and degrade both single and double stranded RNA duplexes.29a,b We hypothesized that nanoparticle superlattices would become more accessible to the enzyme as interparticle distances increased due to larger pores for diffusion.30a–c To eliminate a purely diffusion-based interaction, a flow-cell setup was utilized, where the enzyme and superlattice were in constant oscillation, as described in SI Materials and Methods. For the RNA superlattices, as the linker length between the nanoparticles was increased, the time span over which the superlattices retained their structure decreased dramatically (6 min for short and medium linkers and 0.25 min for long linkers; Figure 3a and S5). While pure DNA superlattices were stable in the presence of ribonuclease, one might expect that DNA/RNA superlattices would still be able to respond to enzymes due to the presence of RNA. Indeed, the ribonuclease can degrade the DNA/RNA superlattices, though they retain their order over a longer period of time than the RNA superlattices (13 min for the short linker, 8.5 min for the medium linker, and 9 min for the long linker; Figure 3b, S5c). This process is also concentration dependent (Figure S5). Taken together, these data show that the RNA-containing bonds in the nanoparticle superlattices are responsive to enzymes and such responses are dependent on both oligonucleotide identity and length.
Figure 3.
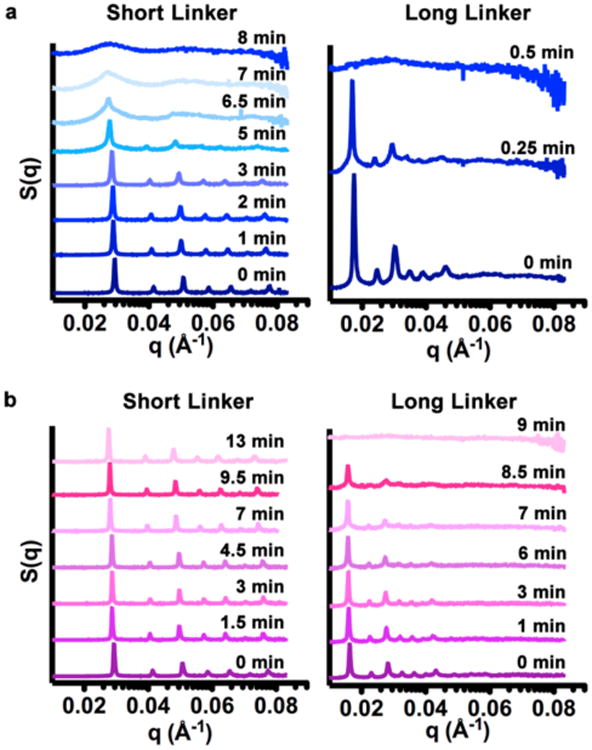
Functional oligonucleotide bonds in nanoparticle superlattices. (a) Time-dependent SAXS scattering patterns for RNA superlattices with short and long linkers upon addition of 1 μg ribonuclease (RNase) A. (b) Time-dependent SAXS scattering patterns for DNA-RNA superlattices with short and long linkers upon addition of 1 μg RNase A. [AuNP] ≈ 45 nM.
In order to better understand the structural changes that occur during enzymatic degradation, several attributes of the time-dependent SAXS data were studied. By examining the breadth and position of the first-order scattering peak (q0), one can begin to quantify how the bond length and identity affects the superlattice's response to enzymes. The position of q0 is used to calculate the interparticle distance. The first analysis involved monitoring changes in the position of q0 as the lattice is degraded by the enzyme, which is manifested by shifts in peak positions to lower values of q and thus larger interparticle distances. The greatest overall change in the position of q0 (0.002 Å-1) is observed for RNA superlattices over the course of 6.5 min, whereas for the hybrid superlattices, a smaller change in q0 (0.0017 Å−1) is observed over a longer period of time (13 min; Figure 4a). Variations in the breadth of the q0 peak, indicating changes in domain size and relative crystal quality, are characterized by the fwhm, where a smaller value of the fwhm indicates a larger domain and higher quality crystal. For RNA superlattices, the fwhm increases over 8 min (0.0014 Å −1) before the structure falls apart, which is in stark contrast to the hybrid superlattices, where almost no change in fwhm is observed (0.0003 Å −1; Figure 4b). Similar trends are observed for the medium and long linkers (Figure S6).
Figure 4.
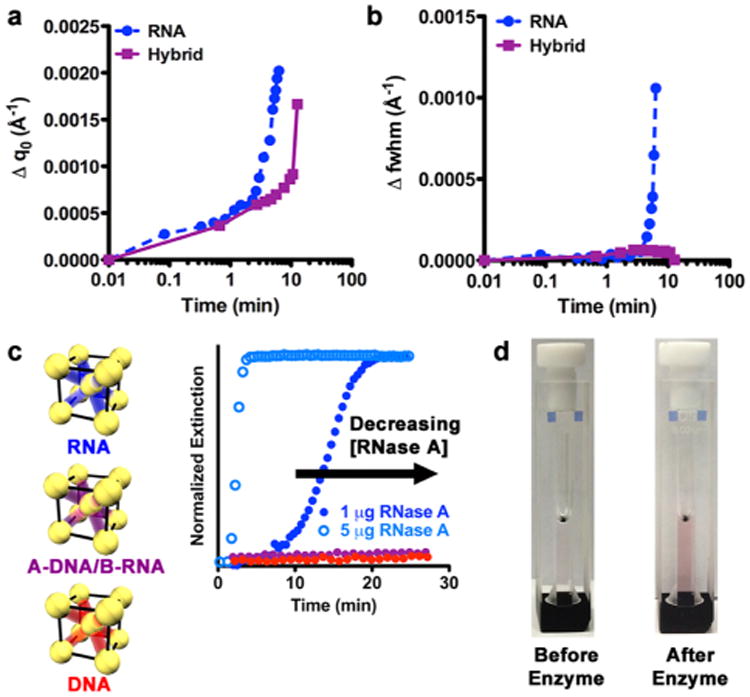
Measurement of the superlattice response to enzymes. (a) Changes in position of the first-order scattering peak (Δq0) versus time extracted from the time-dependent SAXS data for RNA (blue circles) and hybrid (purple squares) superlattices with short linkers. Lines are not fits but rather guides for the eye. The zero point is represented at 0.01 min. (b) Changes in grain size and relative crystal quality indicated by changes in the full width at half-maximum (Δfwhm) during incubation with enzymes extracted from the time-resolved SAXS data. Analysis of the RNA superlattice stopped at 6.5 min, after which long-range order was no longer maintained. Lines are not fits but rather guides for the eye. The zero point is represented at 0.01 min. (c) UV–vis kinetics demonstrating changes in the localized surface plasmon resonance (LSPR) upon incubation with RNase A for superlattices of RNA (blue), A-DNA/B-RNA (purple) and DNA (red). Open circles represent 1 μg RNase A and closed circles represent 5 μg RNase A. [AuNP] ≈ 1.5 nM. (d) Optical images showing the change in RNA nanoparticle superlattices before (left) and after (right) incubation with RNase A.
For RNA superlattices with short linkers, these data indicate that RNA connections are lost as enzyme incubation time increases. This degradation reduces the number of connections holding the RNA superlattice together, thus allowing more conformational degrees of freedom for each nanoparticle. This manifests as an increase in fwhm (Figure 4a, b). Specifically, the structure is able to retain long-range order for 5 min before sufficient RNA–RNA interconnects are lost and the structure rapidly becomes disordered over the next 3 min. In contrast, while a small change in q0 is observed, a minimal change in fwhm is seen for the hybrid superlattices, thus indicating that some DNA/RNA connections may be lost but not enough to result in a change in overall crystal quality. This suggests that having one component of DNA allows the crystal to retain grain size and relative ordering as the interparticle distance is increased. This is because RNase A is only known to cleave single and double stranded RNA,29a,b and thus is only able to recognize and degrade RNA originating from particle B, and not the DNA on particle A or the hybrid sticky end. Almost no change is seen in q0 position or fwhm for the DNA superlattices (Figure S6), which further confirms that the enzyme recognition is oligonucleotide bond specific. For both the RNA and hybrid superlattices, it was observed that crystals with shorter interparticle distances are better able to withstand enzymatic degradation. Taken together, these data suggest that the introduction of RNA into nanoparticle superlattices leads to bonds that are selectively addressable.
Thus, it has been demonstrated that the bond in nanoparticle superlattices is responsive to enzymes and structural changes in the nanoparticle superlattices can be monitored by time-dependent SAXS. This transition also can be monitored using UV–visible (UV–vis) spectroscopy, where RNase A was added to a solution of RNA superlattices and an increase in extinction over time was observed, much like what is observed in a melting experiment (Figure 4c). In this case, the strong extinction by the nanoparticles at 520 nm provides a spectroscopic and colorimetric handle for tracking this process. Again, it is observed that the enzymatic degradation is concentration- and oligonucleotide-bond-dependent, as almost no change in extinction was observed for the hybrid and DNA superlattices. Thus, the strong AuNP absorption can be used as a spectroscopic handle to monitor the function of the oligonucleotide bond quickly on the benchtop, without the need for a synchrotron light source (Figure 4d).
Conclusion
The data presented herein show that design rules for nanoparticle superlattices1d still hold true when using RNA as opposed to DNA as a programmable ligand, and that the identities of the oligonucleotide “bonds” in nanoparticle superlattices can be independently changed without changing the “atoms”. This novel capability provides a pathway for deliberately tailoring superlattice properties, something not possible with conventional atomic and molecular systems. Indeed, the realization of responsive oligonucleotide bonds within such structures dramatically increases the breadth and sophistication of the design space for these materials and creates several challenges for the field moving forward. These challenges include: (1) the study of other specialty oligonucleotides, such as peptide nucleic acids31 and locked nucleic acids, which could allow one to further tailor the charge, stability, and function of oligonucleotide bonds, (2) the creation of mixed superlattice systems with different bonds that can be addressed and modified independently and selectively with enzymes, and (3) the creation of bonds that move beyond linear struts, such as structures that contain catalytic loops. Taken together, the development of a programmable system to utilize oligonucleotides other than DNA to direct the assembly of nanoparticles into three-dimensional crystals shows promise in developing nanoparticle superlattices with dynamic and functional bonds for many areas, including catalysis and sensing.
Supplementary Material
Acknowledgments
This material is based upon work supported by the AFOSR (Award FA9550-11-1-0275) and the Center for Cancer Nanotechnology Excellence (CCNE) initiative of the National Institutes of Health (NIH) (Award U54 CA151880). SNB and RVT acknowledge a National Science Foundation Graduate Research Fellowship. SNB also acknowledges a P.E.O. scholar award. MBR acknowledges a National Defense and Science Engineering Graduate fellowship. Nick Savalia is acknowledged for assistance in the synthesis and purification of RNA. This work made use of the EPIC facility (NUANCE Center-Northwestern University), which receives support from the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); and the State of Illinois. Portions of this work were carried out at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) beamline located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E. I. DuPont de Nemours & Co., Dow Chemical Company, and the state of Illinois. Use of the APS was supported by the U.S. DOE, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-06CH11357.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b07908. Detailed materials and methods, synthesis and characterization of PAEs, melting transitions for medium and long linkers, superlattice stability over time, and enzymatic response to superlattices with other linkers. (PDF)
The authors declare no competing financial interest.
References
- 1.(a) Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]; (b) Alivisatos AP, Johnsson KP, Peng X, Wilson TE, Loweth CJ, Bruchez MP, Schultz PG. Nature. 1996;382:609. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]; (c) Park SY, Lytton-Jean AKR, Lee B, Weigand S, Schatz GC, Mirkin CA. Nature. 2008;451:553. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]; (d) Macfarlane RJ, Lee B, Jones MR, Harris N, Schatz GC, Mirkin CA. Science. 2011;334:204. doi: 10.1126/science.1210493. [DOI] [PubMed] [Google Scholar]; (e) Tan SJ, Campolongo MJ, Luo D, Cheng W. Nat Nanotechnol. 2011;6:268. doi: 10.1038/nnano.2011.49. [DOI] [PubMed] [Google Scholar]; (f) Auyeung E, Cutler JI, Macfarlane RJ, Jones MR, Wu J, Liu G, Zhang K, Osberg KD, Mirkin CA. Nat Nanotechnol. 2012;7:24. doi: 10.1038/nnano.2011.222. [DOI] [PubMed] [Google Scholar]; (g) Nykypanchuk D, Maye MM, van der Lelie D, Gang O. Nature. 2008;451:549. doi: 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- 2.(a) Young KL, Ross MB, Blaber MG, Rycenga M, Jones MR, Zhang C, Senesi AJ, Lee B, Schatz GC, Mirkin CA. Adv Mater. 2014;26:653. doi: 10.1002/adma.201302938. [DOI] [PubMed] [Google Scholar]; (b) Zhang C, Macfarlane RJ, Young KL, Choi CHJ, Hao L, Auyeung E, Liu G, Zhou X, Mirkin CA. Nat Mater. 2013;12:741. doi: 10.1038/nmat3647. [DOI] [PubMed] [Google Scholar]
- 3.(a) Jones MR, Macfarlane RJ, Lee B, Zhang J, Young KL, Senesi AJ, Mirkin CA. Nat Mater. 2010;9:913. doi: 10.1038/nmat2870. [DOI] [PubMed] [Google Scholar]; (b) O'Brien MN, Jones MR, Lee B, Mirkin CA. Nat Mater. 2015;14:833. doi: 10.1038/nmat4293. [DOI] [PubMed] [Google Scholar]
- 4.Brodin JD, Auyeung E, Mirkin CA. Proc Natl Acad Sci U S A. 2015;112:4564. doi: 10.1073/pnas.1503533112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross MB, Ku JC, Vaccarezza VM, Schatz GC, Mirkin CA. Nat Nanotechnol. 2015;10:453. doi: 10.1038/nnano.2015.68. [DOI] [PubMed] [Google Scholar]
- 6.Auyeung E, Morris W, Mondloch JE, Hupp JT, Farha OK, Mirkin CA. J Am Chem Soc. 2015;137:1658. doi: 10.1021/ja512116p. [DOI] [PubMed] [Google Scholar]
- 7.Guo P. Nat Nanotechnol. 2010;5:833. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taanman JW. Biochim Biophys Acta, Bioenerg. 1999;1410:103. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 9.Noller HF. Annu Rev Biochem. 1984;53:119. doi: 10.1146/annurev.bi.53.070184.001003. [DOI] [PubMed] [Google Scholar]
- 10.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 11.Cech TR, Zaug AJ, Grabowski PJ. Cell. 1981;27:487. doi: 10.1016/0092-8674(81)90390-1. [DOI] [PubMed] [Google Scholar]
- 12.Wittmann A, Suess B. FEBS Lett. 2012;586:2076. doi: 10.1016/j.febslet.2012.02.038. [DOI] [PubMed] [Google Scholar]
- 13.Davidson ME, Harbaugh SV, Chushak YG, Stone MO, Kelley-Loughnane N. ACS Chem Biol. 2013;8:234. doi: 10.1021/cb300274g. [DOI] [PubMed] [Google Scholar]
- 14.Behlke MA. Oligonucleotides. 2008;18:305. doi: 10.1089/oli.2008.0164. [DOI] [PubMed] [Google Scholar]
- 15.Jones MR, Seeman NC, Mirkin CA. Science. 2015;347:1260901. doi: 10.1126/science.1260901. [DOI] [PubMed] [Google Scholar]
- 16.Chworos A, Severcan I, Koyfman AY, Weinkam P, Oroudjev E, Hansma HG, Jaeger L. Science. 2004;306:2068. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 17.(a) Severcan I, Geary C, Verzemnieks E, Chworos A, Jaeger L. Nano Lett. 2009;9:1270. doi: 10.1021/nl900261h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dibrov SM, McLean J, Parsons J, Hermann T. Proc Natl Acad Sci U S A. 2011;108:6405. doi: 10.1073/pnas.1017999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaeger L, Chworos A. Curr Opin Struct Biol. 2006;16:531. doi: 10.1016/j.sbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Grabow WW, Jaeger L. Acc Chem Res. 2014;47:1871. doi: 10.1021/ar500076k. [DOI] [PubMed] [Google Scholar]
- 20.Jin R, Wu G, Li Z, Mirkin CA, Schatz GC. J Am Chem Soc. 2003;125:1643. doi: 10.1021/ja021096v. [DOI] [PubMed] [Google Scholar]
- 21.Hurst SJ, Lytton-Jean AK, Mirkin CA. Anal Chem. 2006;78:8313. doi: 10.1021/ac0613582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. J Am Chem Soc. 2009;131:2072. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barnaby SN, Lee A, Mirkin CA. Proc Natl Acad Sci U S A. 2014;111:9739. doi: 10.1073/pnas.1409431111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]; (b) Cutler JI, Auyeung E, Mirkin CA. J Am Chem Soc. 2012;134:1376. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 24.(a) Sugimoto N, Nakano Si, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M. Biochemistry. 1995;34:11211. doi: 10.1021/bi00035a029. [DOI] [PubMed] [Google Scholar]; (b) Lesnik EA, Freier SM. Biochemistry. 1995;34:10807. doi: 10.1021/bi00034a013. [DOI] [PubMed] [Google Scholar]
- 25.(a) Chamberlin MJ, Patterson DL. J Mol Biol. 1965;12:410. doi: 10.1016/s0022-2836(65)80264-9. [DOI] [PubMed] [Google Scholar]; (b) Roberts R, Crothers D. Science. 1992;258:1463. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]; (c) Hung SH, Yu Q, Gray DM, Ratliff RL. Nucleic Acids Res. 1994;22:4326. doi: 10.1093/nar/22.20.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Riley M, Maling B, Chamberlin MJ. J Mol Biol. 1966;20:359. doi: 10.1016/0022-2836(66)90069-6. [DOI] [PubMed] [Google Scholar]; (b) Martin FH, Tinoco I. Nucleic Acids Res. 1980;8:2295. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auyeung E, Li TING, Senesi AJ, Schmucker AL, Pals BC, de la Cruz MO, Mirkin CA. Nature. 2014;505:73. doi: 10.1038/nature12739. [DOI] [PubMed] [Google Scholar]
- 28.Holde KEv. Springer Series in Molecular and Cell Biology. Springer-Verlag; New York: 1989. [Google Scholar]
- 29.(a) Weickmann JL, Olson EM, Glitz DG. Cancer Res. 1984;44:1682. [PubMed] [Google Scholar]; (b) Raines RT. Chem Rev. 1998;98:1045. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 30.(a) Kim Y, Macfarlane RJ, Mirkin CA. J Am Chem Soc. 2013;135:10342. doi: 10.1021/ja405988r. [DOI] [PubMed] [Google Scholar]; (b) Macfarlane RJ, Jones MR, Lee B, Auyeung E, Mirkin CA. Science. 2013;341:1222. doi: 10.1126/science.1241402. [DOI] [PubMed] [Google Scholar]; (c) Maye MM, Kumara MT, Nykypanchuk D, Sherman WB, Gang O. Nat Nanotechnol. 2010;5:116. doi: 10.1038/nnano.2009.378. [DOI] [PubMed] [Google Scholar]
- 31.Lytton-Jean AKR, Gibbs-Davis JM, Long H, Schatz GC, Mirkin CA, Nguyen ST. Adv Mater. 2009;21:706. doi: 10.1002/adma.200801724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.