

Abstract
Background
Folsomia candida is a model in soil biology, belonging to the family of Isotomidae, subclass Collembola. It reproduces parthenogenetically in the presence of Wolbachia, and exhibits remarkable physiological adaptations to stress. To better understand these features and adaptations to life in the soil, we studied its genome in the context of its parthenogenetic lifestyle.
Results
We applied Pacific Bioscience sequencing and assembly to generate a reference genome for F. candida of 221.7 Mbp, comprising only 162 scaffolds. The complete genome of its endosymbiont Wolbachia, was also assembled and turned out to be the largest strain identified so far. Substantial gene family expansions and lineage-specific gene clusters were linked to stress response. A large number of genes (809) were acquired by horizontal gene transfer. A substantial fraction of these genes are involved in lignocellulose degradation. Also, the presence of genes involved in antibiotic biosynthesis was confirmed. Intra-genomic rearrangements of collinear gene clusters were observed, of which 11 were organized as palindromes. The Hox gene cluster of F. candida showed major rearrangements compared to arthropod consensus cluster, resulting in a disorganized cluster.
Conclusions
The expansion of stress response gene families suggests that stress defense was important to facilitate colonization of soils. The large number of HGT genes related to lignocellulose degradation could be beneficial to unlock carbohydrate sources in soil, especially those contained in decaying plant and fungal organic matter. Intra- as well as inter-scaffold duplications of gene clusters may be a consequence of its parthenogenetic lifestyle. This high quality genome will be instrumental for evolutionary biologists investigating deep phylogenetic lineages among arthropods and will provide the basis for a more mechanistic understanding in soil ecology and ecotoxicology.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3852-x) contains supplementary material, which is available to authorized users.
Keywords: Collembola, Intragenomic rearrangement, Gene family expansions, Hox genes, Horizontal gene transfer, Genome collinearity, Carbohydrate metabolism, Palindrome
Background
The soil environment harbors communities that are functionally important and abundant, as well as biologically extremely diverse. In addition, the greater part of soil microbes and invertebrates remains unknown and has been qualified as the “unseen majority” [1]. To reveal the functionalities of soil organisms, genome information may be of help. Until now such information is mostly limited to nematodes, representatives of the (microscopic) microfauna and a few macrofaunal species, such as the earthworm Lumbricus rubellus [2]. Previously, we studied the genome sequence of a representative of the mesofauna, the springtail Orchesella cincta, which is an obligate sexual reproducing species belonging to the family of Entomobryidae [3]. In this paper, we focus on the parthenogenetic species Folsomia candida, belonging to the family of Isotomidae.
F. candida (Fig. 1) is a soil-dwelling invertebrate that belongs to the hexapod subclass Collembola (springtails), which shares the most recent common ancestor with insects [4]. Collembola are one of the most abundant arthropods. They inhabit soil and leaf litter layers and they have radiated into many niches [5]. For many years, F. candida has been used as a model organism for standardized toxicity tests [6, 7]. F. candida has a high rate of reproduction and is easy to culture, which makes it suitable for laboratory testing.
Fig. 1.

A specimen of the parthenogenetic species Folsomia candida. It has a slender, white body shape on average 2.5 mm long. It has a fully developed furca that is used for jumping. The chewing type of mandibula has a molar plate present. F. candida is a euedaphic species, thriving inside the soil. Photo by Jan van Duinen
The genome of F. candida is diploid and consists of seven chromosome pairs [8]. It has been suggested that the sex determination is diplo-diploid (XX/XO) [8, 9], although definite proof of this is still lacking. Overall, F. candida reproduces through parthenogenesis, although bisexual populations have been observed [10]. Riparbelli et al. [11] have shown that the oocyte is able to self-assemble microtubule-based asters to support centrosome formation and successfully complete meiosis I. The highly unusual spindle structures in subsequent rounds of mitosis may cause maintenance of diploidy in the absence of fertilization [11]. Parthenogenesis is most likely imposed by Wolbachia, an endosymbiotic bacterium located in the ovaries and brain of the animal [10]. In fact, the presence of Wolbachia is essential for reproduction: animals cured of Wolbachia by antibiotic treatment lay eggs that fail to hatch and develop [12, 13]. Despite these observations, causal evidence of Wolbachia-mediated induction of parthenogenesis is currently lacking [14].
In the soil ecosystem, desiccation stress is an important selective factor [15]. F. candida is adapted to cope with moderately dry soil conditions through a unique system of water vapor absorption. The animal maintains all body fluids hyperosmotic to its surroundings to allow net water uptake from the atmosphere by passive diffusion along the gradient in water potential. Below a relative air humidity of 95% the animal actively increases the osmotic pressure of its body fluids by a production of sugars and polyols [16]. Indeed, a transcriptome study performed by Timmermans et al. [17] confirmed the involvement of sugar metabolism in drought adaptation.
Collembolans are a globally significant group of organisms that play a major role in soil functioning [18], particularly through their effect on the rate of litter decomposition and nutrient fluxes [19]. F. candida prefers grazing on fungi growing on organic surfaces rather than on soil particles [5], which has been suggested to stimulate the spread of fungal infections among plants [20]. These plants benefit from a moderate amount of grazing activity by having an increased transfer of spores of essential arbuscular mycorrhizal fungi (AMF) from other plants [21]. More recently, plants have been shown to transfer feeding repellents to their mycorrhizae when the fungal hyphae of AMF are grazed by F. candida [22]. This indicates a tritrophic interaction between plant, AMF, and F. candida potentially improving soil fertility and plant production.
In this study, we describe the first high-quality reference genome of F. candida, assembled from Pacific Bioscience SMRT long sequence reads. We pay special attention to features of the genome that may be associated with a parthenogenetic lifestyle inside the soil. We focus on expanded and lineage-specific gene families as well as horizontal gene transfer (HGTs), and genes related to stress tolerance. We also investigated intragenomic rearrangements and discuss potential consequences of parthenogenesis on genome structure and variation. Where possible, we compare data to the already available genome sequence of O. cincta. The presented genome will be highly instrumental for evolutionary biologists investigating deep phylogenetic lineages among arthropods and will provide the basis for a more mechanistic understanding in soil ecotoxicology.
Results and discussion
F. candida Genome features
A total of 1.43 million PacBio reads were generated from 20 SMRT cells with an N50 of 20,147 bp. As such, we used 18.85 Gbp of raw data to assemble a high-quality reference genome of 221.7 Mbp in only 162 scaffolds with an N50 of 6.5 Mbp and a maximum uninterrupted sequence length of 28.5 Mbp (Table 1). Gaps occupied only 0.1% of all bases. In total, 28,734 gene models could be predicted, which were supported by various experimental data (see section below). We identified 254,312 repeat segments that cover 23.3% (bp) of the genome. The most abundant types of repeats were simple repeats, unclassified, low complexity repeats, and TcMar DNA transposons (Additional file 1). Single-nucleotide polymorphisms (SNP) density was very low and predicted to be 0.148 per kbp. The GC content of the F. candida nuclear genome was 37.5%, which is slightly higher than in the other published collembolan genome of Orchesella cincta [3].
Table 1.
Folsomia candida genome properties
| Assembly | |
| Total sequences | 162 |
| Total bases (Mbp) | 221.7 |
| Min sequence length (bp) | 2433 |
| Max sequence length (Mbp) | 28.5 |
| N50 length (Mbp) | 6.5 |
| GC % | 37.5 |
| Structural Annotation | |
| Genes | 28,734 |
| Mean gene length (bp) | 4615 |
| Exon (%) | 31.9 |
| Intron (%) | 28.0 |
| Repeats (%) | 23.3 |
| Functional annotation | |
| Swiss | 16,528 |
| TrEMBL | 19,592 |
| InterPro | 16,840 |
| Gene Ontology | 15,883 |
| Enzyme Codes | 5535 |
| Validation | |
| CEGMA complete | 245 (98.8%) |
| CEGMA complete + partial | 246 (99.2%) |
| BUSCO complete | 363 (84%) |
| BUSCO complete + partial | 377 (87%) |
Blobology analysis revealed 13 contigs with deviating GC content, which could be the result of contamination. The GC content in scaffold 160 was only 29.9%. Blast searches in the NCBI non-redundant database yielded no significant hits to non-metazoan sequences for this scaffold, suggesting that this scaffold is not a result of contamination. Instead, visual inspection revealed stretches of low base pair calling. (N stretches) along this scaffold, which resulted in a biased GC estimate. The remaining 12 scaffolds show GC contents of approximately 50%. Blast searches indicated that these scaffolds comprised long stretches of GC-rich repeat sequences without any indication for contamination.
The high quality of the genome assembly and the 28,734 gene models were supported by various lines of evidence. First, we identified 245 complete genes and one partial gene out of 248 (98.8%) core eukaryotic CEGMA predicted gene models, which is a good indicator of completeness of the assembled gene space. Subsequently, we applied Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis against the eukaryote dataset and identified 363 (84%) complete single copy orthologs, of which 116 (27%) were duplicated, and 14 (3.2%) were fragmented, while 52 (12%) were missing. Also, 37,680 (99.1%) previously assembled de novo transcripts [23] mapped to the genome. Finally, more than 96% of PacBio long reads and Illumina reads mapped to the final genome scaffolds (Table 1).
Gene density in the F. candida genome was 129.6 genes per Mbp, which is 1.8 times higher than the gene density in the genome of the recently sequenced Collembola species O. cincta [3], but less than in Daphnia pulex (192.9 genes per Mbp) [24]. Overall, the coding sequences covered 31.9% of the total genome. Interpro domains were identified for 16,840 (58.6%) predicted proteins and 20,179 (70.2%) predicted proteins showed similarity to proteins from other species in SwissProt or TrEMBL databases. Also, 15,913 (55.4%) genes were supported by RNA-Seq data with a fraction per kilobase of exon per million fragments (FPKM) of at least 0.5. Moreover, gene ontology (GO) terms could be assigned to 15,883 (55.3%) gene models (Additional file 2), of which 5535 (19.3%) were associated with enzyme codes (EC).
The assembly also included a single scaffold for the mitochondrial genome of F. candida. It is 15,147 bp long and consists of 22 tRNA genes, 13 protein-coding genes, and 2 rRNA genes (Additional file 1). The mitochondrial (mt) genome of F. candida showed the highest sequence identity (75%) and coverage (92%) with the mt genome of the collembolan Cryptopygus antarcticus. Moreover, size, AT percentage and gene content were highly comparable to C. antarcticus as well as to the mt genome of O. cincta, suggesting monophyly of mt genomes within this animal group.
Additionally, we assembled the complete genome of the Wolbachia endosymbiont of F. candida (wFol) in a single scaffold with a size of 1,801,583 bp and GC content of 34.84%. A total of 48 ankyrin (ANK) repeat-containing genes, 35 tRNA genes, 1627 protein coding genes and 3 rRNA genes were identified in the wFol genome (Additional file 1). The presence of ANK repeats in facultative intracellular prokaryotes may be explained by their typical involvement in mediating protein-protein and protein-DNA interactions with the host cells. Such interactions can modulate transcriptional regulation, signal transduction, inflammation response and protein transport in such a way that the endosymbiont is retained in the host [25]. The highest number of ANK genes (60) in a prokaryote has been reported for the Wolbachia strain residing in Culex pipiens, where they show sex-specific expression, implying sex-specific interaction with their host [26]. The authors suggest that ANK genes play a role in the feminization process associated with Wolbachia infection. Gene expression profiling of the wFol genome will be necessary to elucidate whether ANK genes play such a role in F. candida. Interestingly, eight Wolbachia ANK genes were identified to be horizontally transferred into the F. candida genome (Additional file 1). However, these ANK genes showed low protein identity with the ones identified in wFol genome, indicating ancient HGT events.
Recently, Gerth et al. [27] showed that wFol belongs to supergroup E, which is a sister group ancestral to all fully assembled Wolbachia genomes represented in supergroups A to D, F and H. Here, we show that wFol has the largest genome size of all Wolbachia genomes analyzed so far. This suggests that there was a genome reduction event in the common ancestor of more recently evolving Wolbachia strains. Alternatively, the wFol genome expanded in size during evolution in the F. candida host. Timmermans and Ellers [13] showed that F. candida reproduction (egg development) is dependent on Wolbachia, while most of the other sequenced Wolbachia strains form a facultative relationship with their host. The genome size and gene repertoire of the wFol genome thus represents an exception to the general trend that a strong interdependency between an endosymbiont and its host leads to a reduction in the size of the genome of the endosymbiont [28].
Gene family analysis
Out of the total number of proteins in F. candida 18,654 (64.9%) grouped into 7738 gene clusters and 10,080 remained unassigned, although about 35% of these are actively transcribed. Of all gene clusters, 74 were specific for F. candida and 1265 for the collembolan lineage (Additional file 1). In total, we predicted 12,030 gene families in F. candida, represented by more than one gene. Orthofinder was used to identify ortholog groups among 12 species, where five were considered to be outgroups species to hexapods (Strigamia maritima, Ixodes scapularis, Tetranychus urticae, D. pulex and Caenorhabditis elegans), while the remaining seven are all hexapod ingroup species (F. candida, O. cincta, and insects Tribolium castaneum, Pediculus humanus, Acyrthosiphon pisum, Drosophila melanogaster, Aedes aegypti). In total, 2444 gene families were shared between F. candida and all other species in our analysis.
We identified 368 gene families that were expanded in the F. candida genome in relation to the above mentioned species (Additional file 1). Gene ontology analysis identified diverse biological functions among them (Additional file 2). The top five largest expanded families included Sec14-like proteins (154 genes), zinc-finger proteins (123 genes), putative diacylglycerol O-acyltransferase (116 genes), and group 2 allergens (78 genes). Several genes within each of 19 expanded gene clusters were associated with differential expression in response to stress [29]. Among these, the largest gene families included sec14-like proteins, group 2 allergens, ATP-binding cassette (ABC) transporters, glutathione S-transferases (GSTs), and DBH-like monooxygenases/cytochrome p450s. Glutathione S-transferases and ABC transporters participate in phase II xenobiotic metabolism (conjugation of xenobiotic metabolites) and phase III (further modification and excretion of conjugated metabolites). These reactions act to detoxify and remove xenobiotics, plant anti-herbivory toxins, lignocellulose breakdown products, and feeding deterrents. Expansion of these gene families could be an important mechanism of adaptation to the soil environment, where potentially toxic organic compounds persist and accumulate in decaying soil organic matter.
Sec14 proteins are involved in the cytosolic transport of secretory proteins from the Golgi apparatus in yeast [30]. More recent research [31] indicates that Sec14 proteins are essential in membrane trafficking, connecting lipid metabolism with phosphoinositide signaling through transport of phosphatidylinositol. Sec14 proteins contain a CRAL-TRIO lipid-binding domain, and protein families with this domain also show expansion in lepidopteran species [32]. However, the number of Sec14 proteins in both the F. candida genome and the genome of a related collembolan, O. cincta, exceeds by far the greatest number observed in any hexapod genome (Additional file 1). The major expansion of this gene family in two collembolans suggests an important role for this gene in the evolution of these collembolan species.
Two other expanded families worth mentioning are both ATP-dependent DNA helicase PIF1 families that consisted of 52 and 28 members respectively. ATP-dependent DNA helicase is essential in homologous recombination and DNA repair, which are important functions for successful incorporation and maintenance of DNA after translocation and horizontal gene transfer [33]. Likewise, Spo11, involved in homologous recombination, is expanded in the genome of the bdelloid rotifer Adineta vaga, which contains the largest number of horizontally transferred genes of all sequenced metazoans so far [34]. Translocation and horizontal gene transfer seem to have played an important role in F. candida’s genome structure (see sections below), and the expanded ATP-dependent DNA helicase PIF1 family could have facilitated abundant horizontal transfer of genes into the F. candida genome.
Horizontal gene transfer analysis
Earlier we reported that F. candida carries an Isopenicillin N synthase (IPNS) gene most likely acquired by HGT [35]. In order to systematically identify all HGTs in the F. candida genome, we assessed 28,734 genes with the horizontal gene transfer index h, calculated as the score difference between the best non-metazoan and the best metazoan match. We identified a total of 809 ‘foreign’ genes in the F. candida genome (2.8%), which were validated by physical linkage with native genes and phylogenetic inference (Additional file 3). This number is among the highest found in metazoan genomes, being only exceeded in rotifers and some nematode species [34, 36]. Interestingly, transposon abundance was significantly correlated with HGT abundance (Spearman rank correlation rho = 0.637, p < 2.2e−16, Additional file 2). Furthermore, 792 HGTs contain a complete intact open reading frame, while 15 HGTs only miss an ATG start codon, and two miss a 3′ stop codon. In total, 466 HGTs (57.6%) were supported by RNA-Seq data with an FPKM value of more than 0.5, suggesting that a large fraction of HGTs are functional and are being transcribed. The majority of HGTs originated from bacteria (39.9%), fungi (32.8%) and protists (24.6%) (Fig. 2a). A subset of 330 F. candida HGTs each share an orthologous HGT in the O. cincta genome, suggesting that a proportion of these genes have been horizontally transferred before divergence of the two species. The evolutionary split between F. candida and O. cincta is substantial. They belong to different families and have evolved very different adaptive traits [37, 38]. The observation that roughly 30% of HGT has persisted in both species after divergence suggests that these genes are important in the ecology of Collembola.
Fig. 2.
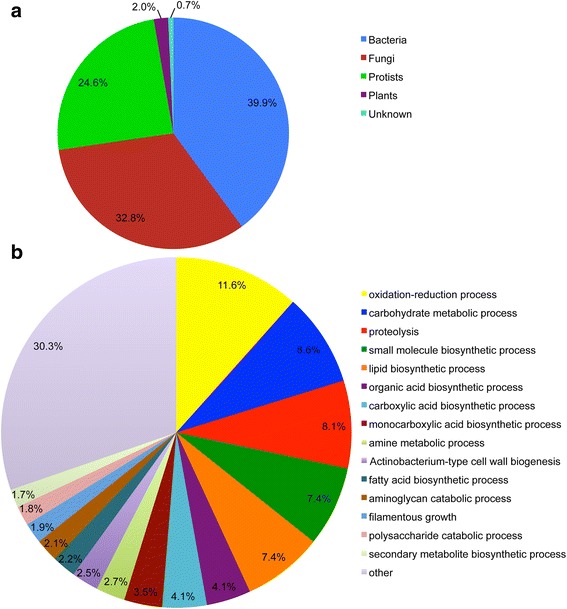
Origin and function of horizontal gene transfer in Folsomia candida. a Origin of foreign genes. b Percentage of enriched gene ontology biological processes associated with foreign genes
Remarkably, the arbuscular mycorrhizal fungus (AMF) Rhizophagus irregularis (Glomus intraradices) is indicated as a source of HGTs in 87 cases. Eighteen of them showed high similarity to heat shock protein (Hsp) 70/90 co-chaperone. Interestingly, three laccases were identified, suggesting the potential of F. candida to break down lignins. F. candida has an intimate association with AMF like Rhizophagus, as it preferably grazes these fungi [22] and the AMF-derived genes may have optimized their grazing capacity. Low springtail abundance is also beneficial for the AMF because grazing induces spreading and fungal inoculation of AMF to other plants [21]. Because AMFs facilitate uptake of phosphorus by plants, while they benefit from the plant’s assimilation products, there is a mutually beneficial tritrophic interaction between F. candida, AMF and the plant, which contributes to soil health and agricultural production [20–22].
Many HGT genes in F. candida were shown to be involved in carbohydrate metabolism (Fig. 2b). A similar pattern was observed in the springtail O. cincta [3]. Statistical analysis showed that HGTs were enriched for CAZymes (229 out of 809 genes have CAZy domains) and that significantly more HGT genes had assigned CAZy domains when compared to the presence of CAZy domains among native genes (two-tailed p-value <0.0001). The largest three families of CAZymes were glycoside hydrolases (GHs), glycosyltransferases (GTs) and auxiliary activity family (AAs) (Fig. 3).
Fig. 3.
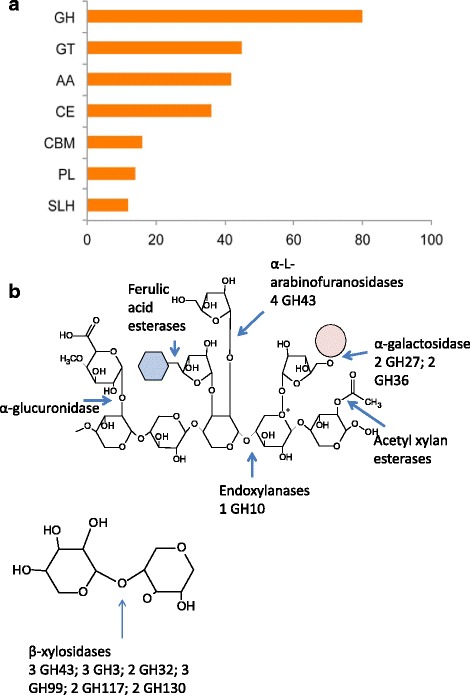
HGT genes with functional domains involved in carbohydrate metabolism as depicted from Carbohydrate-Active enZYmes Database (CAZy). a CAZy general categories among HGTs. Horizontal bars represent the number of unique sequences annotated with CAZyme domains. GH – glycoside hydrolases; GT – glycosyltransferases; AA – auxiliary activity module; CE – carbohydrate esterases; CBM – carbohydrate binding module; PL – polysaccharide lyases; SLH – S-layer homology module. b Hemicellulose degradation. The numbers represent the number of identified HGT enzymes. Pink circle represents D-Galactose; Blue hexagon represents Furelic acid
Glycoside hydrolases and carbohydrate esterases (CEs) are known to degrade cell walls, as was shown in a study on plant cell wall disintegration by fungi and bacteria [39]. Moreover, most enzymes in the AA category belong to the AA7 sub category, which are potentially involved in biotransformation/detoxification of lignocellulose compounds [40]. Based on these findings, we hypothesize that HGTs involved in cell wall degradation could be beneficial for life in the soil. These genes may allow access to important food sources in a habitat rich in plant- and fungal degradation products, which are rich in polysaccharides.
Secondary metabolite and antibiotic biosynthesis gene clusters
Using the antiSMASH tool, we identified 36 biosynthetic gene clusters in the F. candida genome. Most of the genes (52) in these clusters were also identified as HGTs. Functional annotation revealed that 30 clusters were type I polyketide synthases (PKSs), and three were non-ribosomal peptide synthetases (NRPS). Two clusters were related to terpene metabolism and one cluster was unclassified (Additional file 1). Both NRPSs and PKSs are considered to be rare in animals [41].
An interesting feature of F. candida is that it is the first animal discovered with penicillin biosynthesis genes in its genome. One single isopenicillin N synthase (IPNS) gene was reported previously [35]. Our current systemic HGT analysis re-identified IPNS, but the antiSMASH analysis identified a second expressed IPNS gene in the genome together with a second amino adipyl-cysteine-valine synthase (ACVS, antiSMASH cluster 29, Additional file 1), while cluster 30 contained ACVS, and IPNS together with cmcJ and cmcI. This cluster was very recently reconfirmed using Sanger sequencing in another study and is flanked by a retrotransposon [42]. The role of beta-lactam production in the gut epithelium and excretion in the gut lumen could be two-fold. It will allow the animal to control the gut microbial association, stabilizing it compared to its surrounding highly diverse soil microbial community. Indeed, our recent study on gut microbiome diversity in F. candida supports this hypothesis, although differences were observed between animals from a laboratory culture and animals from a natural population [43]. Alternatively, internal beta-lactam production could protect the animal from potentially harmful pathogens in the soil and may explain its lack of susceptibility to pathogens as observed in previous studies [44, 45].
Intragenomic collinearity
Segmental duplications can play a major role in gene and genome evolution because they have been associated with gene innovation and disease-causing rearrangements for instance in humans [46, 47]. To identify syntenic blocks of highly similar gene pairs, a BlastP search was performed using protein sequences within the genome to identify collinear groups of protein-coding gene pairs. We observed 883 (3.2% of all genes) collinear genes, which were organized in 55 syntenic blocks. Most of these were observed on scaffold 4 and 5 (Fig. 4, Additional file 2), while most other large scaffolds (2, 3, 6, and 7) were completely devoid of such structures. These regions were correlated with increased numbers of transposons (Spearman rank correlation rho = 0.637, p < 2.2e−16, Additional file 2), and were also associated with increased numbers of HGT genes (Spearman rank correlation rho = 0.359, p < 1.4e−10, Additional file 2). In contrast, no such collinear blocks were observed within the obligate sexual reproducing species O. cincta.
Fig. 4.
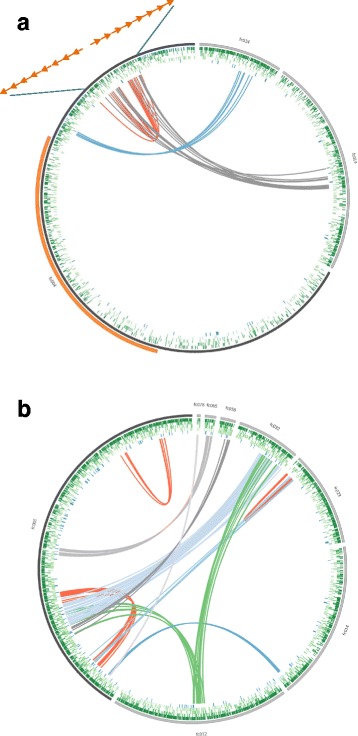
Examples of intragenomic collinearity in the scaffolds 4 (a) and 5 (b) of the Folsomia candida genome. The rings are from outer to inner represent: 1) DNA transposons, 2) LTRs, 3) LINEs, 4) RC (=Helitrons, or rolling circle transposons), 5) HGT genes. The orange bar in Fig. 5a marks the location of the Hox gene cluster
Fourty cases of synteny were observed between collinear blocks across different scaffolds. Some of the collinear blocks suggested allelic variation (scaffold 5 with 58, 65 and 78, Fig. 5a). For instance, scaffold 58 was completely collinear with a gene cluster located on major scaffold 2. Such patterns were previously observed for all collinear gene clusters in the parthenogenetic nematode Meloidogyne incognita [48]. The authors suggested that this might be a consequence of the unusual reproduction system of M. incognita, which includes only mitotic cell division. As such, homologous chromosomes cannot undergo meiotic recombination and therefore diverge independently from each other. However, this explanation cannot be considered for F. candida since it undergoes meiosis I [11]. Moreover, we should observe a division of PacBio coverage between two collinear blocks, if allelic variation is captured in these structures. This was not the case: PacBio coverage did not differ significantly between regions (1 Mb windows) with or without collinear blocks (t259.81 = −0.83, P = 0.41). For instance highly rearranged scaffolds 4 and 5 show highly similar PacBio read coverage when compared with scaffold 2, 3, 6 and 7, which are void of collinear blocks across other scaffolds (Additional file 2). Moreover, several regions showed collinearity with regions on other scaffolds, but their surrounding sequences did not match among the two scaffolds. This was observed for scaffold 5 and 14 (dark blue lines, Fig. 4b), where 12 genes showed collinearity, but their surrounding sequences did not match at all. Such observations may be better explained by translocation events.
Fig. 5.
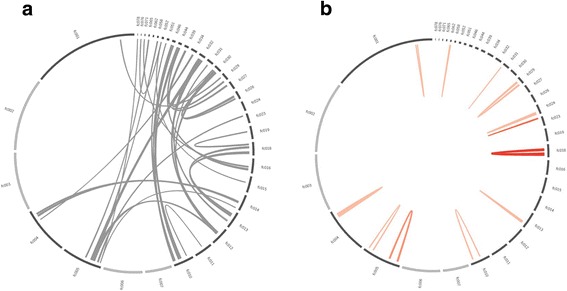
Collinearity of the Folsomia candida genes among and within scaffolds. a inter-scaffold collinear blocks among the seven largest scaffolds (gray); light gray bars depict scaffolds without collinearity. b intra-scaffold collinear blocks; palindromes are depicted in pink; tandem repeats are depicted in red
We identified 15 blocks that were physically linked within a scaffold, of which 11 formed palindromes by inverted duplication (Fig. 5b). Flot et al. [34] suggested that such an arrangement does not allow meiotic pairing. Two palindromes showed substantial copy number variation of a particular open reading frame. The palindrome on scaffold 4 was of special interest because manual inspection showed that it contained two times eight immunoglobin superfamily member Dscam2 genes (schematic representations of Dscam 2 copies are depicted as arrows in Fig. 4a). Dscam2 is proposed to be a tiling receptor in lamina 1 neurons of Drosophila [49]. Drosophila’s visual system is modular with photoreceptor neurons projecting on lamina neurons via a process called tiling. Dscam2 is essential in this tiling process. It is unclear why this particular gene was amplified in this way, especially since F. candida has lost its eye structures as a consequence of evolution in the soil. Another palindrome on scaffold 30 contained two times five copies of a zink finger protein, which is well known to bind DNA and RNA and may be involved in transcriptional regulation.
Palindromic organization of gene synteny was first discovered in the human Y-chromosome [50]. More recently, 17 instances of the palindromic organization were discovered in the genome of the parthenogenetic rotifer A. vaga [34]. Rozen et al. suggested that palindromes are involved in gene conversion, driving the opposite collinear blocks to evolve in concert. This was indeed shown for palindromic sequences in the human Y chromosome [50]. Gene conversion may remove mutations because they are overwritten with the other allelic version. This process can slow down Muller’s ratchet, the irreversible accumulation of detrimental mutations. Alternatively, mutations may become homozygous, and therefore exposed to selection [34]. The Dscam2 genes organized in a palindrome on scaffold 4 (Fig. 4a) showed a low synonymous substitution rate of 0.2, although this was still one magnitude higher than observed in Y-chromosome genes. Subsequently, we compared Ks for segmental duplications regions with inter collinear regions. Figure 6 shows that both palindromic collinear blocks and tandem repeated collinear blocks show a wide range of Ks values. This indicates that a mechanism of concerted evolution does not explain the occurrence of these blocks, despite previous suggestions that such a process would be advantageous for parthenogenetically reproducing animals to counteract accumulation of deleterious mutations.
Fig. 6.
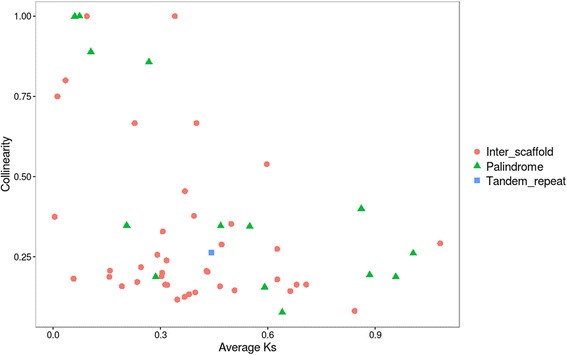
Relation between Ks (synonymous divergence, x-axis) and the fraction of collinear genes for collinear regions (y-axis). Regions with high collinearity and low divergence are probably due to recent duplication events
Rearrangement in the Hox gene cluster
The homeobox domain-containing genes of the Hox gene cluster are important in patterning the anterior-posterior axis during embryonic development. An intact well-ordered Hox gene cluster was observed in the F. candida genome on scaffold 4, spanning 5.36 Mbp. This is extraordinary long when compared to the Hox cluster in a range of invertebrates, which does not exceed 500 Kbp [51]. We checked whether the synteny of Hox cluster in F. candida was according to the ancestral arthropod synteny [51]. Hox genes usually have an anterior-posterior collinearity pattern, where labial/Hox1 coordinates development of anterior/head structures. At the posterior side, Abdominal-B/Hox 11 coordinates more abdominal body parts. The synteny of the ancestral arthropod Hox gene cluster is depicted in Fig. 7.
Fig. 7.
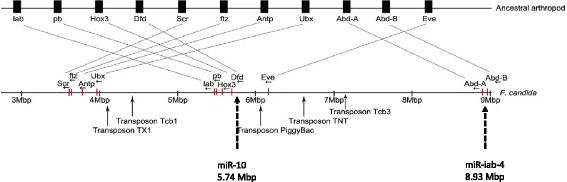
The Hox gene cluster of Folsomia candida on scaffold 4, as compared to consensus arthropod Hox gene cluster. Lab, labial; pb, proboscipedia; Dfd, deformed; Scr, sex combs reduced; ftz, fugi tarazo; Antp, antennapedia; Ubx, ultrabithorax; Abd-A, abdominal-A; Abd-B, abdominal-B; Eve, even-skipped; miR, microRNA (miRNA) positions depicted with dashed arrows. Transposon positions depicted with arrows
Both F. candida and O. cincta contain a full set of 11 functional Hox genes (Fig. 7). In F. candida, they are identified scaffold 4 and surrounded by 577 predicted genes (www.collembolomics.nl). We observed a major rearrangement of this cluster, while in O. cincta the synteny of the Hox gene cluster is completely in accordance with the ancestral arthropod synteny [3]. In F. candida, the segment containing Hox genes Sex combs reduced (Scr), Fushi-tarazu (ftz), Antennapedia (Atnp), and Ultrabithorax (Ubx), (involved in patterning of thoracic segments) were positioned uptream of the Hox genes Labial (lab), Proboscipedia (pb), Hox3, and Deformed (Dfd) (involved in anterior/head patterning), while Even-skipped (Eve) was positioned in between lab, pb, Hox3, Dfd and Abdominal A and B (Abd-A, Abd-B (Fig. 7). This rearrangement can most likely be explained by a translocation event in the region containing Scr-Ubx and violates the general principle of collinearity in the Hox genes cluster. A meta-analysis of Hox genes cluster organization among bilaterian animals by Duboule [52] suggests that collinearity in the Hox genes cluster has strictly been consolidated in vertebrates, but to a lesser extent in arthropods. For instance, the Hox gene cluster is split and distributed over two chromosomal regions in D. melanogaster and Bombyx mori. Furthermore, predatory mites show a completely atomized Hox gene organization [53]. The Hox gene cluster organization of F. candida is more comparable to the disorganized structure in the Hox gene cluster of sea urchins, where the Hox genes 1-3 (lab, pb, Hox3) region is positioned opposite to the posterior Hox cluster next to Hox gene 13 (Abd-B) [54]. This rearrangement was also suggested to be the result of a translocation.
Interestingly, we identified two microRNAs (miR), essential for spatiotemporal Hox gene expression, in F. candida’s Hox gene cluster [55]. MicroRNA miR-iab-4 was positioned in between Abd-A and Abd-B. The experimentally verified targets of miR-iab-4 are Abd-A and Ubx, which are usually positioned next to each other [52]. In contrast, in F.candida Ubx was relocated in front of the anterior gene labial. Whether these genes are still under control of miR-iab-4, needs to be experimentally verified. The other microRNA (miR-10) is usually positioned between Scr and Dfd [55]. In the case of F. candida, it was situated next to Dfd but it seemed disassociated from Scr (Fig. 7), while the latter has been shown to be a target of miR-10 transcriptional control. Temporal and spatial gene expression analysis should elucidate whether these unusual microRNA positions have affected timing of Hox gene activation. Furthermore, we observe five transposons in the Hox cluster, of which two are positioned at break points of the putative translocation event. They could have facilitated the genomic rearrangement of the Hox gene cluster. Additionally, they could have provided novel transcription factor binding sites for correct spatio-temporal expression of the Hox gene cluster [52, 56]. To conclude, we observed a highly unusual Hox gene cluster organization in F. candida. The organization of its body plan, however, contains bilaterian antero-posterior and dorso-ventral axes with normal head, thoracic and abdominal appendages. This suggests that the structural organization of the Hox-cluster bears little relation to the organization of the body plan.
Conclusions
We successfully applied PacBio sequencing to assemble high quality nuclear and mitochondrial genomes of F. candida, including a genome of its endosymbiont Wolbachia. Functional analysis of expanded gene families suggests they are involved in response to environmental stress, recombination and DNA repair, and membrane tracking of lipids. Expansions in such families could be beneficial pre-adaptations to cope with life in the soil as a parthenogenetic species. We also identified a substantial number of HGTs, mainly enriched for carbohydrate active enzymes that may be optimal for unlocking carbohydrates from the plant and fungal decaying matter, which may provide an abundant food source in soil. Moreover, a functional antibiotic biosynthesis cluster was identified, suggesting the production of yet undiscovered antimicrobial compounds in an animal genome.
Analysis of Hox genes indicates a disorganized structure as compared to ancestral arthropod Hox organization. Despite that, F. candida shows a clear anteroposterior axis formation, commonly observed among hexapods. The collinearity analysis of the F. candida genome reflects some extraordinary features that may be related to genetic consequences of a parthenogenetic lifestyle.
Finally, all genome information is organized within the F. candida genome portal (http://www.collembolomics.nl/folsomia/portal) featuring the data and results of our analyses. It will assist researchers interested in the genome and genes of F. candida, and also facilitate further functional studies of this fascinating species.
Methods
Sample preparation and sequencing
F. candida animals (“Berlin strain”, Vrije Universiteit, Amsterdam) were kept at 15 °C in a climate room [57]. For high molecular DNA isolation, we crushed 16 times 100 animals with CTAB lysis buffer (800 μl per 100 animals) and incubated the lysates for 2 h at 65 °C. We extracted DNA from the lysates with phenol:chloroform:isoamyl alcohol (25:24:1) and chloroform:isoamyl alcohol (24:1). After this step, we pooled the water phases into 4 samples and precipitated the DNA with isopropanol. The pellets were dried and solved in 400 μl H2O. To each aliquot, we added 1 μl RNAse (100 mg/ml) and after an incubation of 15 min at 37 °C, we purified the DNA again with chloroform:isoamyl alcohol. We precipitated the DNA with (3 x volume) ethanol 100% and (1/10 x volume) NaAc (3 M, pH = 5.2), washed the pellets with ethanol 70% and after drying we solved the DNA in 200 μl H2O. An extra cleanup of the DNA was necessary for downstream procedures. Therefore, we used the MoBio Power Clean® DNA clean up kit and followed the manufacturer’s protocol. F. candida genome fragments of more than 20 kbp were sequenced using 12 SMRT cells on the PacBio RS II platform (with P6 chemistry) according to the manufacturer’s protocol at the Leiden Genome Technology Center (LGTC).
At the initial stage of the project, we also generated Illumina genomic data. For that, we isolated DNA from 2 × 50 animals from an isofemale line of F. candida. The animals were washed in 70% ethanol and crushed in 200 μl PBS. 200 μl Nuclei Lysis Solution (Promega) and 4 μl Proteinase K (20 mg/ml, Roche) were added before incubation for 15 min at 65 °C. After incubation 340 μl DNA Lysis Buffer (Promega) was added. The lysate was centrifuged for 10 min at 14.000 rpm. DNA was recovered from the supernatant using a DNA spin column (Promega), following the manufacturer’s protocol. DNA of the 2 samples was pooled and precipitated with ice-cold 2,5 x volume 100% ethanol/0,1 x volume NaAc (3 M, pH = 5,2) at −80 °C. After washing with ice-cold 70% ethanol, the DNA pellet was solved in 30 μl H2O. For sequencing, we used 3 μl 500 ng/μl DNA. Next-generation sequencing was performed on the Illumina HiSeq2000 platform at the LGTC.
Assembly of F. candida genome
The assembly of F. candida genome PacBio reads was done with the Falcon assembler [58]. Reads longer than 8 kbp were assembled with the following parameters (output_multi, min_it = 0.70, local_match_count_threshold = 2, max_n_read = 200, max_diff = 100, max_cov = 50, min_cov = 5, bestn = 10) followed by PBJelly [59] scaffolding and gap filling and polishing with Quiver [60]. Afterwards, we performed another scaffolding with SSPACE-LongRead v.1.1 (l = 2) [61] and polished final scaffolds with Pilon v.1.13 (−-fix bases, −-diploid) [62]. Blobology was applied to identify taxon-annotated GC-coverage plots, possibly indicating contamination from external sources [63]. CEGMA pipeline v.2.4 [64] and Benchmarking Universal Single-Copy Orthologs (BUSCO) tool v.1.1 [65] was used to identify universal single copy orthologs in the assembly as a measure of the completeness and contiguity.. Additionally, we mapped F. candida de novo transcripts [23] to the genome scaffolds with Blat and used isoblat v.3.0 [66] to assess assembly completeness. The genome of Wolbachia endosymbiont was identified among assembled scaffolds using BlastN [67] against other Wolbachia genomes. All reads mapping to these two scaffolds were selected and assembled in Falcon [58] (output_multi, min_it = 0.70, local_match_count_threshold = 2, max_n_read = 100, max_diff = 40, max_cov = 60, min_cov = 2, bestn = 10) followed by PBJelly [59]. Strings of N’s were removed from the scaffolds of the original assembly introducing breaks and the two assemblies were merged using Minimus2 [68]. The remaining two gaps were resolved by blasting the trimmed edges of the scaffolds to the PacBio library in search for reads bridging them. Consensuses of these reads were created with PBdagcon (https://github.com/PacificBiosciences/pbdagcon), after which they were manually inserted into the assembly. Thereafter all reads were mapped back to the assembly and a consensus was created with PBdagcon [69] followed by a polishing with Quiver [60].
To reconstruct a mitochondrial genome of F. candida, quality checked Illumina reads were trimmed in Trimmomatic v.0.33 [70] with the following parameters: ILLUMINACLIP:TruSeq2-PE.fa:2:30:10; LEADING:3; TRAILING:3; SLIDINGWINDOW:4:15; HEADCROP:8; MINLEN:50. The reads were then error corrected in Lighter (k = 17; alpha = 0.085) [71]. We used a PacBio read covering the complete mitochondrial genome as a reference for IOGA [72]. The annotation of the mitochondrial genome was performed in MITOS annotation web-tool [73] followed by manual curation.
Genome annotation
Genome annotation was performed using the MAKER v.2.31.8 pipeline [74]. Augustus v.3.1.0. [75] and GeneMark [76] were used as ab initio gene predictors; they were trained using BRAKER1 [77] with raw RNA-Seq data (17.9 Gbp Illumina, SRR935329 [23], 3.3 Gbp Illumina, SRR921597 [4]). The de novo F. candida transcriptome [23], as well as RNA-Seq data mapped with TopHat2 [78] and protein sequences from D. pulex, A. pisum and D. melanogaster from the Ensembl Genomes database [79] were used as transcript-based and protein homology-based evidence, respectively. We used RepeatMasker with RepBase database [80, 81] together with a de novo repeat library, constructed using RepeatModeler [82] to find and mask repeats in the F. candida genome. Functional annotation was assigned with BlastP [67] (E-value threshold of 0.1) using SwissProt, TrEMBL databases, and InterProScan analysis [83] against the Superfamily [84] and Pfam [85] protein databases. Also, we used Blast2GO suite v.3.1 to predict gene ontology (GO) terms in our dataset based on InterPro domains and BlastP hits against Swiss-Prot database (E-value threshold of 1e−5). To identify secondary metabolite biosynthesis gene clusters, we applied antiSMASH [86].
Gene family expansion analysis
OrthoFinder with inflation parameter of 1.5 [87] was used to calculate ortholog clusters between F. candida, O. cincta, T. castaneum, P. humanus, A. pisum, D. melanogaster, A. aegypti, S. maritima, I. scapularis, T. urticae, D. pulex and C. elegans. In order to identify expanded gene families in F. candida, we calculated z-scores as proposed by Cao et al. [88]. In short, the gene number for each family was subtracted by the average gene number of the family among all species in the analysis. This number was then divided by the standard deviation of the average gene number. Gene families with z-scores ≥2 were assumed to be expanded. Additionally, we performed a BlastN search (with E-value threshold of 1e−5) of expanded gene families against genes that were differentially expressed in response to stress [29].
Horizontal gene transfer
To identify foreign genes in the genome of F. candida, we performed calculations of the horizontal gene transfer index h based on the protocol described by Crisp et al. [36] with a number of modifications described by Faddeeva-Vakhrusheva et al. [3]. The final set of foreign genes was predicted based on the h-score, followed by a physical linkage test (PacBio long read evidence of physical linkage of HGT candidate to neighboring native genes). HGT candidates that passed the linkage test, but with a best metazoan Blast bit-score of ≥50 were also subjected to phylogenetic validation with the same parameters as described by Faddeeva-Vakhrusheva et al. [3, 36]. To predict enriched gene ontology biological processes and molecular functions among the foreign genes in F. candida, we performed GO enrichment analysis using the topGO package [89] in R (v.3.2.2) with an elim algorithm (with the p-value threshold of 0.05). To check whether HGT genes were enriched for carbohydrate metabolism a chi-square test was performed. We additionally checked CAZy (Carbohydrate-Active enZYmes Database) domains [90] in the HGT genes and we compared the number of HGT genes with CAZy (with an e-value <0.1) domains to the number of native genes with CAZy domains with a chi-square test.
Hox gene cluster analysis
We used BlastP to identify Hox genes and we assessed the synteny manually using the genomic locations on scaffold 4.
Collinearity analysis
A BlastP search of all-against-all F. candida proteins was performed using an E-value cutoff of 1e−10. Collinear blocks of genes were detected using the package MCScanX [91]. Synteny plots were drawn using Circos [92]. A pearson correlation test was performed to statistically test association between collinear blocks, transposons and horizontally transferred genes.
Additional files
Tables with associated data. (XLSX 4914 kb)
Word file containing gene ontology distribution in Folsomia candida genome, metabolic map for Folsomia candida, correlation plots between HGTs transposons and collinearity; additional collinear scaffolds, distribution of PacBio read coverage along the 7 largest scaffolds. (DOCX 2743 kb)
Phylogenetic trees for confirmed HGTs in Folosmia candida genome. (PDF 1641 kb)
Acknowledgements
We thank Prof. Dr. Johan den Dunnen for facilitating next-generation sequencing at the LGTC, Dr. Marnix Medema for performing antiSMASH analysis, Michael Akam and David Ferrier for helpful discussions on the Hox gene cluster, and Peter H. Neleman for help with python scripts. We also thank 1KITE Consortium, Sara Calçada Novais, and Tiago Filipe da Silva Simões for providing additional RNA-Seq data for F. candida. We acknowledge SURFsara support team for assistance with the calculations on the SURFsara LISA cluster and SURFsara cloud. Finally, we thank Roland den Hollander from VU IT for Research for help with website hosting.
Funding
This work is supported by grant F08.001.03 and F07.003.05 from the BE-BASIC foundation. DR receives additional funding from EU FP7 program Sustainable Nanotechnologies (SUN) grant number 604305. JE and KK are supported by the Netherlands Organisation for Scientific Research, VICI grant 865.12.003. These funding bodies had no role in the design of the study and collection, analysis, and interpretation of data or in writing this manuscript.
Availability of data and materials
Data deposition: raw Illumina and PacBio data are deposited to the NCBI SRA under accessions SRX1356974 and SRX1436141 correspondingly. The draft genome including gene annotations is available at GenBank under BioProject accession: PRJNA299291. The mitochondrial genome is available at GenBank under accession number KU198392. In addition, all genomic data is available via http://www.collembolomics.nl/folsomia/portal/. The complete genome of the Wolbachia endosymbiont of Folsomia candida is available at GenBank under accession number Bioproject accession number PRJNA300838.
Authors’ contributions
DR, AFV and NMvS conceived, designed, coordinated the study and drafted the manuscript. SYA and AFV performed the assembly of the genome. KK, DR and JE performed collinearity analysis and Spearman rank tests. KK provided frequency distribution graphs of PacBio read coverages in scaffolds. MFLD, AFV and SS performed further scaffolding of the initial genome assembly, performed gene annotation and set up the genome portal. AFV performed orthology analysis and gene family expansion analysis, and DR assisted in interpretation of the GO-enriched annotated gene families. VA, WS, AFV and DR performed HGT analysis, including phylogenetic validation and PacBio linkage tests. AAK and JE assembled the wFol genome including initial annotation of the wFol genome. GLN performed CAZy analysis and assisted in the interpretation of GO enrichment of the HGT gene set. CAMvG assisted in the analysis of gene family expansions involved in stress response. JM prepared ultra clean high molecular genomic DNA for PacBio sequencing and assisted in PacBio SMRT library preparation and sequence data acquisition. DR performed Hox gene cluster analysis and microRNA analysis. All authors provided input and comments on subsequent revisions of the manuscript and approved the final version. Finally, each of the authors have confirmed to be accountable for all aspects of the work by appropriately investigating and resolving issues related to accuracy and integrity.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ABC
ABC transporter
- Abd
Abdominal
- ACVS
Amino adipyl-cysteine-valine synthase
- AMF
Arbuscular mycorrhizal fungus
- ANK
Ankyrin
- Antp
Antennapedia
- CAZy
Carbohydrate enzyme
- Dfd
Deformed
- Dscam
Down syndrome cell adhesion molecule
- FPKM
fraction per kilobase of exon per million fragments
- ftz
Fushi-tarazu
- GST
Glutathione S-transferases
- HGT
Horizontal gene transfer
- IPNS
Isopenicillin N synthase
- lab
Labial
- miR
microRNA
- PacBio
Pacific Biosciences Sequencing
- pb
Proboscipedia
- Scr
Sex combs reduced
- SMRT
Single molecule real-time sequencing
- SNP
Single nucleotide polymorphism
- Ubx
Ultrabithorax
- wFol
Wolbachia endosymbiont of F. candida
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3852-x) contains supplementary material, which is available to authorized users.
Contributor Information
Anna Faddeeva-Vakhrusheva, Email: ann.faddeeva@gmail.com.
Ken Kraaijeveld, Email: ken@kenkraaijeveld.nl.
Martijn F. L. Derks, Email: martijn.derks@wur.nl
Seyed Yahya Anvar, Email: S.Y.Anvar@lumc.nl.
Valeria Agamennone, Email: vale_aga@hotmail.com.
Wouter Suring, Email: woutersuring@gmail.com.
Andries A. Kampfraath, Email: a.a.kampfraath@vu.nl
Jacintha Ellers, Email: j.ellers@vu.nl.
Giang Le Ngoc, Email: giangln@gmail.com.
Cornelis A. M. van Gestel, Email: kees.van.gestel@vu.nl
Janine Mariën, Email: janine.marien@vu.nl.
Sandra Smit, Email: sandra.smit@wur.nl.
Nico M. van Straalen, Email: n.m.van.straalen@vu.nl
Dick Roelofs, Phone: +31 20 59 87078, Email: dick.roelofs@vu.nl.
References
- 1.Van Der Heijden MG, Bardgett RD, Van Straalen NM. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett. 2008;11:296–310. doi: 10.1111/j.1461-0248.2007.01139.x. [DOI] [PubMed] [Google Scholar]
- 2.Elsworth BL. Unearthing the genome of the earthworm Lumbricus rubellus. Edinburgh: University of Edinburgh; 2013.
- 3.Faddeeva-Vakhrusheva A, Derks M, Anvar S, Agamennone V, Suring W, Smit S, van Straalen N, Roelofs D. Gene family evolution reflects adaptation to soil environmental stressors in the genome of the collembolan Orchesella cincta. Genome Biol Evol. 2016;8:2106–2117. doi: 10.1093/gbe/evw134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misof B, Liu S, Meusemann K, Peters RS, Donath A, Mayer C, Frandsen PB, Ware J, Flouri T, Beutel RG. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346:763–767. doi: 10.1126/science.1257570. [DOI] [PubMed] [Google Scholar]
- 5.Fountain MT, Hopkin SP. FOLSOMIA CANDIDA (COLLEMBOLA): a “standard” soil arthropod. Annu Rev Entomol. 2005;50:201–222. doi: 10.1146/annurev.ento.50.071803.130331. [DOI] [PubMed] [Google Scholar]
- 6.ISO 11267 guideline: Soil Quality. Inhibition of Reproduction of Collembola (Folsomia candida) by Soil Pollutants. Geneve: International Organization for Standardization; 2014.
- 7.Organisation for Economic Co-operation and Development, OECD. Test No. 232: Collembolan Reproduction Test in Soil. Paris: Publishing; 2009.
- 8.Kiauta B. Review of the germ cell chromosome cytology of Collembola, with a list of chromosome numbers and data on two species new to cytology. Genen en Phaenen. 1970;4:89–99. [Google Scholar]
- 9.Hopkin SP. Biology of the springtails:(Insecta: Collembola). Oxford: OUP Oxford; 1997.
- 10.Frati F, Negri I, Fanciulli PP, Pellecchia M, De Paola V, Scali V, Dallai R. High levels of genetic differentiation between Wolbachia-infected and non-infected populations of Folsomia candida (Collembola, Isotomidae) Pedobiologia. 2004;48:461–468. doi: 10.1016/j.pedobi.2004.04.004. [DOI] [Google Scholar]
- 11.Riparbelli MG, Giordano R, Callaini G. Centrosome inheritance in the parthenogenetic egg of the collembolan Folsomia candida. Cell Tissue Res. 2006;326:861–872. doi: 10.1007/s00441-006-0253-x. [DOI] [PubMed] [Google Scholar]
- 12.Pike N, Kingcombe R. Antibiotic treatment leads to the elimination of Wolbachia endosymbionts and sterility in the diplodiploid collembolan Folsomia candida. BMC Biol. 2009;7:54. doi: 10.1186/1741-7007-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmermans MJ, Ellers J. Wolbachia endosymbiont is essential for egg hatching in a parthenogenetic arthropod. Evol Ecol. 2009;23:931–942. doi: 10.1007/s10682-008-9282-0. [DOI] [Google Scholar]
- 14.Czarnetzki AB, Tebbe CC. Detection and phylogenetic analysis of Wolbachia in Collembola. Environ Microbiol. 2004;6:35–44. doi: 10.1046/j.1462-2920.2003.00537.x. [DOI] [PubMed] [Google Scholar]
- 15.Holmstrup M, Slotsbo S, Rozsypal J, Henriksen PG, Bayley M. Accumulation of free amino acids during exposure to drought in three springtail species. J Insect Phys. 2015;82:114–121. doi: 10.1016/j.jinsphys.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Bayley M, Holmstrup M. Water vapor absorption in arthropods by accumulation of myoinositol and glucose. Science. 1999;285:1909–1911. doi: 10.1126/science.285.5435.1909. [DOI] [PubMed] [Google Scholar]
- 17.Timmermans MJ, Roelofs D, Nota B, Ylstra B, Holmstrup M. Sugar sweet springtails: on the transcriptional response of Folsomia candida (Collembola) to desiccation stress. Insect Mol Biol. 2009;18:737–746. doi: 10.1111/j.1365-2583.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 18.Rusek J. Biodiversity of Collembola and their functional role in the ecosystem. Biodivers Conserv. 1998;7:1207–1219. doi: 10.1023/A:1008887817883. [DOI] [Google Scholar]
- 19.Cragg RG, Bardgett RD. How changes in soil faunal diversity and composition within a trophic group influence decomposition processes. Soil Biol Biochem. 2001;33:2073–2081. doi: 10.1016/S0038-0717(01)00138-9. [DOI] [Google Scholar]
- 20.Lussenhop J. Collembola as mediators of microbial symbiont effects upon soybean. Soil Biol Biochem. 1996;28:363–369. doi: 10.1016/0038-0717(95)00145-X. [DOI] [Google Scholar]
- 21.Klironomos JN, Moutoglis P. Colonization of nonmycorrhizal plants by mycorrhizal neighbours as influenced by the collembolan, Folsomia candida. Biol Fert Soils. 1999;29:277–281. doi: 10.1007/s003740050553. [DOI] [Google Scholar]
- 22.Duhamel M, Pel R, Ooms A, Bücking H, Jansa J, Ellers J, Van Straalen NM, Wouda T, Vandenkoornhuyse P, Kiers ET. Do fungivores trigger the transfer of protective metabolites from host plants to arbuscular mycorrhizal hyphae? Ecology. 2013;94:2019–2029. doi: 10.1890/12-1943.1. [DOI] [PubMed] [Google Scholar]
- 23.Faddeeva A, Studer R, Kraaijeveld K, Sie D, Ylstra B. Mariën J, op den camp H, Datema E, den Dunnen J, van Straalen N: collembolan Transcriptomes highlight molecular evolution of hexapods and provide clues on the adaptation to terrestrial life. PLoS One. 2015;10:e0130600. doi: 10.1371/journal.pone.0130600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaur SJ, Rahman MS, Ammerman NC, Beier-Sexton M, Ceraul SM, Gillespie JJ, Azad AF. TolC-dependent secretion of an ankyrin repeat-containing protein of Rickettsia typhi. J Bacteriol. 2012;194:4920–4932. doi: 10.1128/JB.00793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker T, Klasson L, Sebaihia M, Sanders MJ, Thomson NR, Parkhill J, Sinkins SP. Ankyrin repeat domain-encoding genes in the w pip strain of Wolbachia from the Culex pipiens group. BMC Biol. 2007;5:39. doi: 10.1186/1741-7007-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerth M, Gansauge MT, Weigert A, Bleidorn C. Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nat Comms. 2014;5:5117. doi: 10.1038/ncomms6117. [DOI] [PubMed] [Google Scholar]
- 28.McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- 29.de Boer TE, Janssens TK, Legler J, van Straalen NM, Roelofs D. Combined Transcriptomics analysis for classification of adverse effects as a potential end point in effect based screening. Environ Sci Technol. 2015;49:14274–14281. doi: 10.1021/acs.est.5b03443. [DOI] [PubMed] [Google Scholar]
- 30.Bankaitis VA, Malehorn DE, Emr SD, Greene R. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bankaitis VA, Mousley CJ, Schaaf G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem Sci. 2010;35:150–160. doi: 10.1016/j.tibs.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith G, Briscoe AD. Molecular evolution and expression of the CRAL_TRIO protein family in insects. Insect Biochem Mol Biol. 2015;62:168–173. doi: 10.1016/j.ibmb.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Wu L, Hickson ID. DNA Helicases required for homologous recombination and repair of damaged replication forks. Ann Rev of Genet. 2006;40:279–306. doi: 10.1146/annurev.genet.40.110405.090636. [DOI] [PubMed] [Google Scholar]
- 34.Flot J-F, Hespeels B, Li X, Noel B, Arkhipova I, Danchin EG, Hejnol A, Henrissat B, Koszul R, Aury J-M. Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature. 2013;500:453–457. doi: 10.1038/nature12326. [DOI] [PubMed] [Google Scholar]
- 35.Roelofs D, Timmermans MJ, Hensbergen P, van Leeuwen H, Koopman J, Faddeeva A, Suring W, de Boer TE, Marien J, Boer R, et al. A functional isopenicillin N synthase in an animal genome. Mol Biol Evol. 2013;30:541–548. doi: 10.1093/molbev/mss269. [DOI] [PubMed] [Google Scholar]
- 36.Crisp A, Boschetti C, Perry M, Tunnacliffe A, Micklem G. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 2015;16:50. doi: 10.1186/s13059-015-0607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dell'Ampio E, Meusemann K, Szucsich NU, Peters RS, Meyer B, Borner J, Petersen M, Aberer AJ, Stamatakis A, Walzl MG, et al. Decisive data sets in Phylogenomics: lessons from studies on the Phylogenetic relationships of primarily wingless insects. Mol Biol Evol. 2014;31:239–249. doi: 10.1093/molbev/mst196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D'Haese CA. Were the first springtails semi-aquatic? A phylogenetic approach by means of 28S rDNA and optimization alignment. Proc Roy Soc B-Biol Sci. 2002;269:1143–1151. doi: 10.1098/rspb.2002.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ospina-Giraldo MD, Griffith JG, Laird EW, Mingora C. The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics. 2010;11:525. doi: 10.1186/1471-2164-11-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci U S A. 2014;111:9259–9264. doi: 10.1073/pnas.1401734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suring W, Meusemann K, Blanke A, Mariën J, Schol T, Agamennone V, Faddeeva-Vakhrusheva A, Berg MP, the KBHc, Brouwer A, et al. Evolutionary ecology of beta-lactam gene clusters in animals. Mol Ecol. 2017. In press. [DOI] [PubMed]
- 43.Agamennone V, Jakupovic D, Weedon JT, Suring WJ, van Straalen NM, Roelofs D, Roling WFM: The microbiome of Folsomia candida: an assessment of bacterial diversity in a Wolbachia-containing animal. Fems Microbiol Ecol. 2015;91, article 11: 1–10. [DOI] [PubMed]
- 44.Broza M, Pereira RM, Stimac JL. The nonsusceptibility of soil Collembola to insect pathogens and their potential as scavengers of microbial pesticides. Pedobiologia. 2001;45:523–534. doi: 10.1078/0031-4056-00104. [DOI] [Google Scholar]
- 45.Dromph KM, Vestergaard S. Pathogenicity and attractiveness of entomopathogenic hyphomycete fungi to collembolans. Appl Soil Ecol. 2002;21:197–210. doi: 10.1016/S0929-1393(02)00092-6. [DOI] [Google Scholar]
- 46.Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- 47.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 48.Abad P, Gouzy J, Aury J-M, Castagnone-Sereno P, Danchin EG, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne Incognita. Nat Biotechnol. 2008;26:909–915. doi: 10.1038/nbt.1482. [DOI] [PubMed] [Google Scholar]
- 49.Millard SS, Flanagan JJ, Pappu KS, Wu W, Zipursky SL. Dscam2 mediates axonal tiling in the Drosophila visual system. Nature. 2007;447:720–724. doi: 10.1038/nature05855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature. 2003;423:873–876. doi: 10.1038/nature01723. [DOI] [PubMed] [Google Scholar]
- 51.Chipman AD, Ferrier DE, Brena C, Qu J, Hughes DS, Schröder R, Torres-Oliva M, Znassi N, Jiang H, Almeida FC. The first myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol. 2014;12:e1002005. doi: 10.1371/journal.pbio.1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duboule D. The rise and fall of Hox gene clusters. Development. 2007;134:2549–2560. doi: 10.1242/dev.001065. [DOI] [PubMed] [Google Scholar]
- 53.Pace RM, Grbic M, Nagy LM. Composition and genomic organization of arthropod Hox clusters. EvoDevo. 2016;7:11. doi: 10.1186/s13227-016-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cameron RA, Rowen L, Nesbitt R, Bloom S, Rast JP, Berney K, Arenas-Mena C, Martinez P, Lucas S, Richardson PM. Unusual gene order and organization of the sea urchin hox cluster. J Exp Zool B Mol Develop Evol. 2006;306:45. doi: 10.1002/jez.b.21070. [DOI] [PubMed] [Google Scholar]
- 55.Miura S, Nozawa M, Nei M. Evolutionary changes of the target sites of two MicroRNAs encoded in the Hox Gene cluster of Drosophila and other insect species. Genome Biol Evol. 2011;3:129–139. doi: 10.1093/gbe/evq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daborn P, Yen J, Bogwitz M, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P. A single P450 allele associated with insecticide resistance in Drosophila. Science. 2002;297:2253–2256. doi: 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- 57.de Boer ME, de Boer TE, Mariën J, Timmermans MJ, Nota B, van Straalen NM, Ellers J, Roelofs D. Reference genes for QRT-PCR tested under various stress conditions in Folsomia candida and Orchesella cincta (Insecta, Collembola) BMC Mol Biol. 2009;10:54. doi: 10.1186/1471-2199-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falcon Pacific Biosciences https://github.com/PacificBiosciences/FALCON. Accessed Sep 2015.
- 59.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. Plos ONE. 2012;7:e47768. [DOI] [PMC free article] [PubMed]
- 60.Quiver Pacific Biosciences https://github.com/PacificBiosciences/GenomicConsensus. Accessed Sep 2015.
- 61.Boetzer M, Pirovano W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC bioinformatics. 2014;15:211. doi: 10.1186/1471-2105-15-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Frontiers Genet. 2013;4:237. doi: 10.3389/fgene.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 65.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 66.Ryan JF: Baa. pl: a tool to evaluate de novo genome assemblies with RNA transcripts. arXiv preprint arXiv. 2013;13092087. https://arxiv.org/abs/1309.2087.
- 67.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 68.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC bioinformatics. 2007;8:1. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pbdagcon Pacific Biosciences https://github.com/PacificBiosciences/pbdagcon. Accessed Sep 2015.
- 70.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song L, Florea L, Langmead B. Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014;15:509. doi: 10.1186/s13059-014-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bakker FT, Lei D, Yu J, Mohammadin S, Wei Z, Kerke S, Gravendeel B, Nieuwenhuis M, Staats M, Alquezar-Planas DE. Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline. Biol J Linnean Soc. 2016;117:33–43. doi: 10.1111/bij.12642. [DOI] [Google Scholar]
- 73.Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phyl Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 74.Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M: BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2015:btv661. doi:10.1093/bioinformatics/btv661 [DOI] [PMC free article] [PubMed]
- 78.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ensembl Genomes http://ensemblgenomes.org, 2015. Accessed Sep 2015.
- 80.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cyt Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 81.Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Cur Protocols Bioinformatics. 2009:4-10. doi:10.1002/0471250953.bi0410s25. [DOI] [PubMed]
- 82.RepeatModeler. http://www.repeatmasker.org/RepeatModeler/. Accessed Sep 2015.
- 83.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, Chothia C, Gough J. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009;37:D380–D386. doi: 10.1093/nar/gkn762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014:D222–30. [DOI] [PMC free article] [PubMed]
- 86.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E. Breitling R: antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cao Z, Yu Y, Wu Y, Hao P, Di Z, He Y, Chen Z, Yang W, Shen Z, He X. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nature comm. 2013;4:2602. doi: 10.1038/ncomms3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 90.Terrapon N, Lombard V, Gilbert HJ, Henrissat B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics. 2015;31:647–655. doi: 10.1093/bioinformatics/btu716. [DOI] [PubMed] [Google Scholar]
- 91.Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X. Lee T-h, Jin H, Marler B, Guo H: MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables with associated data. (XLSX 4914 kb)
Word file containing gene ontology distribution in Folsomia candida genome, metabolic map for Folsomia candida, correlation plots between HGTs transposons and collinearity; additional collinear scaffolds, distribution of PacBio read coverage along the 7 largest scaffolds. (DOCX 2743 kb)
Phylogenetic trees for confirmed HGTs in Folosmia candida genome. (PDF 1641 kb)
Data Availability Statement
Data deposition: raw Illumina and PacBio data are deposited to the NCBI SRA under accessions SRX1356974 and SRX1436141 correspondingly. The draft genome including gene annotations is available at GenBank under BioProject accession: PRJNA299291. The mitochondrial genome is available at GenBank under accession number KU198392. In addition, all genomic data is available via http://www.collembolomics.nl/folsomia/portal/. The complete genome of the Wolbachia endosymbiont of Folsomia candida is available at GenBank under accession number Bioproject accession number PRJNA300838.