

Abstract
Pancreatic cancer is a very aggressive disease with a poor prognosis. The majority of them are attributed to sporadic causes, especially to many modifiable risk factors such as tobacco or alcohol abuse. The principal histologic subtype of pancreatic cancer is ductal adenocarcinoma. Pancreatic neuroendocrine tumors, which constitute a more indolent entity, represent second type of pancreatic cancer in terms of incidence. Individuals with a family history of pancreatic cancer carry an increased risk of developing the disease, which may be related to an underlying hereditary component. Unfortunately, in the majority of these families the suspected germline genetic cause responsible of the disease will not be identified, but approximately in a 20% of the cases a hereditary cancer predisposition syndrome with increased risk of pancreatic cancer development can be recognized. This review will be focused on the leading hereditary cancer syndromes related to pancreatic ductal adenocarcinoma and pancreatic neuroendocrine tumors. Additionally, we will try to explain clinical aspects related to the identification of germline mutations in pancreatic cancer patients and their potential implications in oncologic treatment decisions.
Keywords: Pancreatic ductal adenocarcinoma, Pancreatic neuroendocrine tumor, Familial pancreatic cancer, Genetic testing, Hereditary cancer
Background
From a histological point of view [1], almost 95% of pancreatic cancers are pancreatic ductal adenocarcinomas (PDAC). PDAC has the worst prognosis among the major cancers and it constitutes the fourth leading cause of cancer death in the developed world. Such is its relevance that PDAC will be the third cause of cancer mortality in European Union in 2017, with 43,800 expected deaths among women and 43.600 among men [2]. In the United States the prospects also are not looking very hopeful, with 53,670 new cases and 43,090 deaths due to pancreatic cancer predicted this year [3]. The average lifetime risk of pancreatic cancer for both men and women is about 1 in 65 (1.5%) and the five year survival rate is 7% [4], which is related to advanced stage at diagnosis in the majority of the cases.
Pancreatic neuroendocrine tumors (PNETs) are infrequent neoplasms that represent approximately 1-2% of all pancreatic cancers [5]. Estimated incidence for PNETs is less than 1/100,000 per year although their relative indolent nature could underestimate these numbers [5, 6]. Five year survival rate in PNETs is about 42%, which is in concordance with predominant diagnosis of low-intermediate grade tumors [7].
Older age constitutes one of the main risk factors in the development of PDAC. Median age of onset of PDAC is 71 years [8]. Tobacco is the most important recognized toxic risk factor, doubling the risk [9]. Alcohol abuse [10], chronic pancreatitis [11], dietary factors, obesity, exposure to different agents and Diabetes Mellitus type 2 (DM2) also increase risk for PDAC [12]. Apart from these factors, family history also can influence in the probability of PDAC development [13]. Different reports that have evaluated incidence of pancreatic cancer in relatives have found that first degree relatives have almost a two-fold increased risk of developing PDAC and also that this risk seems to be proportional to the number of first-degree relatives with PDAC [14, 15]. It is estimated that a hereditary component may be implicated in nearly 10% of all PDAC cases [16, 17], but currently in less than 20% of them a defined hereditary cancer predisposition syndrome with increased risk of PDAC development can be identified.
Conversely, in PNETs, there are no well recognized modifiable risk factors [18]. The majority of them are considered sporadic tumors and in about 20% of the cases a cancer hereditary syndrome can be recognized [19].
Hereditary cancer syndromes related to PNETs
Main hereditary cancer syndromes related to an increased risk in PNETs development are multiple endocrine neoplasia type 1 (MEN1), Von-Hippel-Lindau disease (VHL) and neurofibromatosis type 1 (NF-1). Although less frequently, a relation between PNETs and Tuberous sclerosis complex (TSC) has been suggested. Predominantly PNETs in hereditary syndromes are grade 1 (PNET G1, Ki-67 index <3%) and grade 2 (PNET G2, Ki-67 index 3-20%) tumors [20].
MEN1, which is also referred as Wermer syndrome [21], is clinically characterized by the classical triad of tumors of the parathyroid glands, the pancreatic islet cells, and the anterior pituitary and it is inherited in an autosomal dominant manner with high penetrance. MEN1 gene is on chromosome 11q13 and encodes menin protein [22]. Parathyroid tumors, resulting in primary hyperparathyroidism are the most common feature of MEN1 and occur in approximately 95% of patients [23]. PNETs occur in 20-75% and anterior pituitary tumors occur in 30% of patients [24]. Multifocality is one of the main features of PNETs in this syndrome [25]. Most of PNETs are non-functioning tumors [26], and insulinomas are the second group in frequency order. More rarely, glucagonomas and vipomas have been described among MEN1 related PNETs [27].
VHL disease is an autosomal dominant condition secondary to mutations in VHL gene, and it is typically associated with pheochromocytomas, renal cell carcinomas, central nervous system hemangioblastomas, endolymphatyc sac tumors and cystic pancreatic lesions [28]. Most of these pancreatic cysts are considered benign in a sense that they do not alter pancreatic function or they do not have an increased risk of malignancy towards PDAC. Presence of PNETs has been described in around 5-17% of patients with VHL disease, and they are almost always single non-functioning tumors [29].
NF type 1 is a disorder inherited in an autosomal dominant manner and it is caused by mutations in NF1 gene [30]. Neurofibromas, café-au-lait spots, Lisch nodules and freckles in the underarms are typical features of this syndrome, and they are included in well defined clinical diagnosis criteria [31]. PNETs have been described in less than 1% of patients with NF type 1, with somatostatin and insulin secreting tumors being the most commonly associated lesions [32, 33]. Data about increased risk of PDAC and NF type 1 are scarce and inconclusive, with isolated cases reported in the literature [34].
TSC is a rare entity inherited in an autosomal dominant manner, characterized mainly by multiple hamartomatous lesions, epilepsy and intellectual disability, and it is produced by mutations in TSC1 and TSC2 genes [35]. Although there is scarcity of data about TSC and increased risk of PNETs, insulinomas and non-functioning tumors have been reported in patients with TSC [36].
Hereditary cancer syndromes related to increased risk of PDAC
The more remarkable hereditary cancer predisposition syndromes with increased risk of PDAC are: hereditary breast and ovarian cancer syndrome (HBOC), familial melanoma (FM), Lynch syndrome (LS), familial adenomatous polyposis (FAP), Peutz-Jeghers syndrome (PJS) and Li-Fraumeni syndrome (LFS) [37]. Also ATM gene has been defined as a potential predisposing factor of PDAC in heterozygous carriers [38]. They account for almost 10-15% of PDAC familial cases, defined by a minimum of two PDAC diagnoses among first degree relatives [39]. The remaining 85-90% of familial cases with pancreatic cancer aggregation lacks these hereditary cancer predisposition cancer syndromes [40] and they are defined as familial pancreatic cancer (FPC). Considering that in the majority of families with PDAC susceptibility a responsible gene mutation will not be identified, several multigene panel and/or whole genome sequencing studies have been designed [41]. Multiple gene panels for PDAC include among others, BRCA1, BRCA2, PALB2, CDKN2A, MLH1, MSH2, MSH6, PMS2, EPCAM, ATM, APC, STK11, PRSS1 and TP53 genes [42]. A PACGENE Consortium study [43] included samples from 727 unrelated probands with PDAC family history (521 met FPC criteria) who were tested for mutations in BRCA1, BRCA2, PALB2 and CDKN2A. They found that the prevalence of pathogenic variants among included probands was: BRCA1, 1.2%; BRCA2, 3.7%; PALB2, 0.6% and CDKN2A 2.5%. As a consequence of this approach, many families that meet classical definition of FPC are now being reclassified into specific hereditary cancer syndromes. Therefore, we could affirm that FPC is currently a diagnosis of exclusion, strictly reserved for those families with 2 or more first-degree relatives with PDAC in the absence of a recognizable syndrome or genetic disorder. Mean age of onset of pancreatic neoplasia in FPC seems to be slightly lower (64-65 years) than sporadic pancreatic cancer cases [44].
Hereditary Breast and Ovarian Cancer syndrome
HBOC syndrome is caused mainly by germline mutations in BRCA1/2 genes. BRCA1 or BRCA2 mutations increase risk of developing breast and ovarian cancer and they are inherited in an autosomal dominant manner. BRCA1 or BRCA2 mutations may be suspected in those families with multiple breast-ovarian cancer cases, especially when they are diagnosed at early age [45]. The estimated prevalence of BRCA1 and BRCA2 mutations is about 1/550 [46], although this number may vary depending on the selected population [47]. Data about prevalence of PDAC among BRCA mutations carriers are heterogeneous, but a study which performed BRCA testing on an unselected collected cohort of 306 patients showed that 4.6% of them had pathogenic BRCA1 or BRCA2 germline variants [48]. Although BRCA1 mutations and risk of PDAC development is debatable, with studies that have shown no risk and others with a relative risk of 2.8 [49, 50], there is consensus about BRCA2 mutations and its relationship with increased risk of PDAC, with a relative risk in a range of 2.3-7 across different published studies [48, 51]. BRCA2 gene mutations constitute the most frequent inherited risk factor for PDAC [52].
PALB2 (Partner and Localizer of BRCA2) gene mutations increase relative risk of breast cancer [53] and also it has been identified as a PDAC susceptibility gene [54]. The prevalence of PALB2 among families with PDAC aggregation is estimated in a range from 3% to 4% [55]. PDAC relative risk associated with PALB2 mutations is not well defined at this time [56]. Germline testing for PALB2 gene should be considered in individuals with striking family history of breast and pancreatic cancers who have non-informative results for mutations in BRCA1/2. A current report yielded a PALB2 mutation frequency of 0.05% among general population [57].
Familial Melanoma
FM is defined by the presence of two cases of invasive melanoma among first degree relatives (rule of two). In geographical areas where melanoma prevalence is higher, three cases among close relatives are necessary to meet the clinical definition (rule of three) [58]. CDKN2A gene germline mutations constitute the main hereditary cause in familial melanoma, although other genes such as CDK4 and BAP1 have been associated to this syndrome [59]. CDKN2A mutations are inherited in an autosomal dominant manner [60]. Estimated prevalence of CDKN2A mutations among general population is 0.01% [61]. An increased risk of pancreatic cancer in FM kindred with a known CDKN2A mutation has widely been documented [62–64], especially in those with a specific 19 base pair p16 pathogenic variant, referred to as p16-Leiden [65]. Retrospective analysis in this founder mutation group estimated a cumulative risk of 17% in suffering from pancreatic cancer [66]. Recently, a new CDKN2A pathogenic variant, p.D84V (c.251A > T) has been described in an Italian study which included patients with multiple primary cutaneous melanomas or with primary cutaneous melanoma associated with family history of melanoma and/or PDAC [67].
Lynch Syndrome
LS (hereditary nonpolyposis colorectal cancer) is caused by germline mutations in mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) and EPCAM gene. One of the hallmarks of tumors in LS is high microsatellite instability. It represents the most common cause of hereditary colorectal cancer. Other cancer risks includes: endometrial cancer, ovarian cancer, gastric cancer, urothelial cancer, skin cancer, brain cancer and PDAC [68]. Population prevalence of LS is estimated at 1:440 [69]. There is a near 9-fold increase in risk of developing pancreatic cancer among families with pathogenic MMR gene variants compared to the general population [70]. Medullary carcinoma of the pancreas is an infrequent type of pancreatic adenocarcinoma which has been suggested to be related to LS [71].
Familial Adenomatous Polyposis
FAP syndrome is an autosomal dominant entity characterized by hundreds to thousands of adenomas throughout the colon and also a variety of common signs, such as polyps of the gastric fundus and duodenum, osteomas, dental anomalies, congenital hypertrophy of the retinal pigment epithelium (CHRPE), thyroid, brain and periampullary carcinomas, and desmoids tumors. Individuals with FAP have a risk in developing colorectal carcinoma by the fourth decade of life that is near 100% [72]. FAP syndrome is caused by mutations in the APC gene [73]. Estimated prevalence of FAP among general population is over 1/10,000 [74]. In patients with FAP, the relative risk for PDAC is estimated to be 4.5 times higher than for the general population [75].
Peutz-Jeghers syndrome
PJ syndrome is a rare and autosomal dominant entity caused by mutations in the STK11 (serine/threonine kinase 11) /LKB1 gene. This disorder is characterized by mucocutaneous pigmentation, typically in oral mucosa and around the lips, and pathognomonic intestinal hamartomatous polyps [76]. PJ syndrome patients have an increased risk for cancers of the colon, stomach, small intestine, pancreas, breast, and other organs [77]. Prevalence of PJ is estimated from 1 in 8300 to 1 in 280 000 individuals [78]. Relative risk for PDAC in PJ syndrome is the highest of all known hereditary cancer predisposition syndromes, being estimated as high as 132 [79]. A study about cancer risk in PJ syndrome published in 2006 [80] yielded that the cumulative lifetime risk of pancreatic cancer for PJ patients was 11%. A Dutch study [81] including 144 PJ syndrome patients showed a cumulative risk for PDAC of 26% at the age of 70 and a relative risk of 76.
Li-Fraumeni syndrome
LF syndrome is an autosomal dominant cancer predisposition condition characterized by the development of a wide spectrum of childhood and adult-onset malignancies and it is caused by germline mutations in TP53 gene [82]. It is estimated that around 50% of the individuals with LFS will develop cancer by the age of 30 years [83]. The core cancers associated with LFS are breast cancer, sarcomas, brain tumors, adrenocortical carcinomas and leukemia. Individuals with this syndrome have also increased risk of suffering from lymphoma, melanoma, lung, PDAC, prostate and ovarian cancers [84]. Estimated prevalence of TP53 gene mutations among general population is 0.005-0.01% [85]. The relative risk for developing PDAC is near 7-fold increased [86].
ATM gene
ATM (ataxia telangiectasia mutated) gene causes ataxia telangiectasia syndrome when biallelic pathogenic variants are inherited [87]. The reported monoallelic carrier frequency of pathogenic ATM variants in the population is 0.5–1% [88]. Recent studies have suggested a plausible relationship between ATM heterozygous status and an increased risk in PDAC development. Some authors have published a 2-3 fold increased risk for PDAC [89, 90], whereas others have not found this effect [91]. A study which included 166 familial pancreatic cancer probands showed that 2.4% (4/166) carried pathogenic ATM variants [92]. Other study has supported this association between ATM heterozygous pathogenic variants carriers and PDAC risk [38].
Hereditary pancreatitis
Repeated pancreatic injury can lead to chronic pancreatitis, increasing risk of malignant transformation. Hereditary pancreatitis (HP) is an extremely rare condition with an estimated prevalence of 3 in 1,000,000 people [93]. HP patients have 50 to 70 times relative risk for PDAC compared with general population [94] and they usually develop PDAC about 20 years earlier [95]. It is estimated that 30% to 40% of HP affected individuals will develop PDAC by the age of 70 [94]. PRSS1 (protease, serine 1) gene, which encodes cationic trypsinogen, is the main gene related to HP. In fact, it is calculated that near 80% of patients with HP have pathogenic variants in PRSS1 [96]. PRSS1 gene mutations are inherited in an autosomal dominant manner, generating a scenario where activated trypsin cannot be degraded and/or activation of trypsinogen into trypsin is stimulated, leading to inflammation and pancreas self-destruction [97]. SPINK1 (serine peptidase inhibitor, Kazal type 1) gene, which encodes a trypsin inhibitor that is secreted by the pancreatic acinar cells, is also related to HP [98]. Since the majority of SPINK1 mutations are inherited in an autosomal recessive pattern, some of them may be inherited in an autosomal dominant manner [99]. Besides, we must consider cystic fibrosis, caused by mutations in CFTR gene, in differential diagnosis of HP. It is estimated that 1.5% of all patients with cystic fibrosis will suffer from pancreatitis, potentially increasing risk of PDAC development [100, 101].
An algorithm with an approximation to differential diagnosis of pancreatic tumors in familial or individuals with suspected inherited/germline component is shown in Fig. 1.
Fig. 1.

Proposed algorithm in differential diagnosis of pancreatic tumors (PDACs and PNETs) in a gene by gene strategy
Translational oncology: germline genetic testing in pancreatic cancer and potential impact on treatment decisions
Metastatic PDAC patients are usually treated with chemotherapy [102]. Current options in patients with good performance status are FOLFIRINOX (a platinum containing regimen) or gemcitabine/nab-paclitaxel in first line setting [103, 104] and treatment decision depends on patients’ comorbidities or expected toxicities profiles. Tumors harboring somatic or germline pathogenic variants in genes related to DNA double strand damage repair, such as BRCA1, BRCA2, PALB2 or ATM, have been associated to better responses to platinum-based chemotherapy schedules [105]. Platinum compounds generate double DNA strand breaks that cannot be repaired when homologous recombination related genes are affected [106]. This benefit in platinum based schedules has also been reported in patients with PDACs related to HBOC syndrome and constitutes the basis for tailored therapy clinical trials [107–109].
Poly-ADP ribose polymerases (PARP) are involved in single DNA strand break repair. Those tumors harboring homologous recombination genes mutations can draw upon this salvage pathway in order to repair DNA damage [110]. Therefore, inhibition of PARP mediated pathway could lead to tumor cell destruction (synthetic lethality concept) in the presence of a pathogenic BRCA variant. Olaparib was the first PARP inhibitor approved as maintenance therapy in advanced high grade serous ovarian carcinomas that have platinum sensitive recurrences [111]. Olaparib and its copartners (veliparib, rucaparib) alone or in combination with platinum based chemotherapy have shown high activity in BRCA/PALB2 mutated pancreatic cancers and they are object of study in current open randomized clinical trials [108, 112–114].
It is hypothesized that tumors with high genomic instability may benefit more from immunotherapy checkpoint inhibitors, especially from the program death 1/program death-ligand 1 (PD1/PD-L1) axis agents (nivolumab, pembrolizumab) [115]. Tumors harboring high genomic instability are related to a higher mutational load which potentially can increase the number of neoepitopes that are exposed and virtually generate a specific anticancer immune response [116]. Genomic instability is one of the main characteristic of Lynch syndrome related tumors and it has also been correlated with tumors with somatic alterations in BRCA and PALB2 genes [117]. It has to be emphasized that a phase II study of pembrolizumab in patients with colorectal cancer showed no tumor responses in the group with mismatch repair proficient tumors, and an impressive 50% of objective responses in patients with mismatch repair deficient tumors [118]. Also, PD-1 inhibition treatment approximation has shown promising results in a phase II study including patients with different gastrointestinal cancers: those with mismatch repair deficient non colorectal tumors had an immune-related objective response rate of 71%. This fact should be taken into consideration when designing clinical trials of immunotherapy in pancreatic cancer and also in treatment decision in patients with metastatic PDACs related to hereditary cancer syndromes [119]. Figure 2 summarizes these features of PDAC in the context of hereditary cancer syndromes and their potential implication in targeted therapies development.
Fig. 2.
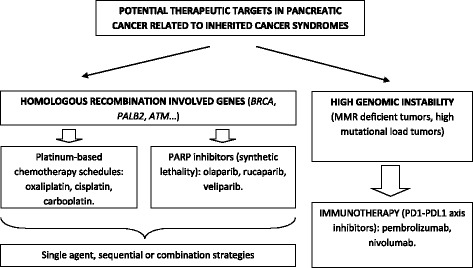
Approximation to customized PDAC treatment in the context of hereditary cancer syndromes
The majority of neuroendocrine tumors have somatic mutations in MEN1, ATRX, DAXX and/or in genes involved in phosphoinositide 3-kinase, AKT, and mammalian target of rapamycin (PI3K/AKT/mTOR) pathway [120, 121]. The presence of specific somatic or germline mutations in PNETs and their correlation with better responses to different multitargeted inhibitors is object of research. This way is being explored in a prospective phase II trial [122], which is recruiting patients diagnosed of low-intermediate grade neuroendocrine tumors; patients with germline/somatic MEN1 mutations are assigned to sunitinib (multityrosine kinase inhibitor) treatment and those with germline/somatic NF1/VHL/TSC mutations are treated with everolimus (mTOR pathway inhibitor).
Conclusions
PDAC is the more frequent histological subtype of pancreatic cancer. Even though the majority of pancreatic cancer cases are considered as sporadic, it is estimated that about 10% of them have a familial component. FPC is defined as a family with 2 or more first-degree relatives with pancreatic cancer. The majority of families with multiple cases of pancreatic cancer do not have an identifiable causative gene or syndrome and strictly they meet FPC definition. A small subset of hereditary pancreatic cancer (20%) is attributable to known inherited cancer predisposition syndromes. PNETs are the second type of pancreatic cancer in terms of incidence and about 20% of them have an involved inherited condition. Recognition of germline mutations in patients with pancreatic tumors does not only suppose an impact in genetic counseling process, since it also may affect treatment decisions and predict response to specific therapies.
Acknowledgements
Not applicable.
Funding
Not applicable. No funding.
Availability of data and materials
Not applicable.
Authors’ contributions
All the authors have reviewed the literature and contribute to the review. CS has written the review. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sergio Carrera, Phone: +34946006333, Email: sergio.carrerarevilla@osakidetza.net.
Aintzane Sancho, Email: aintzane.sanchogutierrez@osakidetza.net.
Eider Azkona, Email: eider.azkonauribelarrea@osakidetza.net.
Josune Azkuna, Email: josune.azcunasagarduy@osakidetza.net.
Guillermo Lopez-Vivanco, Email: guillermo.lopezvivanco@osakidetza.net.
References
- 1.Gold EB, Goldin SB. Epidemiology of and risk factors for pancreatic cancer. Surg Oncol Clin N Am. 1998;7:67–91. [PubMed] [Google Scholar]
- 2.Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C et al. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann Oncol 2017 mdx033. doi:10.1093/annonc/mdx033. [DOI] [PubMed]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Pancreatic Cancer: statistics. Available from: http://www.cancer.net/cancer-types/pancreatic-cancer/statistics. Accessed 20 Feb 2017.
- 5.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19(10):1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frilling A, Akerström G, Falconi M, Pavel M, Ramos J, Kidd M, Modlin IM. Neuroendocrine tumor disease: an evolving landscape. Endocr Relat Cancer. 2012;19(5):R163–85. doi: 10.1530/ERC-12-0024. [DOI] [PubMed] [Google Scholar]
- 7.Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1766–73. doi: 10.1158/1055-9965.EPI-05-0120. [DOI] [PubMed] [Google Scholar]
- 8.National Cancer Institute. SEER Stat fact sheets: pancreas cancer. Available from: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed 20 Feb 2017.
- 9.Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393:535–545. doi: 10.1007/s00423-007-0266-2. [DOI] [PubMed] [Google Scholar]
- 10.Lucenteforte E, La Vecchia C, Silverman D, Petersen GM, Bracci PM, Ji BT, et al. Alcohol consumption and pancreatic cancer; a pooled analysis in the International Pancreatic Cancer case-control Consortium (PanC4) Ann Oncol. 2012;23:374–382. doi: 10.1093/annonc/mdr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen DK, Andren-Sandberg Å, Duell EJ, Goggins M, Korc M, Petersen GM, et al. Pancreatitis-diabetes-pancreatic cancer: summary of an NIDDK-NCI workshop. Pancreas. 2013;42:1227–1237. doi: 10.1097/MPA.0b013e3182a9ad9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosetti C, Rosato V, Li D. Diabetes, antidiabetic medications, and pancreatic cancer risk: an analysis from the International Pancreatic Cancer Case-Control Consortium. Ann Oncol. 2014;25(10):2065–72. doi: 10.1093/annonc/mdu276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemminki K, Li X. Familial and second primary pancreatic cancers: a nationwide epidemiologic study from Sweden. Int J Cancer. 2003;103(4):525–30. doi: 10.1002/ijc.10863. [DOI] [PubMed] [Google Scholar]
- 14.Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus G, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64(7):2634–38. doi: 10.1158/0008-5472.CAN-03-3823. [DOI] [PubMed] [Google Scholar]
- 15.Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer. 2009;8(2):109–117. doi: 10.1007/s10689-008-9214-8. [DOI] [PubMed] [Google Scholar]
- 16.Klein AP, Beaty TH, Bailey-Wilson JE, Brune KA, Hruban RH, Petersen GM. Evidence for a major gene influencing risk of pancreatic cancer. Genet Epidemiol. 2002;23(2):133–149. doi: 10.1002/gepi.1102. [DOI] [PubMed] [Google Scholar]
- 17.Petersen GM. Familial Pancreatic Adenocarcinoma. Hematol Oncol Clin North Am. 2015;29(4):641–653. doi: 10.1016/j.hoc.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halfdanarson TR, Bamlet WR, McWilliams RR, Hobday TJ, Burch PA, Rabe KG, et al. Risk factors for pancreatic neuroendocrine tumors: a clinic-based case-control study. Pancreas. 2014;43(8):1219–22. doi: 10.1097/MPA.0000000000000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crona J, Skogseid B. Genetics of neuroendocrine tumors. European Journal of Endocrinology. 2016;174(6):275–290. doi: 10.1530/EJE-15-0972. [DOI] [PubMed] [Google Scholar]
- 20.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of Tumours of the Digestive System. 4. Lyon: International Agency for Research on Cancer (IARC); 2010. [Google Scholar]
- 21.Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. 1954;16(3):363–371. doi: 10.1016/0002-9343(54)90353-8. [DOI] [PubMed] [Google Scholar]
- 22.Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 23.Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Am J Med. 1987;82(4):731–737. doi: 10.1016/0002-9343(87)90008-8. [DOI] [PubMed] [Google Scholar]
- 24.Calender A, Giraud S, Lenoir GM, Cougard P, Chanson P, Proye C. Hereditary multiple endocrine neoplasia. New genetic data and clinical applications in type 1 multiple endocrine neoplasia. Presse Med. 1995;24:542–546. [PubMed] [Google Scholar]
- 25.Triponez F, Dosseh D, Goudet P, Cougard P, Bauters C, Murat A, et al. Epidemiology data on 108 MEN1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg. 2006;243:265–272. doi: 10.1097/01.sla.0000197715.96762.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jensen RT, Berna MK, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113(suppl 7):1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30(5):560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 28.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. Von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 29.Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087–95. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- 30.Reynolds RM, Browning GG, Nawroz I, Campbell IW. Von Recklinghausen’s neurofibromatosis: neurofibromatosis type 1. Lancet. 2003;361:1552–54. doi: 10.1016/S0140-6736(03)13166-2. [DOI] [PubMed] [Google Scholar]
- 31.DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health Criteria for diagnosis of Neurofibromatosis 1 in children. Pediatrics. 2000;105:608–14. doi: 10.1542/peds.105.3.608. [DOI] [PubMed] [Google Scholar]
- 32.Evans DGR, Komminoth P, Scheithauer BW, Peltonen J. Neurofibromatosis type 1. Pathology and genetics: tumors of endocrine organs. WHO classification of tumor. Lyon: IARC. 2004;243–48.
- 33.Hamy A, Heymann MF, Bodic J, Visset J, Le Borgne J, Lenéel JC, et al. Duodenal somatostatinomas. Anatomic clinical study of 12 operated cases. Ann Chir. 2001;126(3):221–6. doi: 10.1016/S0003-3944(01)00493-X. [DOI] [PubMed] [Google Scholar]
- 34.Niv Y, Abu-Avid S, Oren M. Adenocarcinoma of pancreas and duodenum associated with cutaneous neurofibromatosis. Am J Med. 1987;82(2):384–85. doi: 10.1016/0002-9343(87)90096-9. [DOI] [PubMed] [Google Scholar]
- 35.Dworakowska D, Grossman AB. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer. 2009;16(1):45–58. doi: 10.1677/ERC-08-0142. [DOI] [PubMed] [Google Scholar]
- 36.Arva NC, Pappas JG, Bhatla T, Raetz EA, Macari M, Ginsburg HB, Hajdu CH. Well-differentiated pancreatic neuroendocrine carcinoma in tuberous sclerosis--case report and review of the literature. Am J Surg Pathol. 2012;36(1):149–53. doi: 10.1097/PAS.0b013e31823d0560. [DOI] [PubMed] [Google Scholar]
- 37.Whitcomb DC, Shelton CA, Brand RE. Genetics and Genetic testing in pancreatic cancer. Gastroenterology. 2015;149:1252–1264. doi: 10.1053/j.gastro.2015.07.057. [DOI] [PubMed] [Google Scholar]
- 38.Roberts NJ, Klein AP. Genome wide sequencing to identify the cause of hereditary cancer syndromes: with examples from familial pancreatic cancer. Cancer Lett. 2013;340(2):227–233. doi: 10.1016/j.canlet.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hruban RH, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, Falatko F, et al. Familial pancreatic cancer. Ann Oncol. 1999;10(Suppl 4):69–73. doi: 10.1093/annonc/10.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- 40.Matsubayashi H, Takaori K, Morizane C, Maguchi H, Mizuma M, Takahashi H, et al. Familial pancreatic cancer: Concept, management and issues. World J Gastroenterol. 2017;23(6):935–948. doi: 10.3748/wjg.v23.i6.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6(2):166–175. doi: 10.1158/2159-8290.CD-15-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;148(3):556–564. doi: 10.1053/j.gastro.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2 and CDKN2A mutations in familial pancreatic cancer (FPC): a PACGENE study. Genet Med. 2015;17(7):569–577. doi: 10.1038/gim.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James TA, Sheldon DG, Rajput A, Kuvshinoff BW, Javle MM, Nava HR, et al. Risk factors associated with earlier age of onset in familial pancreatic carcinoma. Cancer. 2004;101:2722–26. doi: 10.1002/cncr.20700. [DOI] [PubMed] [Google Scholar]
- 45.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The breast cancer linkage consortium. Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paluch-Shimon S, Cardoso F, Sessa C, Balmana J, Cardoso MJ, Gilbert F, ESMO Guidelines Committee et al. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann Oncol. 2016;27(suppl 5):v103–v110. doi: 10.1093/annonc/mdw327. [DOI] [PubMed] [Google Scholar]
- 47.Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14:185–187. doi: 10.1038/ng1096-185. [DOI] [PubMed] [Google Scholar]
- 48.Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33(28):3124–3129. doi: 10.1200/JCO.2014.59.7401. [DOI] [PubMed] [Google Scholar]
- 49.Axilbund JE, Argani P, Kamiyama M, Palmisano E, Raben M, Borges M, et al. Absence of germline BRCA1 mutations in familial pancreatic cancer patients. Cancer Biol Ther. 2009;8:131–135. doi: 10.4161/cbt.8.2.7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365–72. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- 51.Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. Hereditary breast cancer study Group. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107(12):2005–2009. doi: 10.1038/bjc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(2):342–346. doi: 10.1158/1055-9965.EPI-06-0783. [DOI] [PubMed] [Google Scholar]
- 53.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancers families. Clin Genet. 2010;78:490–494. doi: 10.1111/j.1399-0004.2010.01425.x. [DOI] [PubMed] [Google Scholar]
- 56.Janatová M, Borecká M, Soukupová J, Kleiblová P, Stříbrná J, Vočka M, et al. PALB2 as Another Candidate Gene for Genetic Testing in Patients with Hereditary Breast Cancer in Czech Republic. Klin Onkol. 2016;29(Suppl 1):S31–34. doi: 10.14735/amko2016S31. [DOI] [PubMed] [Google Scholar]
- 57.Kotsopoulos J, Sopik V, Rosen B, Fan I, McLaughlin JR, Risch H, et al. Frequency of germline PALB2 mutations among women with epithelial ovarian cancer. Fam Cancer. 2017;16:29–34. doi: 10.1007/s10689-016-9919-z. [DOI] [PubMed] [Google Scholar]
- 58.Leachman SA, Lucero OM, Sampson JE, Cassidy P, Bruno W, Queirolo P, et al. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017 Mar 10 doi:10.1007/s10555-017-9661-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 59.Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74(3):395–407. doi: 10.1016/j.jaad.2015.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counseling. J Am Acad Dermatol. 2016;74(3):411–420. doi: 10.1016/j.jaad.2015.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94(12):894–903. doi: 10.1093/jnci/94.12.894. [DOI] [PubMed] [Google Scholar]
- 62.Lynch HT, Shaw TG. Familial atypical multiple mole melanoma (FAMMM) syndrome: history, genetics, and heterogeneity. Fam Cancer. 2016;15(3):487–491. doi: 10.1007/s10689-016-9888-2. [DOI] [PubMed] [Google Scholar]
- 63.Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66(20):9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- 64.Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333(15):970–974. doi: 10.1056/NEJM199510123331504. [DOI] [PubMed] [Google Scholar]
- 65.De Snoo FA, Bishop DT, Bergman W, van Leeuwen I, van der Drift C, van Nieuwpoort FA, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res. 2008;14(21):7151–7. doi: 10.1158/1078-0432.CCR-08-0403. [DOI] [PubMed] [Google Scholar]
- 66.Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87(6):809–811. doi: 10.1002/1097-0215(20000915)87:6<809::AID-IJC8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 67.Borroni RG, Manganoni AM, Grassi S, Grasso M, Diegoli M, Giorgianni C, et al. Genetic counselling and high-penetrance susceptibility gene analysis reveal the novel CDKN2A p.D84V (c.251A > T) mutation in melanoma-prone families from Italy. Melanoma Res. 2017;27(2):97–103. doi: 10.1097/CMR.0000000000000324. [DOI] [PubMed] [Google Scholar]
- 68.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, et al. Colon cancer family registry. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–1487. doi: 10.1001/jama.296.12.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. The risk of pancreatic cancer in families with lynch syndrome. JAMA. 2009;302(16):1790–1795. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banville N, Geraghty R, Fox E, Leahy DT, Green A, Keegan D, et al. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol. 2006;37(11):1498–1502. doi: 10.1016/j.humpath.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 72.Waller A, Findeis S, Lee MJ. Familial Adenomatous Polyposis. J Pediatr Genet. 2016;5(2):78–83. doi: 10.1055/s-0036-1579760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3(2):121–125. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- 74.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moussata D, Senouci L, Berger F. Familial adenomatous polyposis and pancreatic cancer. Pancreas. 2015;44(3):512–513. doi: 10.1097/MPA.0000000000000295. [DOI] [PubMed] [Google Scholar]
- 76.Jelsig AM, Qvist N, Brusgaard K, Nielsen CB, Hansen TP, Ousager LB. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis. 2014;9:101. doi: 10.1186/1750-1172-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Resta N, Pierannunzio D, Lenato GM, Stella A, Capocaccia R, Bagnulo R, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45(7):606–611. doi: 10.1016/j.dld.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 78.Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15(43):5397–5408. doi: 10.3748/wjg.15.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hearle NC, Rudd MF, Lim W, Murday V, Lim AG, Phillips RK, et al. Exonic STK11 deletions are not a rare cause of Peutz-Jeghers syndrome. J Med Genet. 2006;43(4):e15. doi: 10.1136/jmg.2005.036830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 81.Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJ, Krak N, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. Med Genet. 2013;50(1):59–64. doi: 10.1136/jmedgenet-2012-101277. [DOI] [PubMed] [Google Scholar]
- 82.Valdez JM, Nichols KE, Kesserwan C. Li-Fraumeni syndrome: a paradigm for the understanding of hereditary cancer predisposition. Br J Haematol. 2017;176(4):539–552. doi: 10.1111/bjh.14461. [DOI] [PubMed] [Google Scholar]
- 83.Correa H. Li-Fraumeni Syndrome. J Pediatr Genet. 2016;5(2):84–88. doi: 10.1055/s-0036-1579759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. 2014;11(5):260–271. doi: 10.1038/nrclinonc.2014.41. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 86.Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FB, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47(6):421–428. doi: 10.1136/jmg.2009.073429. [DOI] [PubMed] [Google Scholar]
- 87.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signaling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 88.Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009–1015. doi: 10.1136/jcp.2005.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Geoffroy-Perez B, Janin N, Ossian K, Laugé A, Croquette MF, Griscelli C, et al. Cancer risk in heterozygotes for ataxia-telangiectasia. Int J Cancer. 2001;93:288–293. doi: 10.1002/ijc.1329. [DOI] [PubMed] [Google Scholar]
- 90.Hu C, Hart SN, Bamlet WR, Moore RM, Nandakumar K, Eckloff BW, et al. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol Biomarkers Prev. 2016;25(1):207–211. doi: 10.1158/1055-9965.EPI-15-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laitman Y, Boker-Keinan L, Berkenstadt M, Liphsitz I, Weissglas-Volkov D, Ries-Levavi L, et al. The risk for developing cancer in Israeli ATM, BLM, and FANCC heterozygous mutation carriers. Cancer Genet. 2016;209(3):70–74. doi: 10.1016/j.cancergen.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 92.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rebours V, Boutron-Ruault MC, Schnee M, Férec C, Le Maréchal C, Hentic O, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009;58(1):97–103. doi: 10.1136/gut.2008.149179. [DOI] [PubMed] [Google Scholar]
- 94.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, et al. European Registry of Hereditary Pancreatitis and Pancreatic cancer (EUROPAC). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252–261. doi: 10.1016/S1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 95.Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK, Jr, Perrault J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(6):442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 96.Howes N, Greenhalf W, Socken DD, Neoptolemos JP. Cationic trypsinogen mutations and pancreatitis. Gastroenterol Clin North Am. 2004;33(4):767–787. doi: 10.1016/j.gtc.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 97.Raphael KL, Willingham FF. Hereditary pancreatitis: current perspectives. Clin Exp Gastroenterol. 2016;26(9):197–207. doi: 10.2147/CEG.S84358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kume K, Masamune A, Ariga H, Hayashi S, Takikawa T, Miura S, et al. genetic variants in the SPINK1 gene affect the level of serum PSTI? J Gastroenterol. 2012;47(11):1267–1274. doi: 10.1007/s00535-012-0590-3. [DOI] [PubMed] [Google Scholar]
- 99.LaRusch J, Barmada MM, Solomon S, Withcomb DC. Whole exome sequencing identifies multiple, complex etiologies in an idiopathic hereditary pancreatitis kindred. JOP. 2012;13(3):258–262. [PMC free article] [PubMed] [Google Scholar]
- 100.De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics. 2005;115(4):e463–e469. doi: 10.1542/peds.2004-1764. [DOI] [PubMed] [Google Scholar]
- 101.Cohn JA, Mitchell RM, Jowell PS. The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis. Clin Lab Med. 2005;25(1):79–100. doi: 10.1016/j.cll.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 102.Vera R, Dotor E, Feliu J, González E, Laquente B, Macarulla T, et al. SEOM Clinical Guideline for the treatment of pancreatic cancer (2016) Clin Transl Oncol. 2016;18(12):1172–1178. doi: 10.1007/s12094-016-1586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 104.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II) Cancer Res. 1998;58(6):1120–1123. [PubMed] [Google Scholar]
- 106.Tassone P, Di Martino MT, Ventura M, Pietragalla A, Cucinotto I, Calimeri T, et al. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol Ther. 2009;8:648–653. doi: 10.4161/cbt.8.7.7968. [DOI] [PubMed] [Google Scholar]
- 107.O’Reilly EM, Lowery MA, Segal MF, Smith SC, Moore MJ, Kindler HL, et al. Phase IB trial of cisplatin (C), gemcitabine (G), and veliparib (V) in patients with known or potential BRCA2 or PALB2-mutated pancreas adenocarcinoma (PC) J Clin Oncol. 2014;32(suppl; abstr 4023):5 s. [Google Scholar]
- 108.A Randomized Phase II Study of Gemcitabine, Cisplatin +/- Veliparib in Patients with Pancreas Adenocarcinoma and a Known BRCA/ PALB2 Mutation (Part I) and a Phase II Single Arm Study of Single-Agent Veliparib in Previously Treated Pancreas Adenocarcinoma (Part II) (NCI #8993). ClinicalTrials.gov Identifier: NCT01585805
- 109.O’Reilly EM: BRCA-mutated pancreas adenocarcinoma: Emerging therapeutic implications. AACR Special Conference on Pancreatic Cancer. Abstract IA28. Presented May 21, 2014.
- 110.Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer. 2011;105:1114–22. doi: 10.1038/bjc.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–61. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 112.Domchek SM, Hendifar AE, McWilliams RR. RUCAPANC: An open-label, phase 2 trial of the PARP inhibitor rucaparib in patients (pts) with pancreatic cancer (PC) and a known deleterious germline or somatic BRCA mutation. J Clin Oncol 34, 2016 (suppl; abstr 4110)
- 113.Olaparib in gBRCA Mutated Pancreatic Cancer Whose Disease Has Not Progressed on First Line Platinum-Based Chemotherapy (POLO). ClinicalTrials.gov Identifier: NCT02184195
- 114.A Study of Rucaparib in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. ClinicalTrials.gov Identifier: NCT02042378
- 115.Yaghmour G, Pandey M, Ireland C, Patel K, Nunnery S, Powell D, et al. Role of genomic instability in immunotherapy with checkpoint inhibitors. Anticancer Res. 2016;36(8):4033–4038. [PubMed] [Google Scholar]
- 116.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;16:e542–51. doi: 10.1016/S1470-2045(16)30406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Le DT, Uram JN, Wang H, Kemberling H, Eyring AD, Bartlett BR, et al. PD-1 blockade in mismatch repair deficient non-colorectal gastrointestinal cancers. J Clin Oncol. 2016;34 (suppl 4S; abstr 195).
- 120.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neychev V, Steinberg SM, Cottle-Delisle C, Merkel R, Nilubol N, Yao J, et al. Mutation targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: protocol for a phase II clinical trial. BMJ Open. 2015;5(5):e008248. doi: 10.1136/bmjopen-2015-008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.