

Abstract
Recent advances in cancer genomics have revolutionized the characterization and classification of medulloblastomas. According to the current WHO guidelines, medulloblastomas are now classified into the following molecularly defined groups: Wnt signaling pathway (WNT)-activated, sonic hedgehog signaling pathway (SHH)-activated and tumor suppressor protein p53 (TP53)-mutant, SHH-activated and TP53-wildtype, and non-WNT/non-SHH (i.e. group 3 and group 4). Importantly, genomic, epigenomic, and proteomic advances have created a potential paradigm shift in therapeutic options. The challenge now is to (i) translate these observations into new therapeutic approaches and (ii) employ these observations in clinical practice, utilizing the classification following a molecular analysis for diagnosis and application of new subgroup-specific targeted therapeutics.
Keywords: Medulloblastoma, specific targeted therapeutics, WNT, SHH, TP53
Introduction
Medulloblastomas account for 12% of childhood brain tumors 1. Approximately 80% of medulloblastomas occur in children under the age of 15. In adults, medulloblastomas are rare (1–2%). To date, the 5-year survival rate for children with average and high-risk disease as defined by clinical criteria is 80% and 60–65%, respectively 2. Recent advances in cancer genomics have led to a fundamental change in medulloblastoma classification. Based on genome-wide transcription profiling, it has been shown that medulloblastomas comprise at least four molecular subgroups ( Table 1), each with unique transcription profiles, mechanism of tumorigenesis, and clinical outcome 3– 6. Each of these subgroups, Wnt signaling pathway (WNT) (10% of medulloblastomas), sonic hedgehog signaling pathway (SHH) (30%), group 3 (15%), and group 4 (45%), will be discussed in the following sections.
Table 1. Features of medulloblastoma subgroups.
| Subtype | Molecular characteristics | Mutations | Age group |
|---|---|---|---|
| WNT activated | WNT pathway activation | CTNNB1 DDX3X Chromatin-remodeling genes TP53 |
Least common of subgroups Found in children and adults, not infants |
|
SHH activated and TP53 wild-
type |
SHH pathway activation | PTCH1 SMO SUFU TERT promoter Chromatin-remodeling genes |
Infants, children, and adults |
|
SHH activated and TP53
mutant |
SHH pathway activation | TP53 | 5–18 years old |
| Group 3 | Elevated expression of MYC GABRA5 over-expression |
SMARCA4 Chromatin-remodeling genes Genes of TGF-β pathway |
Infants and children, not adults
More common in boys than in girls |
| Group 4 | Lmx1A expression | Chromatin-remodeling genes |
More common in children than in
adults Least common in infants |
CTNNB1, catenin beta 1; DDX3X, DEAD-box helicase 3; Lmx1a, LIM homeobox transcription factor; PTCH1, Patched-1; SHH, sonic hedgehog; SMARCA4, SWI (switching)/SNF (sucrose non-fermenting)-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4; SMO, smoothened receptor; SUFU, suppressor of fused homolog protein; TERT, telomerase reverse transcriptase; TGF-β, transforming growth factor beta; TP53, tumor suppressor protein p53.
Clinical behavior
Medulloblastomas typically occur in the cerebellum and are primarily a pediatric brain cancer. Historically, medulloblastomas were described as “small round blue cell tumors” 7. The tumors are highly cellular with minimal cellular differentiation and have been defined by histological features that do not accurately predict clinical outcome (classic, desmoplastic/nodular, large-cell/anaplastic, medulloblastoma with neuroblastic features, medulloblastoma with glial differentiation, medullomyoblastoma, and melanotic medulloblastoma). Approximately one-third of the tumors have metastatic spread through CSF pathways, which puts the tumor into a “high-risk” group and is associated with poor outcome. In terms of MRI imaging characteristics, an adult medulloblastoma patient exhibits disseminated leptomeningeal disease in the brain and spinal cord ( Figure 1A), while pediatric medulloblastoma patients of the WNT subgroup tend to be in the cerebellar peduncle, patients of the SHH subgroup tend to be in the cerebellar hemisphere, but can be centrally located, as shown in Figure 1B, and midline cerebellar tumors tend to belong to groups 3 and 4 or SHH 8.
Figure 1. Imaging of pediatric and adult medulloblastomas.

( A) Magnetic resonance imaging of an adult woman who has medulloblastoma in the brain and spine with leptomeningeal spread: ( a) axial T1 of the brain post-gadolinium contrast; ( b) coronal T1 of the brain post-gadolinium contrast; ( c) sagittal T1 of the cervical spine post-gadolinium contrast. ( B) Brain magnetic resonance imaging of pediatric medulloblastomas: ( a) sagittal post-gadolinium WNT tumor; ( b) axial T2 of a SHH tumor. Red arrows delineate the tumor/leptomeningeal disease.
Treatment: current standard of care
Medulloblastomas are clinically categorized as average-risk and high-risk disease 9 ( Table 2). Maximal surgical resection, in tumors that are amenable to surgery, is the first step in all cases. However, cerebellar mutism (severely diminished or absent speech output) can be an acute post-surgical complication in up to one-quarter of patients, which usually partially recovers, although survivors typically are left with dysarthria and neurocognitive issues 10. For average-risk disease, patients receive craniospinal radiotherapy (23.4 Gy in 30 fractions, followed by conformal tumor bed boost to 54–56 Gy over 6 weeks) with or without vincristine. After the radiation, children older than 3 years with non-disseminated medulloblastoma receive eight cycles of vincristine (mitotic inhibitor), cisplatin (DNA cross-linker), and two alkylating agents – cyclophosphamide and CCNU (lomustine) chemotherapy – for approximately 1 year. For poor-risk disease, craniospinal radiotherapy is given at a higher dose (36–39.6 Gy in 30 fractions, followed by posterior fossa boost to 54–56 Gy over 6 weeks) and chemotherapy (agents used include cisplatin, cyclophosphamide, and vincristine) 9, 11. Sometimes, stem cell transplants are also offered prior to the initiation of therapy. The craniospinal radiation and chemotherapy regimens described are also for the most part used in adult patients, but the use of post-adjuvant chemotherapy has not been shown to improve survival. In addition, vincristine and cisplatin cause significant toxicity. Infants younger than 3 years are often treated with high-dose chemotherapy and stem cell rescue regimens to delay the time to or avoid completely the administration of craniospinal irradiation 9, 11.
Unfortunately, these treatment regimens come with considerable morbidity. For example, the majority of children treated with intensive chemotherapy and irradiation (especially infants and those exposed to higher doses) are at risk for significant hearing loss, endocrine and neurocognitive deficits, and secondary benign and malignant tumors 11.
Table 2. Staging and risk stratification of medulloblastomas.
| Modified Chang Staging | |||
|---|---|---|---|
| T stage | M stage | ||
| T1 | Tumor <3 cm in diameter | M0 | No evidence of gross subarachnoid or
hematogenous metastasis |
| T2 | Tumor ≥3 cm in diameter | M1 | Microscopic tumor cells found in CSF |
| T3a | Tumor >3 cm and with extension
into aqueduct of Sylvius or foramen of Luschka |
M2 | Gross nodular seeding intracranially
beyond the primary site (in cerebellar/cerebral subarachnoid space or in third or lateral ventricle) |
| T3b | Tumor >3 cm and with
unequivocal extension into brainstem |
M3 | Gross nodular seeding in spinal
subarachnoid space |
| T4 | Tumor >3 cm with extension past
aqueduct of Sylvius or down past foramen magnum |
M4 | Metastasis outside cerebrospinal axis |
| Risk Stratification | |||
| Standard (Average) Risk (66%) | High Risk (34%) | ||
| >3 years old | <3 years old | ||
| <1.5 cm 2
residual disease after resection |
Subtotal resection, >1.5 cm 2 residual tumor | ||
| M0 by craniospinal MRI and CSF | M+, leptomeningeal seeding,
and location outside of the posterior fossa |
||
CSF, cerebrospinal fluid; MRI, magnetic resonance imaging.
Potentially new treatments and approaches
With the advent of genomics, it has become increasingly clear that medulloblastoma is not a discrete entity, as shown by the recent WHO classification 8. Currently, there is a planned clinical study looking at the feasibility of surgery and chemotherapy in children with Wnt-positive medulloblastoma (NCT02212574). A Pediatric Brain Tumor Consortium study evaluated the use of GDC-0449 (vismodegib, Genentech Corporation, USA), which blocks a key protein (Smoothened, or SMO) in the SHH signaling pathway in medulloblastoma, and, as anticipated, patients with the SHH subtype who had the SMO/PTCH mutation responded to this drug 12. However, even in this group who responded to the vismodegib, it was transient with resistance developing quickly. Vismodegib has had some success in the recurrent SHH subgroup setting 12, 13. A proposed consensus for the design of next-generation clinical trials was discussed by Ramaswamy et al. 14 and is summarized in Table 3.
Table 3. Adapted table of a proposed consensus for designing the next generation of clinical trials in medulloblastoma (Ramaswamy et al. 14).
|
Medulloblastoma patient subgroups → genome-wide
methylation array → molecularly informed clinical trial or other validated methods |
|
Tissue collection from all patients → snap-frozen
and
paraffin-embedded tumor tissue, blood, and CSF |
|
All patients require tumor board planning for a clinical
trial
registry: neuroimaging, neuropathology, and radiotherapy |
|
Treatment-related side effects in all patients in the short
and
long term: quality-of-life measures and neuropsychological outcomes |
|
Recurrent disease: tumors should be re-biopsied if the
diagnosis was unclear, or 2 years after the initial diagnosis, or before using targeted therapy for 2 years |
|
Extent of resection: neurosurgeons should aim for
maximal
safe removal |
CSF, cerebrospinal fluid.
WNT subgroup of medulloblastoma
WNT medulloblastomas have evidence of WNT pathway ( Figure 2A) activation in their transcription profiles and almost uniformly have oncogenic mutations of CTNNB1, the proto-oncogene that encodes β-catenin. This subgroup comprises 10–15% of all medulloblastomas, found mostly in females aged 6–10. Their expression profiles map to multipotential progenitor cells of the lower rhombic lip 15. According to Phoenix et al. 16, there is a “signaling paradox” identified in which mutant catenin beta 1 (CTNNB1) protein drives constitutive, oncogenic WNT signaling in medulloblastoma 17, 18; this in turn silences normal WNT signaling in surrounding endothelial cells by producing inhibitors such as those of the secreted Frizzled-related protein (sFRP) family and WNT inhibitor factor 1 (WIF-1) that are secreted in situ ( Figure 2A). CTNNB1 mutations are found in approximately 90% of WNT medulloblastomas, and nuclear accumulation of β-catenin is a biomarker for WNT pathway activation. WNT medulloblastomas form a highly hemorrhagic vasculature that lacks a blood–brain barrier 16. This may explain why these tumors are highly susceptible to chemotherapy, especially those that do not typically cross the blood–brain barrier, e.g. vincristine 16. Monosomy 6 is also found in 80–85% of WNT medulloblastomas (incidentally, these do not harbor telomerase reverse transcriptase [TERT] mutations), DEAD-box helicase 3 (DDX3X) mutations are found in 50% of WNT tumors, TERT mutations are found in 31% of WNT tumors 19, and the most common chromatin remodeling mutation found in WNT tumors is SMARCA4. Note that tumor suppressor protein p53 (TP53) mutations present in 15% of WNT tumors have no prognostic impact, unlike SHH TP53-mutant tumors that are associated with poor prognosis 20.
Figure 2. WNT and SHH signaling pathways.
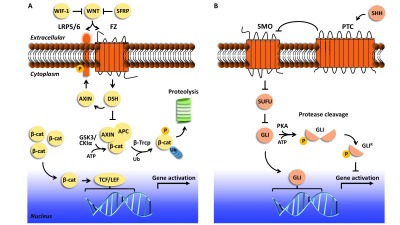
( A) The WNT signaling pathway is mediated by the receptor Frizzled (FZ) and single-pass low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6). In the pathway’s “off” state (in the event of low, no, or WNT ligand function inhibited by WNT inhibitor factor 1 [WIF-1] or secreted frizzle-related protein [SFRP]), β-catenin (β-cat) is targeted for phosphorylation by glycogen synthase kinase 3 (GSK3) and casein kinase I alpha (CKIα), aided by proteins AXIN and adenomatous polyposis coli (APC). β-catenin is then ubiquitinated and targeted for proteolysis by the proteasome. In the pathway’s “on” state, WNT ligand is recognized by FZ and LRP5/6, and LRP5/6 is phosphorylated. The WNT-FZ-LRP5/6 trimeric complex triggers the recognition of Dishevelled (DSH) and AXIN. β-catenin is not phosphorylated, translocates to the nucleus, and functions as a transcriptional coactivator to activate TCF/LEF family transcription factors. Prominent drug targets that aim to regulate WNT-responsive gene expression 22 include those that target (1) extracellular events, such as recognition of WNT by FZ and/or LRP5/6 (vantictumab and ipafricept), (2) cytoplasmic events, such as inhibition of DSH or stabilization of the AXIN/APC interaction (IWR-1; XAV939; 3289-8625; FJ9; NSC 668036; JW74), and (3) transcriptional activation, such as perturbing β-catenin function (PFK115-584; CGP049090; iCRT-3, -5, and -14; PRI-724). There are still other drugs that target events involved in WNT secretion to the extracellular space as well as other enzymes that regulate the pathway, but they are not shown in this schematic. Ub, ubiquitin. ( B) The sonic hedgehog (SHH) pathway is mediated by the receptors Smoothened (SMO) and Patched (PTC). In the pathway’s “off” state (in the event of low or no SHH ligand), SMO transport from intracellular vesicles to the membrane and its activity at the membrane are inhibited, in part by PTC. Members of transcription factor family GLI are inhibited by suppressor of fused (SUFU). Protein kinase A (PKA) phosphorylates the GLI transcription factors, which undergo proteasomal cleavage to yield a functional repressor form (GLI R). GLI R translocates to the nucleus and inhibits target gene expression. In the pathway’s “on” state, SHH binds to and inhibits PTC and SUFU is inhibited. SMO levels at the membrane increase, leading to activation of GLI transcription factors, which translocate to the nucleus to activate SHH-responsive genes. Prominent drug targets that aim to regulate SHH-responsive gene expression 13 include those that target (1) extracellular events, such as SMO function, including by inhibition of SHH (purmorphamine, cyclopamine, vismodegib 12; sonidegib or Odomzo®, jervine; saridegib, CUR 61414, BMS-833923, glasdegib, PF-5274857, TAK-441, Taladegib, and SANT-1) and its binding to PTC (5E1, a monoclonal antibody), and (2) transcription activation, such as regulating GLI transcriptional activation (GANT61 and arsenic trioxide).
Patients with WNT medulloblastomas tend to have the most favorable outcomes; hence, current treatment protocols for WNT subgroup tumors are designed to minimize radiation and standard chemotherapy (refer to NCT01878617) and seek new treatments that target oncogenic mechanisms. Northcott et al. 21 suggested the use of panobinostat (a non-selective histone deacetylase inhibitor) (Novartis, USA) since the disruption of chromatin remodeling is thought to play a pivotal role in WNT medulloblastoma 22, 23. Anastas and Moon 22 also discuss a number of other potential inhibitors to the WNT pathway in their review.
SHH subgroup of medulloblastoma
The SHH medulloblastoma group is a complex heterogeneous group of tumors, and the pathway is delineated in Figure 2B. SHH medulloblastomas have an intact blood–brain barrier and are less responsive to chemotherapy compared to WNT medulloblastomas 16. Infants (0–4 years) are more likely to have SUFU mutations than other age groups, and 42% of the infant samples have PTCH1 alterations, while children (4–17 years) have a higher incidence of MYCN and GLI2 amplifications, and 36% have PTCH1 alterations 24, 25. Adults (anyone over the age of 17) are more likely to have SMO mutations than other age groups, and 54% of the adult samples have PTCH1 alterations 25. Since there is a higher prevalence of PTCH1 and SMO mutations in adult SHH medulloblastomas, this predicts responsiveness to inhibitors of the receptor SMO. Some tumors that arise from SMO mutations are sensitive to SMO inhibitors, but for others the SMO mutation renders the tumor insensitive 24. MYCN and GLI2 amplifications or mutations also have been shown to be insensitive to SMO inhibitors 12, 13. SHH-inhibiting drugs that act downstream of SMO are currently in development 24. TERT promoter mutations are present in 38% of SHH medulloblastomas and, interestingly, are present in 80% of adult SHH tumors 19. Gorlin syndrome (also known as nevoid basal-cell carcinoma syndrome), caused by inherited germline PTCH1 mutations or de novo (60% cases), is an autosomal dominant developmental disorder, and 5% of these individuals develop medulloblastomas during infancy 26. The outcome tends to be favorable as long as the patient does not have a PTEN or GNAS alteration 26. SHH-activated, TP53-mutant is a recent genetically defined WHO classification 8. TP53 mutations occur in 13% of SHH tumors, and many of these are germline mutations (Li-Fraumeni syndrome) 23, 24. The SHH medulloblastomas with TP53 mutations have extremely poor outcomes, and patients with these tumors should be selected for more intensive therapies and parents of those with germline mutations be offered genetic counseling 14. Protocols in development include removing DNA alkylating chemotherapy and minimizing radiation therapy in TP53-mutant tumors and relying instead on antimetabolite, microtubule-disrupting, or other types of chemotherapy 25. The SJMB12 study (NCT01878617) is currently prospectively evaluating treatment of SHH medulloblastoma in molecularly and clinically defined low, average, and high-risk patients and post-chemotherapy maintenance treatment with GDC0449 in children >12 years of age. Also, PI3K, mTOR, arsenic trioxide, and AKT inhibitors are potentially valuable in controlling specific targets in the SHH pathway and its interaction and links with the PI3K, mTOR, and AKT pathways 25.
Non-WNT/non-SHH: group 3 medulloblastoma
Patients with group 3 medulloblastoma have a poor prognosis, and more than 50% of cases are metastatic at the time of diagnosis 4. Interestingly, older children with group 3 medulloblastomas have a 50% survival in 5 years if they have risk-adapted therapy. These tumors are more common in males and infants. This subgroup is notable for MYC over-expression, with MYC amplification observed in 17% of cases 3. Isochromosome 17q is a predictor of poor outcome in group 3 medulloblastomas 27. A large proportion of these group 3 medulloblastomas overexpress GABRA5, which may have therapeutic implications 3, 28– 31. Mutations in a number of genes involving chromatin remodeling affect 28.5% of group 3 tumors 31. Copy number changes that target genes in the transforming growth factor beta (TGF-β) signaling pathway affect approximately 20% of group 3 tumors 31. In addition, PTV1 alterations are present in 12% of tumors, often as a fusion with MYC that drives its expression 31. The Wechsler-Reya group has shown HDAC and PI3K inhibitor combinations are promising in models of group 3 medulloblastoma 30. The SJMB12 study (for all medulloblastoma subgroups, mentioned in the previous section) is also prospectively evaluating the use of pemetrexed, gemcitabine, vincristine, cisplatin, and cyclophosphamide in the high-risk medulloblastoma cases, and this study is currently open.
Non-WNT/non-SHH: group 4 medulloblastoma
Group 4 medulloblastoma is also known as the glutamatergic subgroup, and it is the commonest molecular subgroup. For specific characteristics of this subgroup, please refer to Table 1. The average-risk patients in this subgroup have excellent survival with the current standard-of-care treatment options 11. It has a phototransduction and neuronal signature in its transcription profile, as initially described by Cho et al. 3. However, more recently, the homeobox transcription factor Lmx1A has been identified as a master regulator transcription factor of group 4 medulloblastomas 32. Lmx1A is important in the normal development of cells in the upper rhombic lip and cerebellum, and it is also critical for the development of midbrain dopaminergic neurons 31, 32, which are thought to be where group 4 tumors originate. Interestingly, this subgroup of medulloblastoma is three times more common in males than in females 4. More recently, the presence of metastatic disease at diagnosis or chromosome 11 loss and chromosome 17 gain appear to dictate the prognosis in this subgroup of medulloblastoma patients 27. In addition, copy number changes in target genes that are important in the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) signaling pathway are found in this subgroup 29, 31. The most common chromosomal aberration found in group 4 tumors is isochromosome 17q; it is also found to a lesser degree in group 3 tumors 4.
Recurrent medulloblastomas
Despite the subgroup designation of medulloblastomas, at recurrence (tumor relapse with or without leptomeningeal dissemination), there is substantial divergence of the dominant clone 33. Interestingly, however, subgroup classification is maintained at recurrence or metastasis 33. Most medulloblastomas that recur post-cytotoxic therapies are fatal, and the patterns of relapse tend to be subgroup specific. The concept of intratumor heterogeneity was initially described by Gerlinger et al. 34 for renal cell cancer, and more recently for medulloblastoma 35. In addition, the microenvironment of the tumor will have implications for drug design, including immunotherapies 36. This might be why, despite the design of targeted clinical trials, there is failure of these targeted agents 37.
Secondary tumors following treatment of medulloblastomas
There is the added caveat of medulloblastoma patients who survive radiation and chemotherapy who then go on to develop other tumors such as meningiomas and gliomas 37, etc. Packer et al. 37 in the Children’s Oncology Group A9961 trial described a cohort of 379 patients (aged 3 to 21), and 15 of these patients went on to develop secondary tumors 5.8 years after their initial diagnosis of medulloblastoma. Thus, recurrence and secondary cancers post-standard treatment of medulloblastoma patients makes monitoring of this pediatric population well into adulthood a necessity.
Many groups are looking at targeted next-generation sequencing approaches in neuro-oncology in the initial diagnosis and recurrent setting to improve treatment options 38, since there is a lack of validated targets for non-SHH/WNT medulloblastoma that sequencing may help unravel. In addition, many cancers, including medulloblastomas, have DDX3X mutations, as briefly discussed in the WNT section, and recent work has shown that mutations in this gene result in global reduced translation 39. This may confer certain survival advantages, perhaps in certain microenvironments, which may aid in designing alternative therapeutic options 36.
Future directions
Despite dissecting medulloblastomas in the “genomic sense”, it is clear that therapy cannot be dictated by the subgroups alone. There are other key players, and epigenetics has a big role to play 40. Indeed, it is becoming increasingly clear that global changes in the epigenetic architecture are signatures of cancer and tumorigenesis. It was described in 2014 by Diede et al. 41 that DNA methylation probably prevents normal differentiation in pediatric cancers. It is known that focal regions of low methylation linked to transcription-factor-binding sites shed light on differential transcriptional networks between subgroups; however, increased methylation correlates with gene expression 41, 42.
Although in its infancy in medulloblastoma, proteomics is another strategy being utilized to analyze the tumor microenvironment, since other metabolites, such as small peptides and lipids, can be crucial in regulating tumor development 43. Summarizing the future of medulloblastoma treatment, numerous strategies for designing and tailoring treatment for medulloblastoma will evolve to harness the different technologies, such as genomics, methylomics, and proteomics. Even though personalized medicine is not de rigueur in medulloblastoma management, it is something that is on the horizon.
Abbreviations
CTNNB1, catenin beta 1; DDX3X, DEAD-box helicase 3; SHH, sonic hedgehog signaling pathway; TERT, telomerase reverse transcriptase; TP53, tumor suppressor protein p53; WNT, Wnt signaling pathway.
Acknowledgements
We would like to thank Dr Tobey MacDonald for a critical review of the manuscript and the reviewers for their constructive feedback. We would also like to thank Drs Richard Robertson (Radiology, Boston Children’s Hospital) and Chad Holder (Radiology and Imaging Sciences, Emory University Hospital) for producing publication-quality MRI images for this review.
Editorial Note on the Review Process
F1000 Faculty Reviews are commissioned from members of the prestigious F1000 Faculty and are edited as a service to readers. In order to make these reviews as comprehensive and accessible as possible, the referees provide input before publication and only the final, revised version is published. The referees who approved the final version are listed with their names and affiliations but without their reports on earlier versions (any comments will already have been addressed in the published version).
The referees who approved this article are:
Charles Eberhart, Johns Hopkins Hospital, Baltimore, MD, USA
Sridharan Gururangan, Preston A. Wells Center for Brain Tumor Therapy, Department of Neurosurgery, McKnight Brain Institute, University of Florida, Gainesville, FL, USA
James T Rutka, Division of Neurosurgery, The Hospital for Sick Children, Toronto, ON, Canada
Funding Statement
Soma Sengupta is supported by the National Institute of Neurological Disorders and Stroke grant (K08 NS083626), and the Winship Cancer Institute Institutional Research grant (IRG-14-188-01) from the American Cancer Society.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 1; referees: 3 approved]
References
- 1. Ostrom QT, Gittleman H, Liao P, et al. : CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–63. 10.1093/neuonc/nou223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gajjar A, Packer RJ, Foreman NK, et al. : Children's Oncology Group's 2013 blueprint for research: central nervous system tumors. Pediatr Blood Cancer. 2013;60(6):1022–6. 10.1002/pbc.24427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cho YJ, Tsherniak A, Tamayo P, et al. : Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29(11):1424–30. 10.1200/JCO.2010.28.5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kool M, Korshunov A, Remke M, et al. : Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–84. 10.1007/s00401-012-0958-8 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 5. Taylor MD, Northcott PA, Korshunov A, et al. : Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–72. 10.1007/s00401-011-0922-z [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 6. Jones DT, Jäger N, Kool M, et al. : Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100–5. 10.1038/nature11284 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 7. Bailey P, Cushing H: Medulloblastoma cerebelli: a common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14(2):192–223. 10.1001/archneurpsyc.1925.02200140055002 [DOI] [Google Scholar]
- 8. Louis DN, Ohgaki H, Wiestler OD, et al. : WHO classification of tumors of the central nervous system. Revised 4th edition.2016;184–200. Reference Source [Google Scholar]
- 9. Packer RJ, Vezina G: Management of and prognosis with medulloblastoma: therapy at a crossroads. Arch Neurol. 2008;65(11):1419–24. 10.1001/archneur.65.11.1419 [DOI] [PubMed] [Google Scholar]
- 10. Tamburrini G, Frassanito P, Chieffo D, et al. : Cerebellar mutism. Childs Nerv Syst. 2015;31(10):1841–51. 10.1007/s00381-015-2803-6 [DOI] [PubMed] [Google Scholar]; F1000 Recommendation
- 11. MacDonald TJ, Packer RJ: Pediatric Medulloblastoma. Medscape Drugs & Diseases. 2014. Reference Source [Google Scholar]
- 12. Robinson GW, Orr BA, Wu G, et al. : Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–54. 10.1200/JCO.2014.60.1591 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 13. Samkari A, White J, Packer R: SHH inhibitors for the treatment of medulloblastoma. Expert Rev Neurother. 2015;15(7):763–70. 10.1586/14737175.2015.1052796 [DOI] [PubMed] [Google Scholar]; F1000 Recommendation
- 14. Ramaswamy V, Remke M, Bouffet E, et al. : Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131(6):821–31. 10.1007/s00401-016-1569-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robinson G, Parker M, Kranenburg TA, et al. : Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–8. 10.1038/nature11213 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 16. Phoenix TN, Patmore DM, Boop S, et al. : Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell. 2016;29(4):508–22. 10.1016/j.ccell.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 17. Gibson P, Tong Y, Robinson G, et al. : Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–9. 10.1038/nature09587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clevers H, Nusse R: Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 19. Remke M, Ramaswamy V, Peacock J, et al. : TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013;126(6):917–29. 10.1007/s00401-013-1198-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiang JC, Ellison DW: Molecular pathology of paediatric central nervous system tumours. J Pathol. 2017;241(2):159–72. 10.1002/path.4813 [DOI] [PubMed] [Google Scholar]; F1000 Recommendation
- 21. Northcott PA, Jones DT, Kool M, et al. : Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12(12):818–34. 10.1038/nrc3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anastas JN, Moon RT: WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. 10.1038/nrc3419 [DOI] [PubMed] [Google Scholar]
- 23. Kahn M: Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–32. 10.1038/nrd4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhukova N, Ramaswamy V, Remke M, et al. : Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–35. 10.1200/JCO.2012.48.5052 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 25. Kool M, Jones DT, Jäger N, et al. : Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. 10.1016/j.ccr.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gururangan S, Robinson G, Ellison DW, et al. : Gorlin syndrome and desmoplastic medulloblastoma: Report of 3 cases with unfavorable clinical course and novel mutations. Pediatr Blood Cancer. 2015;62(10):1855–8. 10.1002/pbc.25560 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 27. Shih DJ, Northcott PA, Remke M, et al. : Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–96. 10.1200/JCO.2013.50.9539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sengupta S, Weeraratne SD, Cho Y, et al. : Could α5-GABA-A receptor activation be used as a target for managing medulloblastomas? CNS Oncol. 2014;3(4):245–7. 10.2217/cns.14.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pugh TJ, Weeraratne SD, Archer TC, et al. : Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488(7409):106–10. 10.1038/nature11329 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 30. Pei Y, Liu KW, Wang J, et al. : HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell. 2016;29(3):311–23. 10.1016/j.ccell.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 31. Northcott PA, Shih DJ, Peacock J, et al. : Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56. 10.1038/nature11327 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 32. Lin CY, Erkek S, Tong Y, et al. : Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530(7588):57–62. 10.1038/nature16546 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 33. Ramaswamy V, Remke M, Bouffet E, et al. : Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14(12):1200–7. 10.1016/S1470-2045(13)70449-2 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 34. Gerlinger M, Rowan AJ, Horswell S, et al. : Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 35. Morrissy AS, Garzia L, Shih DJ, et al. : Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529(7586):351–7. 10.1038/nature16478 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 36. Pham CD, Mitchell DA: Know your neighbors: Different tumor microenvironments have implications in immunotherapeutic targeting strategies across MB subgroups. Oncoimmunology. 2016;5(11): e1144002. 10.1080/2162402X.2016.1144002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Packer RJ, Zhou T, Holmes E, et al. : Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children's Oncology Group trial A9961. Neuro Oncol. 2013;15(1):97–103. 10.1093/neuonc/nos267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kline CN, Joseph NM, Grenert JP, et al. : Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. 2016; pii: now254. 10.1093/neuonc/now254 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 39. Valentin-Vega YA, Wang YD, Parker M, et al. : Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci Rep. 2016;6:25996. 10.1038/srep25996 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation
- 40. Batora NV, Sturm D, Jones DT, et al. : Transitioning from genotypes to epigenotypes: why the time has come for medulloblastoma epigenomics. Neuroscience. 2014;264:171–85. 10.1016/j.neuroscience.2013.07.030 [DOI] [PubMed] [Google Scholar]
- 41. Diede SJ, Guenthoer J, Geng LN, et al. : DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proc Natl Acad Sci U S A. 2010;107(1):234–9. 10.1073/pnas.0907606106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hovestadt V, Jones DT, Picelli S, et al. : Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510(7506):537–41. 10.1038/nature13268 [DOI] [PubMed] [Google Scholar]; F1000 Recommendation
- 43. Staal JA, Lau LS, Zhang H, et al. : Proteomic profiling of high risk medulloblastoma reveals functional biology. Oncotarget. 2015;6(16):14584–95. 10.18632/oncotarget.3927 [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Recommendation