

Abstract
The renin-angiotensin system (RAS) is a complex circulating and tissue-based system. There are multiple pathways for the formation and degradation of peptides. In order to understand the functions of the system, characterization of angiotensin peptides (products and substrates) is important. Radioimmunoassays with the requisite specificity and sensitivity have been developed to allow for the characterization and quantification of circulating and tissue angiotensins. Here, we describe the appropriate methods for collecting the tissue and blood, the extractions steps required to partially purify and remove larger molecular weight-interfering proteins from tissue and plasma, and the radioimmunoassay of three of the peptides of this system (Ang I, Ang II, and Ang-(1–7)), as well as the verification of immunoreactive identity for Ang II and Ang-(1–7) by combined high performance liquid chromatography—RIA analysis.
Keywords: Radioimmunoassay, Angiotensin I, Angiotensin II, Angiotensin-(1-7), HPLC, Extraction, Homogenization
1 Introduction
Since 1987 our group has been involved in the determination of angiotensin peptides in plasma and tissue [1]. This characterization included a detailed assessment of the necessity of using a cocktail of protease inhibitors to prevent the endogenous generation and degradation of peptides during sample collection and sample handling [2]. The original inhibitor cocktail was modified when it was reported by Campbell et al. [3] that the aspartyl protease inhibitor pepstatin was not sufficient to prevent the exogenous generation of angiotensin I (Ang I) from renin, requiring the inclusion of a specific renin inhibitor in addition to pepstatin to block this pathway.
The ongoing characterization of the bioactive peptides of the RAS, angiotensin II (Ang II), and Ang-(1–7), has revealed contrasting actions on blood pressure [4], cell growth [5], angiogenesis [6], and inflammation [7], Thus, to determine their physiological role, it is important to quantify these peptides in tissue and plasma as a first step toward understanding the agonist/antagonistic balance of the RAS. Indeed, we recently published an evaluation of angiotensin quantification methods and the expected peptide values in plasma, urine, and various tissues [8]. The predominant method used to characterize the peptides involves direct radioimmunoassays (RIA) characterized by the selective immunoreactivity against the three C terminal products, Ang I [Ang-(1–10)], Ang II [Ang-(1–8)]), and Ang-(1–7) that differ by 1–3 amino acids (Fig. 1). With the specificity of these C-terminally directed antibodies, which do not cross react with each other, the use of direct RIAs is capable of demonstrating the overall pattern of metabolism among the major peptides of the system in various tissues. Moreover, the cross reactivity of each antibody with its amino-terminal fragments allowed for the complete characterization of the angiotensin profile by combining high-performance liquid chromatography (HPLC) separation with RIAs. The validity, however, of using the direct RIAs became apparent when it was discovered that the predominant peptide of each peptide is the parent peptide with the amino-terminal fragments comprising a minority of the overall immunoreactivity [9,10] (Fig. 4).
Fig. 1.

Scheme for the processing of the precursor protein angiotensinogen to the bioactive peptides angiotensin II (Ang II) and Ang-(1–7). The enzyme renin cleaves angiotensinogen at the N-terminus to form the decapeptide Ang I. Processing of Ang I to the octapeptide Ang II involves angiotensin converting enzyme (ACE) and chymase (CHYM). Subsequent processing to the heptapeptide Ang-(1–7) from Ang II occurs through ACE2 and prolyl oligopeptidase (POP). Note the shared N-terminal sequence among all three peptides but the unique C-terminal sequence that dictates the specificity of C-terminus directed antibodies utilized for radioimmunoassays
Fig 4.
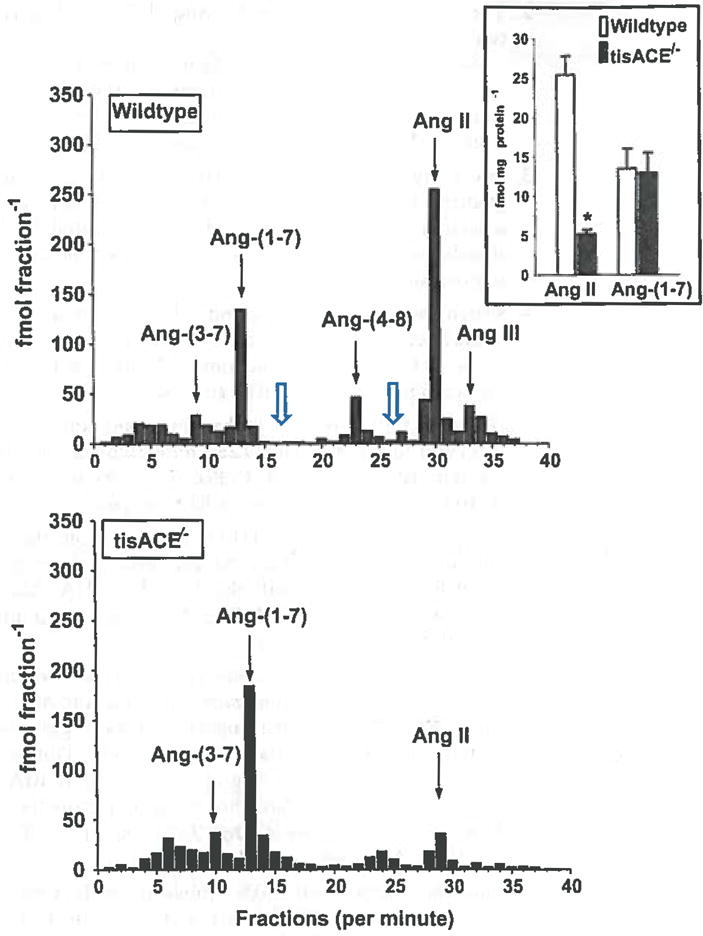
Combined high-performance liquid chromatography (HPLC) separation and RIA analysis of pooled kidney extracts from wildtype and tissue ACE knockout (tisACE−/−) mice. The collected HPLC fractions were completely evaporated and assayed by the Ang-(1–7) RIA (fractions 1–20) and the Ang Il RIA (fractions 21–40). The HPLC solvent system is 0.1 % HFBA (mobile phase A) and 80% acetonitrile/0.1 % HFBA (mobile phase B). Gradient conditions for Ang-(1–7) and Ang II separation were: 15–40 % B linear over 20 min; 40 % B isocratic for 20 min; 40–15 % B linear for 10 min, 15 % isocratic for 20 min at a flow rate of 0.35 ml/min at 25 °C. The arrows indicate the elution peaks for Ang-(3–7), Ang-(1–7), Ang-(4–8), Ang II, and Ang-(2–8) (Ang III). The open arrows indicate expected elution times for Ang-(2–7) and Ang-(3–8), respectively. Inset intrarenal concentration of Ang II and Ang-(1–7) (fmol/mg protein) in wildtype (n=8) and f/sACE−/−mice (n=8); *P<0.001 versus wildtype. Figure adapted with permission from Modrall et al. [9]
2 Materials
1,10-Phenanthroline monohydrate (o-PT, Sigma-Aldrich, St. Louis, MO, #P-1294), pepstatin (Peninsula Labs, San Carlos, CA, #4039, Na p-hydroxymcrcuribenzoate (NaHMB, Sigma-Aldrich, Milville NY, #H0642), rat renin inhibitor (AnaSpec Inc. Fremont CA, #WFML-1), sodium (tetra) ethylenediamine tetraacetate acid (EDTA, Fisher, Pittsburgh, PA, #S657–500), Tris base (Sigma-Aldrich, #T1503), Na Azide (Sigma-Aldrich, #S-2002); NaCl (Sigma-Aldrich, #S-3014), glacial acetic acid (Fisher #A35-500), Sep-Pak C18 3 cc Vac cartridges (Waters, Milford, MA, #WAT020805); n-heptafluorobutyric acid (Fisher #25003), bovine serum albumin (BSA, Sigma #A7888), Ang I radioimmunoassay kit (Peninsula Laboratories, Inc. San Carols, CA 94070 #2067), Ang II radioimmunoassay kit (ALPCO, Windham, NH # RK-A22), Iodine-125 10 mCi per 0.1 ml (Perkin Elmer, Waltham, MA, NEZ033A010MC), trifluoroacetic acid (TFA, HPLC grade, Pierce, Dallas TX, CAS 76-05-01), chloramine T (Sigma #C9887), sodium metabisulfite (Sigma-Aldrich, #S-9000), Methanol HPLC grade (Fisher #A412), phosphoric acid (Sigma-Aldrich #215104), acetonitrile HPLC grade (Fisher, #A998), Ang-(1–7), Ang-(2–7), Ang-(3–7), Ang II, Ang-(2–8), Ang-(3–8), Ang-(4–8) (Bachem, King of Prussia, PA).
3 Methods
3.1 Blood
3.1.1 Materials for Blood Sample Collection
Preparation of inhibitor cocktail for collection of blood and tissue samples. In one beaker, dissolve 0.36 g 1,10-phenanthroline monohydrate (o-PT, Sigma-Aldrich #P-1294) in 115 ml ultrapure water (New England Reagent Laboratory #0015). In another beaker, dissolve 0.302 g pepstatin (Peninsula Labs #4039, mol weight 686 g/l) in 60 ml pure ethyl alcohol. Mix both solutions on magnetic stirrer for 4–5 h at room temperature. Add contents of beakers and continue mixing. Add 1.32 g Na p-hydroxymercuribenzoate (NaHMB, Sigma-Aldrich, #H0642) and allow to dissolve for 10–15 min. Mixture will become less cloudy. Aliquot 10 ml into large glass tubes, cap and store in a refrigerator.
Vortex inhibitor cocktail and then add to EDTA tubes prior to collection: 50 μl per ml of blood to be collected.
For rat and mice blood, RAT RENIN INHIBITOR is added to the blood. Dissolve 0.001 g rat renin inhibitor (AnaSpec Inc. #WFML-1) in 1 ml ultrapure water (1 mM) (New England Reagent Laboratory #0015). Aliquot into 125 μl portions and store at −20 °C. Dilute×10 with ultrapure water (0.1 mM). Unused diluted inhibitor can be aliquoted and stored at−20 °C.
Add 100 μl of 0.1 mM rat renin inhibitor to EDTA tube prior to 3 ml blood collection.
For human blood, no rat renin inhibitor is added to blood.
15% EDTA: Dissolve 15 g EDTA (Fisher #S657-500) in 100 ml distilled water and store in a refrigerator. The EDTA is used to rinse funnels when collecting blood by decapitation.
3.1.2 Method for Collection of Blood
Add the appropriate amount of well-mixed inhibitor cocktail and rat renin inhibitor to a lavender (EDTA) top sample tube according to the following proportions: 3 ml of blood, 0.150 ml of inhibitor cocktail, and 0.100 ml of 0.1 mM rat renin inhibitor.
Prechill the sample tube in an ice water slush bath prior to collection.
Collect the sample and gently invert the tube to mix a number of times. Immediately return tube to the ice bath.
Centrifuge sample at 2000×g for 10 min in refrigerated centrifuge.
Transfer plasma into a prechilled conical centrifuge tube and centrifuge at 2000×g again for 10 min under refrigeration.
Harvest plasma into polypropylene tubes, label and store frozen at −80 °C.
Note: If collecting samples with syringe or by decapitation, rinse syringe or funnel with 15% EDTA solution prior to use.
3.1.3 SepPak Separation of Plasma Peptides
Materials for SepPak for Plasma Samples
Elution solvent. Add 7 ml Ultra Pure Water to 43 ml ethanol, pour into bottle and mix well. Mix 48 ml of this solution with 2 ml glacial acetic acid. The final mixture is used for elution. Keep at room temperature.
4% Glacial Acetic Acid. Add 2 ml glacial acetic acid to 48 ml ultra pure water and mix. Keep at room temperature while seppaking, store remaining solution in refrigerator (can be stored for up to 2 weeks).
125 I Ang II for spiking samples—Add radioactivity to Pro Buffer in amounts needed to achieve a count rate of approximately 500 cpm/50 μl. Ensure adequate amount of radioactivity for TC (total count) tubes to use to count the recovery of your seppak samples.
NOP Buffer (no protein buffer). Weigh and dissolve the following in approximately 900 ml room temperature distilled water: 12.1102 g of Tris base (Sigma-Aldrich, #T1503), 0.5000 g Na Azide (Sigma-Aldrich, #S-2002); 5.0000 g NaCl (Sigma-Aldrich, #S-3014), 4.3800 g EDTA (Fisher #BP 120–500). Adjust the pH to 7.4 using glacial acetic acid (Fisher #A35-500) (approximately 4.4 ml are needed). Bring the final volume to 1000 ml with distilled water, mix and store in the refrigerator.
Pro Buffer (Protein buffer). Add 0.1 g BSA to 100 ml NOP buffer. (Freeze remainder in small aliquots).
SepPak columns: Sep-Pak C18 3 cc Vac cartridges. From Waters cat#WAT020805.
Methods for SepPak for Plasma (1 ml Total Volume Applied to Column)
-
1
Thaw samples in ice water and centrifuge at 4 °C for 30 min, then aliquot 1 ml samples into glass prechilled 16×100 tubes. 1 ml is sufficient to use for the single determination of Ang I, Ang II, and Ang-(1–7). If sample volume is less than 1 ml, use as much sample as possible and record actual volume.
-
2
Add Ang II radioactivity to sample.
-
3
Place Sep-Pak columns on manifold equipped with stopcocks. Unless noted otherwise, the reagents arc applied to the columns in a manner that allows the reagents to drip through the column without drying the column. Allow each solution to go through all of the columns on the manifold before applying the next solution.
-
4
Apply 5 ml elution solvent to each column.
-
5
Apply 5 ml methanol solvent to each column.
-
6
Empty waste in reservoir into used solvent container.
-
7
Apply 5 ml water to each column. (Procedure may be stopped at this point if needed, leave some water on column).
The next steps should continue without stopping.
-
8
Apply 5 ml 4% acetic acid to each column.
-
9
Add sample to column.
-
10
Add 4 ml ultra pure water to the cold sample tubes, rinse tubes, and add water to column.
-
11
Remove sample tubes from ice and add another 4 ml ultra pure water, rinse and add to column.
-
12
Push water through column and apply 2 ml acetone to each column. When acetone has gone through, turn the vacuum on slightly and remove the remaining acetone from each column. (Turn on vacuum to columns one at a time to approx. 5-mmHg for 5 s.) DO NOT ALLOW THE COLUMN TO DRY.
-
13
Collect Eluates in appropriate glass tubes after applying elution solvent in the following manner.
-
14
Apply 1 ml elution solvent and let absorb on the column.
-
15
Apply 1 ml elution solvent and let absorb on the column.
-
16
Apply additional 1.3 ml elution solvent to each column to each column.
-
17
After the final application has been absorbed onto the column, increase vacuum slighly to allow the release of the elution solvent from the columns.
-
18
Weigh the elution fraction collected and record weight.
-
19
Mix content of tubes thoroughly and quantitatively pipette 0.5 ml of the elution fraction into appropriately labeled small (12 × 75) glass tubes for Ang II, 0.5 ml for Ang I, and 2 ml for Ang-(1–7).
-
20
Dry tubes in Savant without heat.
-
21After samples are dry, reconstitute as follows:
- For Ang II—0.6 ml Alpco Kit RLA Buffer (assay volume = 0.5 ml).
- For Ang I—0.125 ml fresh Pro Buffer (assay volume = 0.100 ml).
- For Ang-(1–7)—0.225 ml fresh Pro Buffer (assay volume =0.200 ml). These volumes allow for a single RIA determination for each peptide.
-
22After allowing time for samples to dissolve, count tubes in the gamma counter along with total count tubes.
- Freeze samples and calculate extraction efficiencies.
-
23
FOR TOTAL COUNT TUBES: Add the appropriate amount of radioactivity (i.e., 50 μl) into three tubes and bring volume to 1.333 ml.
3.2 Tissue
3.2.1 Materials for Tissue Homogenization
0.1 N HC1: Add 0.17 ml HC1 to volume of 20 ml ultra pure water.
Acid Ethanol: 80 ml ethanol+20 ml ultra pure water + 0.83 ml 0.1 N hydrochloric acid.
15% EDTA: add 150.0 g sodium (tetra) ethylenediamine tetraacetate technical powder to volume of 1000 ml distilled H2O.
Inhibitor: 150 μl cocktail inhibitor (sec above) + 100 μl rat renin inhibitor (1 mM) in each tube.
Radioactivity: 125I-Ang II for spiking samples—Add radioactivity to Pro Buffer in amounts needed to achieve a count rate of approximately 500 cpm/100 μl (450–550 μl).
1% HFBA: Add 1 ml n-heptafluorobutyric acid (HFBA Fisher Cat #25003) to 99 ml ultra pure water.
-
NOP Buffer (no protein buffer): Weigh and dissolve the following in approximately 900 ml room temperature distilled water (do not use water that has been sitting overnight or longer at room temperature): Tris base (Sigma-Aldrich, #T1503) 12.1102 g; Na Azide (Sigma-Aldrich, #S-2002) 0.5000 g; NaCl (Sigma-Aldrich, #S-3014) 5.0000 g; EDTA (Fisher #BP 120–500) 4.38 g.
Adjust the pH to 7.4 using glacial acetic add (Fisher #A35—500). (Approximately 4.4 ml is needed). Bring the final volume to 1000 ml with distilled water, mix and store in the refrigerator.
Pro Buffer. Dissolve 0.1 g BSA (Sigma Cat # A7888) per 100 ml NOP Buffer. Make fresh each use.
3.2.2 Methods for Tissue Homogenization
Day 1
Prepare dry ice container and an ice water slush bath.
Place 50 ml plastic conical tubes in ice bath labeled for each sample and add the following: 10 ml Acid Ethanol, 100 μl 15 % EDTA, 100 μl Inhibitor Cocktail; 50 μl rat renin inhibitor 1 mM; 100 μl Ang II radioactivity—Store remainder to use for counting recovery.
Label weight boats and place them in dry ice (Tare the weight of the boat).
Place tissue in boats: cut tissue if large (~300 mg for duplicates, ~100 mg for single determination).
Weigh frozen tissue and record weight. Place back on dry ice to keep frozen until sample is homogenized.
Place clean homogenizer blade in a homogenizer and rinse with 50/50 mixture of ethanol/water mixture.
Add tissue to an appropriately labeled 50 ml tube and immediately homogenize; Homogenize at 20,000×g for 30 immersions.
Between each sample homogenizer blade must be rinsed with methanol to remove any remaining tissue.
Remove 500 μl of the sample and transfer to a 12×75 mm tube and store at −20 °C for protein determinations.
Transfer remaining sample to a centrifuge tube (16 ml Nalgene) and spin at 12,000×g for 20 min at 4 °C (Sorvall Super-speed R.C-2B automatic refrigerated centrifuge).
After spin put samples at −20 °C overnight (do not discard supernatant).
Day 2
-
All steps are done on ice.
Rccentrifuge samples at 12,000×g for 20 min at 4 °C.
Transfer supernatant to a 15 ml conical tube and add 5 ml of 1 % HFBA.
Discard pellet.
Place samples at −20 °C for 24 h.
Day 3
Spin samples at 3000×g for 15 min at 4 °C.
Pour off about 4 ml of the supernatant into a 12×75 glass tube and dry in Savant down to 1 ml.
Repeat this step until 1 ml of solution is left. Transfer supernatant to a 15 ml conical tube. (Do not transfer any pellet).
Add 9 ml of 0.1% HFBA to the remaining sample.
Vortex.
Sep-Pak as soon as possible.
3.2.3 Sep-Pak Protocol for Tissue
Materials Solutions for Tissue SepPak
Acid MeOH: add 80 ml of MeOH, 20 ml of H2O, and 100 μl of HFBA.
0.1 % HFBA: Add 100 μl and 99.9 ml of H20.
SepPak columns: Sep-Pak CIS 3 cc Vac cartridges. From Waters cat#WAT020805.
SepPak Methods for Tissues
Label columns and tubes (Waters sep-pak vac 3 cc [200 mg] part #wat 854945).
Prepare column by applying the following solutions: (allow to gravity feed) 5 ml of acid MeOH-Close column after application: 10 ml of 0.1 % HFBA (do not let the column dry out); close column after application; 10 ml of 0.1% HFBA (do not let the column dry out). Close column after application.
Apply the sample (allow to gravity feed). Close column after application.
Wash column with 10 ml of 0.1% HFB. Add 5 ml to tube sample in ice bath and rinse then apply cold to column. Remove from ice bath.
Close column after application. Add remaining 5 ml to the same tube and apply to column. Close column after application.
Wash column with 5 ml of H20. Close column after application. Gently push air through the column to remove any remaining water. (I use a 5 cc syringe and push air 2×).
Discard waste.
Place glass tubes (16 × 100) in sep-pak manifold labeled appropriately. Sequentially add 1 ml of acid MeOH (apply very slight vacuum only to get eluate started to flow then allow to gravity to elute solvent). Close column, add 1 ml of acid MeOH Close column; Add 1.3 ml of acid MeOH. Close column. Allow to gravity feed.
After all the solution has been absorbed onto the column, push air through to completely empty to column.
Weigh samples.
- Vortex and separate as follows:
- For duplicate determination: Label 6 12×75 PS conical tubes for each sample. Pipetter 0.5 ml into each tube.
- For single determination: Label 3 12 × 75 PS conical tubes for each sample. Pipette 1 ml into each tube.
Dry in savant without heat.
When dry, count recovery.
Store at 4 °C until assayed.
3.3 Radio Immunoassays
3.3.1 Ang l Radioimmunoassay
The tissue and plasma extracts are separated and measured by three radioimmunoassays.
Commercially available kit. Peninsula Laboratories, Inc. San Carols, CA 94070 Catalog # 2067. 125 tubes.
-
Standard Curve: 0.25 pg/tube–640 pg/tube.
Assay volume: 100 μl.
Sensitivity: 0.250 pg/tube.
C-terminal directed antibody, thus, cross reacts with Ang I, Ang-(2–10), Ang-(3–10). Little or no cross reactivity with Ang II or Ang-(1–7).
To convert to fmol/ml, multiply pg/ml by 0.772. (MW Ang I = 1296).
3.3.2 Ang II Radioimmunoassay
Commercially available kit. ALPCO, Windham, NH 03087 Catalog # RK-A22. 100 tubes.
-
Standard Curve: 2–500 pg/ml.
Assay volume: 500 μl.
Sensitivity: 0.8 pg/ml.
2 day assay.
C-terminal directed antibody, thus, cross reacts with Ang II, Ang-(2–8), Ang-(3–8). Little or no cross reactivity with Ane I or Ang-(1–7).
To convert to fmol/ml, multiply pg/ml by 0.953 (MW Ang II = 1049).
3.3.3 Ang-(1–7) Radioimmunoassay
Hypertension Core Laboratory developed assay.
-
Standard Curve: Ang-(1–7) (Bachem, King of Prussia PA).
Range of Standard Curve: 2.5–2000 pg/tube.
Assay volume: 100 μl.
Sensitivity: 1.25 pg/tube.
2 day assay.
C-terminal directed antibody, thus, cross reacts with Ang-(1–7), Ang-(2–7), and Ang-(3–7). Little or no cross reactivity with Ang I or Ang-(1–7).
To convert to fmol/ml, multiply the corrected pg/ml by 1.115 (MW for Ang-(1–7) = 897 g/l).
- Values obtained from the RIAs are corrected for recovery, assay volume, concentration and dilution of extraction and homogenization procedures.
- For plasma, values are expressed for plasma as pg/ml (fmol/ml).
- For tissue, values are pg/mg protein (fmol/mg protein) or pg/g tissue weight (fmol/g tissue weight).
3.4 lodination of Peptides
3.4.1 Materials forAng-(1–7) lodination
The radioimmunoassay requires preparation of 125I-labeled peptides.
Iodine-125 10 mCi per 0.1 ml (Perkin Elmer NEZ033A010MC).
MilliQ Plus ultra pure water system with distilled water input (Millipore Q Pak cartridges #CPMO004D2).
Chloramine T (Sigma #C9887) 1 mg/ml in MilliQ ultra-pure water, prepare fresh.
Sodium metabisulfite (Sigma #S-9000) 3 mg/ml in MillQ water, prepare fresh.
Phosphate buffered saline (PBS).
Ang-(1–7) (Bachem) 1 mM in MillQ water.
SepPak columns: Sep-Pak C18 3 cc Vac cartridges (Waters #WAT020805).
Trifluoroacetic acid (TFA, HPLC grade, Pierce CAS 76 05-01) 0.1 % solution. 0.1 ml TFA in 100 ml MillQ water.
Methanol (HPLC grade, Fisher #A412), 80% solution in 0.1 % TFA. Add 0.1 ml TFA to 80 ml methanol and bring to a final volume of 100 ml with MilliQ water.
15 ml conical tubes (Starstedt #62.554.002).
3.4.2 Method of lodination
Peptide iodination and purification on SepPak C18 column is performed in a certified chemical hood. Use multiple pairs of gloves and a lead apron during the iodination procedure. Store iodine behind lead bricks or lead shielding.
- Activate SepPak column.
- Flush 5 ml of 80% methanol/0.1 % TFA through column using a 10 ml syringe that fits into column at approximately one drop per second.
- Flush 10 ml 0.1% TFA through column at one drop per second. Cap the column outflow and leave approximately 200 μl 0.1 % TFA in column bed.
- To a 0.5 ml microccntrifuge tube at room temperature, add in the following order:
- 10 μl Ang-(1–7).
- 20 μl PBS.
- 10 μl Iodine-125. Pierce septum of container with a Hamilton 50 μl syringe (#81460) to transfer iodine-125. Avoid opening container to air.
- 10 μl Chloramine T to initiate iodine-125 incorporation. Incubate for 30 s.
- 50 μl sodium metabisulfite to terminate reaction.
- 200 μl 0.1 % TFA to transfer reaction contents.
- SepPak Purification
- Uncap column and place activated SepPak within the 1st 15 ml tube.
- Transfer iodinated material to column. Rinse reaction vial twice with 200 μl 0.1% TFA and transfer to column. Add 3.0 ml 0.1% TFA to column and allow content to flow through column into tube.
- Transfer column to 2nd 15 ml tube. Wash column with 10.0 ml 0.1% TFA using a syringe.
- Transfer column to 3rd 15 ml tube and wash with 10.0 ml MillQ water using a syringe. Completely push all the Water through the column prior to the elution step.
- Elute 125I-peptide with 80% methanol/0.1% TFA into 1.5 ml centrifuge tubes. Collect 1.0 ml per tube.
- Determine iodination efficiency
- Remove 5 μl from each collection tube and count in a gamma counter. Calculate the total counts per ml based on the volume of each collection tube.
- Unreacted iodine-125 is not retained by the column and is collected in the 1st 15 ml tube. The 2nd and 3rd 15 ml collection tubes should contain little radioactivity.
- The 125I-peptide is completely eluted by methanol in the 1st and 2nd 1.5 ml microcentrifuge tubes. The highest counts per ml should be in the 1st 1.5 ml collection tube.
- Evaporate the methanol solution in a vacuum centrifuge (Savant SpeedVac #100H with Welch Dry Fast pump #2044 and activated carbon filter to trap evaporated iodine-125) prior to final purification by HPLC.
3.5 HPLC Purification of 125I-Peptides
3.5.1 HPLC Equipment and Buffers (See Fig. 2)
Fig. 2.
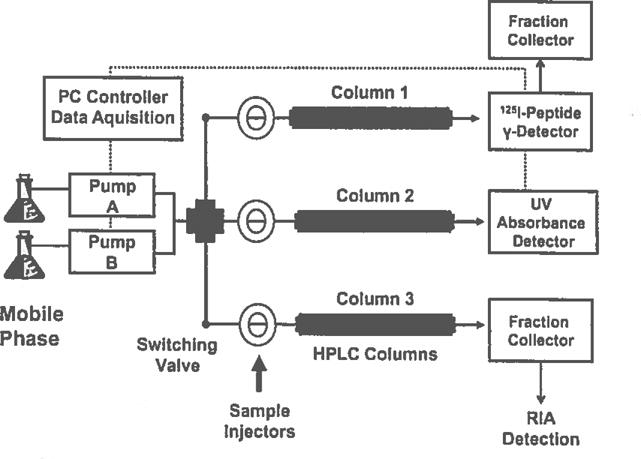
High-performance liquid chromatography (HPLC) system for separation and detection of angiotensin peptides. The system consists of a PC-based controller for operation of the solvent gradient and data acquisition/analysis from the radioactive and UV detectors (….); a switching valve to input solvent flow to the sample injectors and their respective HPLC columns #1–3 that output into an inline 125I-peptide gamma detector, a UV absorbance detector or directly into a fraction collector. Column 1 is utilized for purification of 125I-labeled peptides or analysis of 125I-peptide metabolism products and Identification of peptidase activities [11,12]; column 2 Is exclusively for standardization and analysis of unlabeled angiotensin peptides; column 3 is exclusively utilized for separation of endogenous angiotensins and subsequent detection by RIA
HPLC purification is necessary to separate the unlabeled and the mono- and di-125I forms of the peptide. These forms are not separated by the SepPak C18 extraction column. The SepPak column efficiently removes the unreacted iodine-125 and iodination reagents.
Shimadzu LC20AD system with binary pumps, CTO-20A column oven, and DGU-20A3 degassing system.
Shimadzu #SPD-10A UV-VIS Absorbance Detector, 220 nm for pep tides.
Rheodyne ceramic manual injectors #9125 with 500 μl PEEK injector loop.
Rheodyne switching valves #7025 to direct solvent flow to multiple injectors and columns.
Bioscan Dual Channel Flow Counter #FC-2000 with PMT detector #B-FC-3100 for detection of 125I-peptidcs.
NovaPak C18 narrow bore columns, 2.1 × 150 mm (Waters #WAT023655) with Synergy guard columns 2.1 × 10 mm (Waters #WATO23655).
Gilson FC204 fraction collectors.
Mobile phase A: Phosphoric acid (Sigma Aldrich #215104), 0.1 % solution. Add 1 ml phosphoric acid to 1000 ml degassed MillQ water and filter (GE #R02SP04700, 0.2 μm, 47 mm Nylon membrane).
Mobile phase B: Acetonitrile (Fisher, HPLC Grade #A998), 80% solution. Add 1 ml phosphoric acid to 800 ml acetonitrile and bring to a final volume of 1000 ml with degassed MillQ water and filter (GE #R02SP04700).
Tris–HCl, 1 M, pH 8.0 at 4 °C (Sigma, #Tl503); titrate pH with HCl.
Bovine serum albumin (BSA, Sigma #A6003), 0.1 % in 1 M Tris–HCl, pH 8.0.
3.5.2 HPLC Purification (See figs. 2 and 3)
Fig. 3.
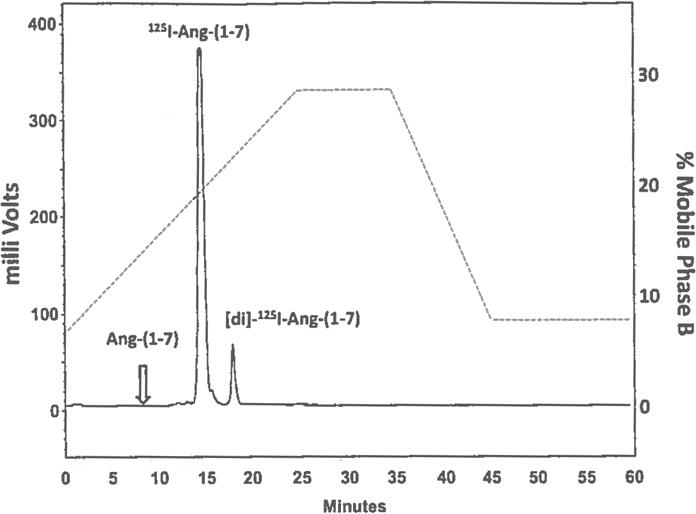
Chromatograph of 125I-Ang-(1–7) purification by high-performance liquid chromatography (HPLC) following peptide iodination and SepPak C18 extraction. The major peak of radioactivity corresponds to mono-125I-Ang-(1–7) that elutes at a retention time of 15 min. The second radioactive peak corresponds to di-125I-Ang-(1–7) that elutes later in the gradient. Although not detected by the flow-through gamma detector, the unlabeled peak of Ang-(1–7), designated by the open arrow, elutes earlier than the iodinated forms of the peptide. The right axis illustrates the gradient change (….) for mobile phase B (80% acetonitrile/0.1 % phosphoric acid) versus time in the HPLC purification. Gradient conditions are 8–28 % B linear over 25 min, 28% B Isocratic for 10 min, 28 to 8 % B linear for 10 min, 8 % isocratic for 15 min at a flow rate of 0.35 ml/min at 25 °C
Equilibrate HPLC system at a flow rate of 0.35 ml/min 8.0% mobile phase B at 25 °C. Direct solvent flow through injector and column 1 via switching valve (see Fig. 2).
- The programmed gradient for 125I.Ang-(1–7) purification is:
- Step 1: 8–28 % B linear gradient for 25 min.
- Step 2: 28% B isocratic conditions for 10 min.
- Step 3: 28 to 8 % B linear gradient for 15 min.
- Step 4: 8 % B isocratic for 10 min.
Reconstitute 125I-peptide with 0.5 ml 8.0% mobile phase B and inject solution onto the HPLC. Initiate gradient and fraction collector for 1 min (0.35 ml) fractions.
Pool the two fractions that contain the major peak of radioactivity corresponding to mono- 125I-Ang-(1–7). Typical elution time for the mono- 125I-form is 15 min. The second smaller peak corresponds to di-125I.Ang-(1–7) and is discarded.
Dilute the pooled fractions 1:1 with 1 M Tris–HCl/0.1 % BSA and store at 4 °C in a lead container.
Flush the injector with 80% mobile phase B and run a gradient to 80% B to remove residual radioactivity from the column. Store the column between purifications in 100% acetonitrile.
3.6 HPLC Verification of Endogenous Angiotensins
3.6.1 HPLC Equipment and Buffers (See Fig. 2)
The HPLC separation of angiotensins can be combined with RIA detection of endogenous peptides in collected fractions to verify the identity of immunoreactive peptides.
Shimadzu HPLC system as described in Subheading 3.5.
Mobile phase A: HFBA (Pierce Sequanal Grade, #25003), 0.1 % solution. Add 1 ml HFBA to 1000 ml degassed MillQ water and filter (GE #R02SP04700, 0.2 μm, 47 mm Nylon membrane).
Mobile phase B: Acetonitrile (Fisher, HPLC Grade #A998), 80% solution. Add 1 ml HFBA to 800 ml acetonitrile and bring to a final volume of 1000 ml with degassed MillQ water and filter (GE #R02SP04700).
Chromtech 0.45 μm PTFE filter vial system (#FV-2045).
Starstedt conical 4.5 ml collection and RIA tubes (#57.477).
- Angiotensin standards, Bachem 1 mM in MillQ water.
- Ang-(1–7).
- Ang-(2–7).
- Ang-(3–7).
- Ang II.
- Ang-(2–8).
- Ang-(3–8).
- Ang-(4–8).
3.6.2 HPLC Standardization
Equilibrate HPLC system at a flow rate of 0.35 ml/min 15.0% mobile phase B at 25 °C. Direct solvent flow through injector, column 2 and UV absorbance detector via switching valve (see Fig. 1).
- The programmed gradient for Ang-(1–7) and Ang II separation is:
- Step 1: 15–40% B linear gradient for 20 min.
- Step 2: 40% B isocratic conditions for 20 min.
- Step 3: 40–15 % B linear gradient for 10 min.
- Step 4: 15% B isocratic for 20 min.
Inject angiotensin standards (10 nmol each) and initiate the gradient. Ensure that the column and gradient achieve baseline separation of angiotensin peptides as indicated by the UV absorbance profile. Note the elution times of the standard angiotensin peptides.
Switch the valve to injector and column 3 connected directly to the fraction collector. Inject 500 μl of 15% B and initiate the gradient. Collect 1 min fractions (0.35 ml) for 40 min (@ 40 fractions) for subsequent RIA analysis.
Once the HPLC returns to baseline conditions, inject Ang-(1–7) and Ang II standards (250 finol each) in 500 μl of 15%. B and initiate the gradient. Collect 1 min fractions for 40 min (@ 40 fractions) for subsequent RIA analysis.
Completely evaporate the HPLC fractions from the previous runs in a vacuum centrifuge (Savant SpeedVac). Assay tubes 1 to 20 from each run with the Ang-(1–7) RIA. Assay tubes 21–40 with the Ang II RIA. The RIA is performed directly in the HPLC collection tubes.
Ensure that the collected fractions from the blank solvent injection do not exhibit immunoreactive peaks by the Ang-(1–7) or Ang II RIAs. Establish that injection of the angiotensin standards reveals baseline separation of the peptides. Plot the immunoreactive values for the Ang-(1–7) and Ang II RIAs versus time to construct a standard chromatograph. Note the order of elution for the peptides is Ang-(3–7), Ang-(1–7), Ang-(2–7), Ang-(4–8), Ang-(3–8), Ang II, Ang-(2–8).
Rinse the injector with 80% mobile phase B. Once HPLC equilibrates, inject 500 μl 80% B and run gradient to remove residual peptide standards.
3.6.3 HPLC Procedure: Sample (Fig. 4)
The immunoreactive content of the sample should be determined prior to the HPLC step to ensure detection of the peptide. We recommend that the sample for HPLC contain a minimum of 250 fmol of immunoreactive Ang-(1–7) or Ang II. Either an individual sample or a pooled sample can be fractioned by HPLC.
Equilibrate HPLC system at a flow rate of 0.35 ml/min 15% mobile phase B at ambient temperature. Inject 500 μl of 15% B, initiate gradient, and collect 40 × 1 min fractions. These fractions from the blank solvent are assayed to ensure there is no background material prior to the HPLC analysis of the sample.
Reconstitute the sample in 15% B to completely dissolve material. To facilitate this process, sample vials can be placed in a sonicating water bath (Branson Ultrasonic Bath, B200R-1). Centrifuge samples at 30,000×g for 10 min to pellet insoluble material and filter on a Chromtech 0.45 μm PTFE filter vial (#FV-2045).
Inject 500 μl of sample onto the HPLC, begin gradient, and collect 40×1 min fractions.
Following equilibration of the HPLC, inject 500 μl of 80% B and run gradient to remove residual sample contaminants.
Either additional samples can be injected on the HPLC (with blank solvent injections between each run) or the mixture of angiotensin peptides for RIA detection is run to calibrate the current separation.
The HPLC fractions are evaporated in the vacuum centrifuge and subjected to the Ang-(1–7) and Ang II RIAs.
Plot the immunoreactive content of the HPLC fractions versus time for each sample to display the profile of angiotensin peptides (see Fig. 4).
Store the HPLC and column in 100% acetonitrile when not in use.
Acknowledgments
Support was provided in part by the National Institutes of Health, NHLBI-P01 HL51952, HL56932, HD084227, Unifi, Inc. Greensboro, NC and Farley-Hudson Foundation, Jacksonville; NC.
Contributors
Amaya Albalat • School of Natural Sciences, University of Stirling, Stirling, UK
Natalia Alenina • Max-Delbrück-Center for Molecular Medicine (MDC), Berlin, Germany
Johny Al-Khoury • Department of Anatomy and Celt Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC, Canada
Samantha Alvarez-Madrazo • Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK
Angelica Amanso • Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA
Michael Bader • Max-Delbrück-Center for Molecular Medicine (MDC), Berlin, Germany
Tlili Barholjmi • Lady Davis Institute for Medical Research, Jewish General Hospital, McGill University, Montreal, QC, Canada
Ghassan Bkaily • Department of Anatomy and Cell Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC, Canada
Thiago Bruder-Nascimento • Kidney Research Centre, Department of Medicine, Ottawa Hospital Research Institute, University of Ottawa, Ottawa, ON, Canada; Department of Pharmacology, Medical School of Ribeirao Preto, University of Sao Paulo, Sao Paula, Brazil
Mercedes L. de Bold • Department of Pathology and Laboratory Medicine, Faculty of Medicine, Ottawa Heart Institute, University of Ottawa and the Cardiovascular Endocrinology Laboratory, Ottawa, ON, Canada
Adolfo J. de Bold • Department of Pathology and Laboratory Medicine, Faculty of Medicine, Ottawa Heart Institute, University of Ottawa and the Cardiovascular Endocrinology Laboratory, Ottawa, ON, Canada
Ana M. Briones • Department of Pharmacology, School of Medicine, Instituto de Investigación Hospital Universitario La Paz (IdiPAZ), Universidad Autónoma de Madrid, Madrid, Spain
K. Bridget Brosnihan • Department of Surgery, Hypertension & Vascular Research, Cardiovascular Sciences Center, Wake Forest School of Medicine, Winston-Salem, NC, USA
Dylan Burger • Kidney Research Centre, Ottawa Hospital Research Institute, University of Ottawa, Ottawa, ON, Canada
Kevin D. Burns • Division of Nephrology, Department of Medicine, Kidney Research Centre, Ottawa Hospital Research Institute, University of Ottawa, Ottawa, ON, Canada
G.E. Callera • Kidney Research Centre, Department of Medicine, Ottawa Hospital Research Institute, University of Ottawa, Ottawa, ON, Canada
R.M. Carey - University of Virgina, School of Medicine, Fontaine Research Park, Charlottesville, VA, USA
Aurelie Nguyen Dinh Cat • Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, Scotland, UK
Mark C. Chappell • Department of Surgery, Hypertension & Vascular Research, Cardiovascular Sciences Center, Wake Forest University School of Medicine, Winston-Salem, NC, USA
Allen W. Cowley Jr • Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, USA
Eleanor Davies • Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK
Christian Delles • Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular, Research Centre, Medical Sciences University of Glasgow, Glasgow, UK
Satoru Eguchi • Department of Physiology, Cardiovascular Research Centre, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, USA
Katherine Elliott • Department of Physiology, Cardiovascular Research Centre, Lewis Katz School of Medicine at Temple University, Philadelphia, PA, USA
R.A. Felder • University of Virgina, School of Medicine, Charlottesville, VA, USA
Denise C. Fernandes • Vascular Biology Laboratory, Heart Institute (InCor), University af São Paulo School of Medicine, São Paulo, Brazil
Anderson J. Ferreira • national Institute of Science and Technology in Nanobiopharmaceutics, Federal University of Minas, Gerais, Brazil; Department of Morphology, Biological Science Institute, Federal University of Minas, Gerais, Brazil
Rodrigo A. Fraga-Silva • National Institute of Science and Technology in Nanobiopharmaceutics, Federal University of Minas, Gerais, Brazil; Institute of Bioengineering, Elcole Polytechnique Federale De Lausanne, Lausanne, Switzerland
Yamil Gerena • School of Pharmacy, Medical Sciences Campus UPR, San Juan, PR, USA
J.J. Gildea - Department of Pathology, University of Virginia, Charlottesville, VA, USA
Renata C. Gonsalves • Vascular Biology Laboratory, Heart Institute (InCor), University of São Paulo School of Medicine, São Paulo, Brazil
Kathirvel Gopalakrishnan • Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology, University of Toledo College of Medicine, Toledo, OH, USA; Program in Physiological Genomics, Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology, University of Toledo College of Medicine and Life Sciences, Toledo, OH, USA
Kathy K. Griendling • Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA
Caryl E. Hill • Department of Neuroscience, John Curtin School of Medical Research, Australian National University, Canberra, ACT, Australia
Holger Husi • School of Natural Sciences, University of Stirling, Stirling, UK
Danielle Jacques • Department of Anatomy and Cell Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC, Canada
Bina Joe • Center for Hypertension and Personalized Medicine, Bioinformatics, Proteomics and Genomics Program, Department of Surgery, University of Toledo College of Medicine, Toledo, OH, USA; Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology, University of Toledo College of Medicine, Toledo, OH, USA; Program in Physiological Genomics, Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology, University of Toledo College of Medicine and Life Sciences, Toledo, OH, USA
Footnotes
The iodination procedure is applicable for angiotensin peptides, their peptide receptor antagonists, and other peptides provided they contain a tyrosine residue.
The short reaction time (30 s) with Chloramine T and the high peptide to iodine ratio ensures predominant monoiodination of the peptide. Longer reaction times will result in greater formation of di-125I-Ang-(1–7).
The efficiency of iodination is typically 60–80%. Reduced efficiency or absence of peptide iodination usually indicates the need to replace the Chloramine T reagent.
Note that the Tris–HCl buffer is temperature sensitive and the final pH of the buffer should be adjusted at the appropriate temperature.
We find that the immediate use of the iodine-125 results in optimal iodination of angiotensin peptides. We recommend obtaining the iodine-125 from the supplier as soon as the product is synthesized.
The 125I-Ang-(1–7) can be repurified by HPLC after 1 to 2 months storage. The 125I-peptide must be initially extracted on the SepPak column to remove the Tris–HCl/BSA buffer that interferes with the HPLC purification.
The use of other HPLC C18 columns configured in narrow or conventional bore (2–4 mm; internal diameter) may be applicable in the purification of 125I-Ang-(1–7) or other peptides; however, the gradient conditions should be optimized for each column with the appropriate peptide standards to ensure resolution and recovery of the 125I-peptide.
Alternative HPLC buffers such as TFA or HFBA (0.1%) can be utilized for the purification of 125I-Ang-(1–7) and other angiotensin peptides. These buffers may necessitate modification of the gradient conditions.
It is imperative that the HPLC fractions are completely evaporated and dry prior to assay by RIA. Although both HFBA and acetonitrile are volatile, any residual amount of the acid or solvent will significantly interfere with the RIA and may yield false-positive values.
The use of two injectors and identical columns ensures that the UV absorbance calibration with the angiotensin standards on one column does not contribute to immunoreactive carryover on the endogenous peptide column. The column calibration by UV absorbance requires markedly higher peptide concentrations than RIA detection (nmol vs. fmol range—a 106-fold difference in sensitivity). For example, 0.001% carryover of 10 nmol Ang II standard would be detected as a significant immunoreactive peak of 100 fmol the by Ang II RIA.
Given the high sensitivity of the angiotensin RIAs, all reagents should be of the highest quality and used solely for the HPLC-RIA application to ensure no interference with the assay.
Other HPLC C18 columns configured in narrow or conventional bore (2–4 mm internal diameter, respectively) may be applicable for the characterization of endogenous angiotensins. The gradient separation conditions must be optimized for each column with the appropriate peptide standards.
Peptide analysis by HPLC should be performed on samples that have undergone extensive extraction to remove potential contaminating material that may interfere with the RIAs employed and/or compromise the HPLC separation.
For a large number of samples, we typically perform direct RIAs to obtain the mean values and subsequently analyze a sample pool to characterize their immunoreactive identity. As shown in Fig. 4 (inset), direct RIAs of Ang II and Ang-(1–7) revealed a lower Ang II content in kidneys of ACE knockout mice, but no change in Ang-(1–7). The HPLC-RIA analysis of pooled kidney samples demonstrates lower immunoreactive peaks corresponding to Ang II and Ang-(4–8) in the ACE knockout mice; however, the peak of Ang-(1–7) is not reduced. The HPLC analysis confirms the direct RIA results for kidney content of Ang II and Ang-(1–7), as well as demonstrates that Ang II and Ang-(1–7) are the predominant immunoreactive forms in the kidney as detected by their respective RIAs.
References
- 1.Chappell MC, Brosnihan KB, Diz DI, Ferrario CM. Identification of angiotensin-(1–7) in rat brain. Evidence for differential processing of angiotensin peptides. J Biol Chem. 1989;264:16518–16523. [PubMed] [Google Scholar]
- 2.Kohara K, Tabuchi Y, Senanayake P, Brosnihan KB, Ferrario CM. Reassessment of plasma angiotensins measurement: effects of protease inhibitors and sample handling. Peptides. 1991;12:1135–1141. doi: 10.1016/0196-9781(91)90070-6. [DOI] [PubMed] [Google Scholar]
- 3.Campbell DJ, Kladis A, Valendjn AJ. Effects of losartan on angiotensin and bradykinin peptides and angiotensin-converting enzyme. J Cardio Pharm. 1995;26:233–240. doi: 10.1097/00005344-199508000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Sampaio WO, Nascimento AAS, Santos RA. Regulation of cardiovascular signaling by kinins and products of similar converting enzyme systems. Systemic and regional hemodynamic effects of angiotensin-(1–7) in rats. Am J Phvsiol Heart Circ Physiol. 2003;284:H1985–H1994. doi: 10.1152/ajpheart.01145.2002. [DOI] [PubMed] [Google Scholar]
- 5.Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin- (1–7) Hypertension. 2003;42:574–579. doi: 10.1161/01.HYP.0000090322.55782.30. [DOI] [PubMed] [Google Scholar]
- 6.Machado RD, Santos RA, Andrade SP. Opposing actions of angiotensins on angiogenesis. Life Sci. 2000;66:67–76. doi: 10.1016/s0024-3205(99)00562-7. [DOI] [PubMed] [Google Scholar]
- 7.Capettini LS, Montecucco R, Mach R, Stergiopulos N, Santos RA, da Silva RF. Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des. 2012;18:963–970. doi: 10.2174/138161212799436593. [DOI] [PubMed] [Google Scholar]
- 8.Chappell MC. Biochemical evaluation of renin-angiotensin system: the good, bad and absolute? Am J Physiol Heart Circ Physiol. 2016;310:H137–H152. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modrall JG, Sadjadi J, Brosnihan KB, Gallagher PE, Yu CH, Bernstein KE, Chappell MC. Depletion of tissue angiotensin-converting enzyme differentially influences the intrarenal and urinary expression of angiotensins. Hypertension. 2004;43:849–853. doi: 10.1161/01.HYP.0000121462.27393.f6. [DOI] [PubMed] [Google Scholar]
- 10.Kohara K, Brosnihan KB, Ferrario CM. Angiotensin-(1–7) in the spontaneously hypertensive rat. Peptides. 1993;14:883–891. doi: 10.1016/0196-9781(93)90063-m. [DOI] [PubMed] [Google Scholar]
- 11.Westwood BM, Chappell MC. Differential metabolism of angiotensin-(I–12) in the circulation and kidney of the mRen2, Lewis congenic rat. Peptides. 2012;35:190–195. doi: 10.1016/j.peptides.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaleyeva L, Gilliam-Davis S, Almeida I, Brosnihan KB, Lindsey SH, Chappell MC. Differential regulation of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis with early onset diabetes. Am J Physiol Renal Physiol. 2012;302:F1374–F1384. doi: 10.1152/ajprenal.00656.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]