

Abstract
In CASP11 we generated protein structure models using simulated ambiguous and unambiguous nuclear Overhauser effect (NOE) restraints with a two stage protocol. Low resolution models were generated guided by the unambiguous restraints using continuous chain folding for alpha and alpha-beta proteins, and iterative annealing for all beta proteins to take advantage of the strand pairing information implicit in the restraints. The Rosetta fragment/model hybridization protocol was then used to recombine and regularize these models, and refine them in the Rosetta full atom energy function guided by both the unambiguous and the ambiguous restraints. Fifteen out of 19 targets were modeled with GDT-TS quality scores greater than 60 for Model 1, significantly improving upon the non-assisted predictions. Our results suggest that atomic level accuracy is achievable using sparse NOE data when there is at least one correctly assigned NOE for every residue.
Keywords: protein structure prediction, Rosetta, NMR, contact assisted, CASP11
Introduction
NMR structure determination and nuclear Overhauser effect spectroscopy (NOESY) data collection has primarily focused on small proteins due to ambiguity of peak assignment for larger proteins. The new “Ts” contactassisted category in CASP11 (11th critical assessment of techniques for protein structure prediction) was designed to test whether structure prediction tools can be used to disambiguate these sparse ambiguous NMR contacts and use them for high-resolution modeling. For this category, simulated sparse NOESY atom pair restraints similar to data available in the initial stages of NMR studies were provided for 19 targets, most lacking PDB homologs and with sizes ranging from 110 to 534 residues.
Rosetta has been demonstrated to speed up and improve NMR structure determination with a range of available data. The basic strategy in all cases is to use the NMR data to guide the search for the lowest energy structure. For small proteins <120 amino acids, incorporation of chemical shift information into the Rosetta fragment selection process drastically improves the Rosetta de novo structure prediction protocol: high accuracy structures (<2 Å RMSD) can quite consistently be determined.1 This method, called CS-Rosetta, has been quite widely used. For proteins with lengths in the 120– 200 residue range which have much larger conformational spaces to be searched, chemical shift information is not sufficient, but with incorporation of residual dipolar coupling information and backbone–backbone NOE's atomic accuracy structures (1.6–4.3 Å RMSD) can again be achieved.2 For proteins up to 400 amino acids, backbone–backbone NOE's are not sufficient, but accurate models (1.1–4.1 Å RMSD) can be generated with the incorporation of methyl–methyl NOEs from I, L, and V residues obtained on selectively labeled samples (where only I, L, and V residues are isotope labeled).3
The data provided for CASP11 differed from that utilized in the above calculations in that simulated NOE data was provided for all residues (except for proline) but no chemical shift information was provided. This modeling scenario is not very realistic given that some NOEs may be quenched for large proteins by short transverse relaxation rates due to the large rotational correlations times, exchange broadening due to internal motions, and other factors causing resonance line-broadening,3 and due to the omission of chemical shift data which is a huge boost to modeling. However, how best to use ambiguous sets of restraints is an interesting modeling challenge. As in CASP10 contact-guided prediction category, we used a two-stage modeling approach involving an initial fold level conformational search followed by refinement using Rosetta with restraints in both stages.4 For all-β proteins, the unambiguous contacts were primarily long-range and between strands, while for all-alpha proteins the unambiguous contacts were typically short-range and within helices, hence different protocols were used in the fold-level conformational search. The results are consistent with our previous structure prediction studies using sparse experimental data,3,5.6 and the modeling accuracy was higher than in previous contact-assisted CASP experiment where only a small number of accurate contacts were provided.
Materials and Methods
Overall procedure: Comparison to CASP10
There were two major differences in the contact information provided for the “Ts” category compared to the previous CASP contact-assisted experiment. First, the number of contacts increased from one contact for every 12 residues to one contact for every residue on average. Second, the provided contacts, simulating NOESY restraints, were ambiguous.
As in the previous CASP contact-assisted experiment, we used a two-stage approach consisting of fold-level modeling and hybridization/refinement4 [Fig. 1(A)]. In the last CASP experiment, significant efforts were made in the fold-level modeling stage to sample the correct fold using both Rosetta de novo continuous chain and broken chain protocols, and for most targets, a large amount of conformational sampling was required. In this experiment, the amount of information provided was sufficient for fold-level modeling with significantly less effort, but denoising and optimal use of the ambiguous data along the modeling path was important. In the first stage, we used only unambiguous data for all-β proteins and included data from peak groups with low ambiguities for the remaining targets. In the following hybridization and refinement stage, starting from these fold-level models, ambiguous data were introduced as restraints through multiple iterations, and eventually all the included data were utilized.
Figure 1.
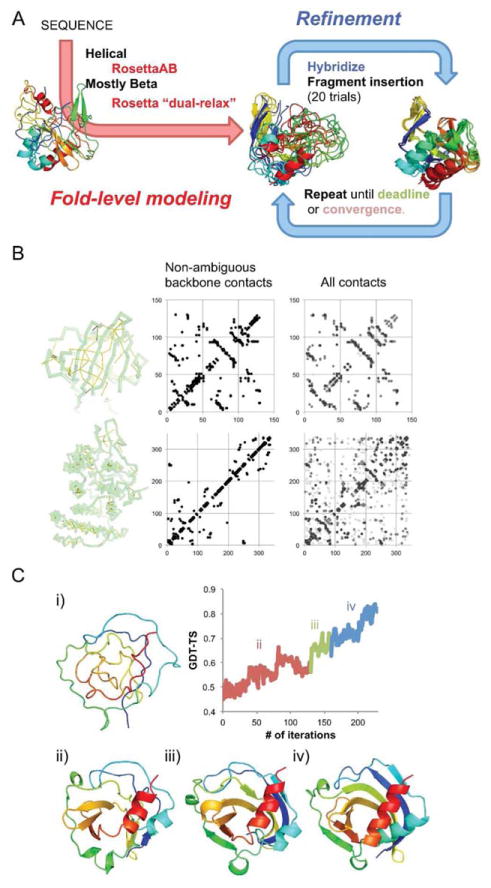
Overview of the methods used in the CASP11 contact-assisted “Ts” category. A: Overall flowchart. B: Contacts provided by organizers for top: all-β protein (Ts763) and bottom: all-α protein (Ts777). Non-ambiguous contacts are represented as yellow bands within the native structure, and non-ambiguous and all contacts are represented as contact maps. C: Change in models as iterative hybridize protocol proceeds for Ts763. (i) starting models (from iterative annealing, GDT-TS5 46.9), (ii) optimization with non-ambiguous restraints (GDT-TS5 56.7), (iii) after maximum ambiguity increased to 5 (GDT-TS5 67.5), and (iv) the final structure optimized with full-ambiguity (GDT-TS579.6).
Fold-level modeling
We used two different fold-level modeling strategies depending on the predicted secondary structure. Unambiguous contacts—NOESY peak groups containing only one contact—generally involved backbone–backbone atom pairs and provided unambiguous information about β-strand pairing [Fig. 1(B)]. For all-β proteins, fold-level accuracy was obtained by iterative annealing in both internal and Cartesian space where at each iteration the strength of the repulsive interactions were rescaled. For this process, we used the Rosetta “dual-relax” proto-col7 which was developed for later stage structure refinement but is also effective in generating reasonable topology level models when there is sufficient contact information. For smaller proteins (<200 length) all the contacts were encoded as ambiguous restraints and used with a strong weight (see below for restraints setup), while for larger proteins only the unambiguous data were used. The dual-relax protocol was run 10 times for each of the server models from the regular “TS” structure prediction category. The top five models with the lowest Rosetta energy plus full-restraint score and without structural knots were selected for the refinement stage. Post-CASP experiments for targets Ts763 and Ts785 revealed that the server-models were not necessary; running the same protocol starting from an extended chain produced similar quality models [Fig. 1(C)].
For proteins with other topologies (all-α, α 1 β, and α/β), the unambiguous contacts provided little information about the relative position of secondary structure elements, and thus more aggressive sampling was necessary. The Rosetta ab initio protocol (RosettaAB) 8,9 was used with 1.5× (number of residues) contacts, weighted by level of ambiguity (see restraint section below) to generate up to 10,000 models. Five models with the lowest Rosetta energy plus full-restraint score were selected for the refinement stage. The restraint weight was assigned such that the restraint score contribution was roughly equal to that of the Rosetta energy.
For some targets, models with fold-level accuracy were already sampled in other prediction categories (the “Tc” contact-assisted category and the “TS” category for which co-evolution information was available), therefore we skipped the fold-level modeling stage and simply selected models for the refinement stage using the same criteria described above. This was done for the following targets: Ts824 (from T0824), Ts835 (from T0835), Ts806 (from T0806), Ts767 (from Tc767), and Ts812 (from Tc812).
Refinement of fold-level models
Once fold-level models were selected, they were recombined and refined using an iterative version of the Rosetta hybridize protocol (RosettaCM) originally developed for comparative modeling.10 Structural optimization was accomplished by recombining the secondary structure segments from each of the input models, together with fragment insertion for added diversity. In each iteration, 20 models were produced independently and the top 4 were selected based on the sum of the Rosetta energy and restraint score for the next iteration. The procedure was repeated until structural convergence was reached. After convergence, the maximum restraint ambiguity (see below for ambiguous restraints) was increased and the procedure was continued until all contacts were used [Fig. 1(C)]. Initially, only unambiguous contacts were used, and as the modeling progressed the number of ambiguous contacts was increased. In the final iterations, all contacts were used. If there were still violated contacts (restraint score greater than 0.000), the restraint weight was increased by 10-fold and additional iterations were carried out.
At this stage, the PDB was scanned for proteins with similar structure (TM-align11 > 0.5). If hits were found (in CASP11 this only occurred in one case, Ts785), alignments were created based on the structural superpositions and used as additional starting templates (Fig. 2). For Ts785, this procedure improved the GDT-TS from 70.1 to 78.4 and Rosetta all atom energy from 2172.5 to 2201.3. This is consistent with our previous observation that when homology derived information is incorporated into Rosetta structure prediction calculations and lower energies are achieved, the models generated are more accurate than those obtained in unrestrained calculations.12
Figure 2.
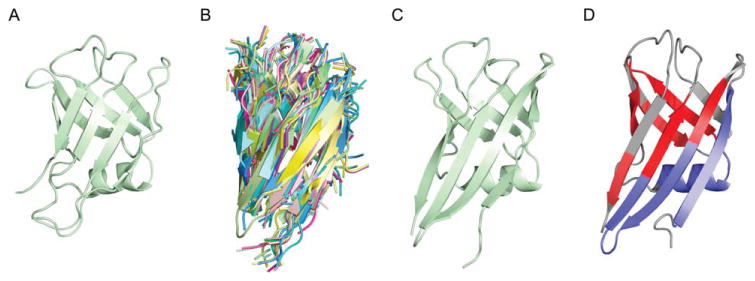
Structural homologs used to refine Ts785. A: Converged model (GDT-TS 70.1) before the addition of structural homologs. B: The top 20 structural homologs detected by TMalign.11 C: Converged model (GDT-TS 78.4) after the addition of structural homologs. D: The native structure colored by PSIPRED secondary structure prediction. The colors are blue for helix, red for sheet, and gray for coil.
Following convergence, models were rescored using the complete set of restraints, and the models with the lowest Rosetta all atom energy and restraint scores were analyzed. If these top models varied significantly, they were clustered, and the five submitted models were selected from the cluster representatives; the model with the best Rosetta energy plus restraint fit was assigned as Model 1. If the top scoring models converged, the five submitted models were selected based on the sum of Rosetta energy and fit to restraint sets with different levels of ambiguity. The best scoring model using all contacts was assigned as Model 1.
Modeling of multi-domain proteins
Proteins larger than 200 amino acids in length were manually parsed into multiple domains guided by the provided contact maps (Ts814, Ts777, Ts826, Ts794). For these targets, domains were modeled and refined separately, and then assembled into full length models. Guided by the provided inter-domain contacts, the global orientation was roughly sampled manually and then refined with local rigid-body docking using Rosetta-Dock.13 Little effort was applied to refining the fit between domains due to time constraints and since it was assumed that targets would be evaluated at the domain level as in CASP10.
Conversion of experimental information to restraints
Multiple restraint sets were generated for different parts of the modeling protocol. The unambiguous contacts were used as strong “bounded” restraints.4 All unambiguous contacts (backbone–backbone, backbone–sidechain, and sidechain–sidechain) were used as restraints for the iterative annealing protocol and the final stages of RosettaAB and RosettaCM. Only the unambiguous contacts between backbone atoms were used for the initial “centroid” (coarse-grained representation in Rosetta) sampling stages of RosettaAB and RosettaCM.
For the “centroid” stages, ambiguous contacts were incorporated as sigmoidal restraints between Cβ–Cβ atoms (Cα in the case of glycine). A contact score for each residue pair was set proportional to the contact confidence value (conf) provided by the CASP organizers and inversely proportional to the ambiguity level. Given that at least one contact is correct in each group of contacts, the probability that any given contact is correct is simply conf/sum(conf_of_group). The final weight for each pair of residues was the sum of this ratio over all groups. Only the highest scoring 3L/2 (L is the sequence length) contacts were used, with sequence separation of three or higher. Contact distances were set to the largest Cβ–Cβ distance found in the PDB for the residue pair when any of the heavy atoms was within 10 Å. For the all-atom stages, ambiguous contacts were converted into ambiguous-bounded restraints as previously described.3 For the latter ambiguous restraints, groups of ambiguous contacts were discarded if the residue sequence separation was <6 to prevent local geometric distortions.
Computational cost
Fold-level modeling of all-β targets using iterative annealing required minutes per run. Fold-level modeling for other topologies required sampling around 1000 to 10,000 models using RosettaAB, which translates to roughly 500 to 5000 core hours for a 200 residue protein. One iteration of the hybridize protocol in the refinement stage took around 20 core hours for a 200 residue protein, and 30–100 iterations, were carried out to reach convergence (or before the submission deadline).
Results
Approach
We found that with the rich contact data provided in this experiment, Rosetta protocols developed for other applications turned out to be surprisingly effective. First, to assemble all beta topologies from the unambiguous backbone–backbone NOEs, the iterative annealing with rescaling of the repulsive interaction followed by quasi-Newton minimization carried out by the Rosetta dual-relax protocol developed for late stage structure refinement proved very effective in generating models with the correct overall topology. Second, the RosettaCM hybridization protocol was found very effective in recombining and regularizing (by fragment insertion) the models generated by iterative annealing, which generally had very poor local geometry and often little regular secondary structure [Fig. 1(C)]. For all targets, all submitted models made all the unambiguous restraints and at least one restraint in each group of ambiguous restraints. The fraction of contacts made in the native structure is nearly identical to the fraction of contacts made in the submitted model (See Supporting Information Table I).
More accurate models were built for the majority of targets
The first submitted models for 15 out of 19 targets have GDT-TS quality scores >60 which is quite encouraging. Models with GDT-TS >45 typically have the native fold (TM-score > 0.5).14 Examples of models with high accuracy are shown in Figure 3(A) (Ts812) and Figure 3(B) (Ts832). The average GDT-TS for Model 1 is 65.3, which is similar to the average of 68.6 for the best among the five submitted models. The fact that even the worst models have similar accuracy (58.4 average GDT-TS) highlights the convergence of all five submitted models; the average similarity among the five models is 0.82 (60.10) in TM-score.11
Figure 3.
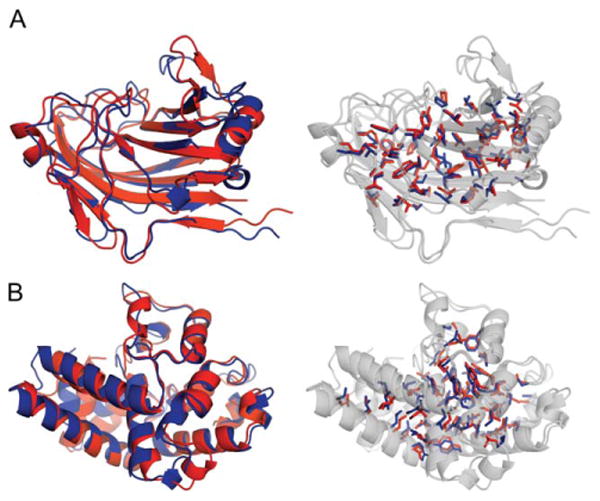
Examples of predictions with accurate side-chain placements. The core side-chains of the native (blue) and submitted model (red) are highlighted. A: Ts812 Model 4 and (B) Ts832 Model 1.
Dependence on secondary structure
The targets consist of 7 all-α, 7 all-β, and 5 α/β or a+β proteins. Targets with more β content were better predicted; the average GDT-TS values are 63.5 (±13.0), 68.7 (±10.6), and 73.6 (±8.0), for all-α, α/β or α+β, and all-β proteins, respectively. Four all-β targets had highly accurate predictions (GDT-TS >75) compared to only one among the all-α targets. In several cases, top predictions were made even for all-α targets [Fig. 4(C, D)] and examples are illustrated in Figure 5(A) (Ts777) and Figure 5(B) (Ts827).
Figure 4.
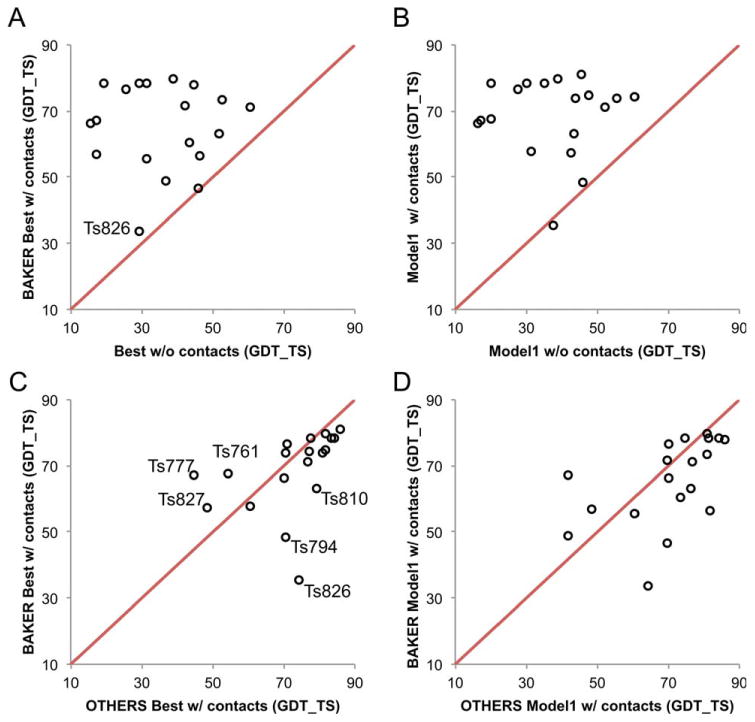
Overview of Baker group models in the Ts category in CASP11. Contact assisted predictions are compared to (A,B) the best non-assisted predictions and (C,D) the best Ts submissions by others. The comparisons in (A) and (C) are between the best of the five submitted models, and in (B) and (D), between the first submitted models.
Figure 5.
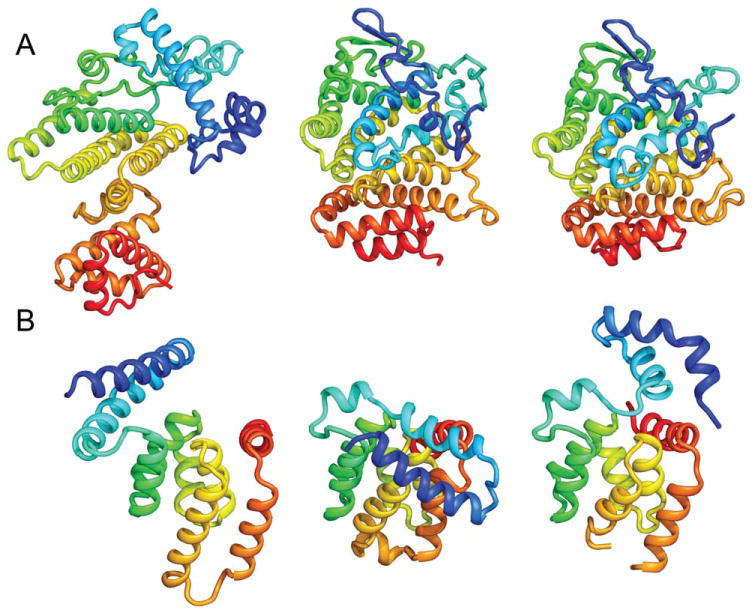
High-accuracy predictions for all alpha proteins. A: Ts777 and (B) Ts827. In each panel, best prediction without information (left), best BAKER Ts prediction (middle), and native (right) are shown, respectively. B: Although our model was the best among all Ts predictions, contact information at the N-terminal region was minimal and our model was more compact than the native structure.
What went wrong: Model bias from fragment library and domain orientation
The best predictions for four targets had GDT-TS values <60, Ts794 (48.6), Ts804 (57.6), Ts826 (35.3), and Ts827 (57.2). The target with lowest model quality, Ts826 [Fig. 6(A)], had fragment quality issues. The success of both fold-level modeling and refinement highly depends on fragment accuracy, which is strongly dependent on the predicted secondary structure. For the transmembrane domain of Ts826, PSIPRED15 over-predicted the helical content, which prevented the modeling methods from sampling critical breaks in helices. For the successfully modeled Ts785, large stretches of beta-sheet were predicted as alpha-helix [Fig. 2(D)] and required the addition of structural homologs to correct for the incorrect fragments [Fig. 2(B)].
Figure 6.
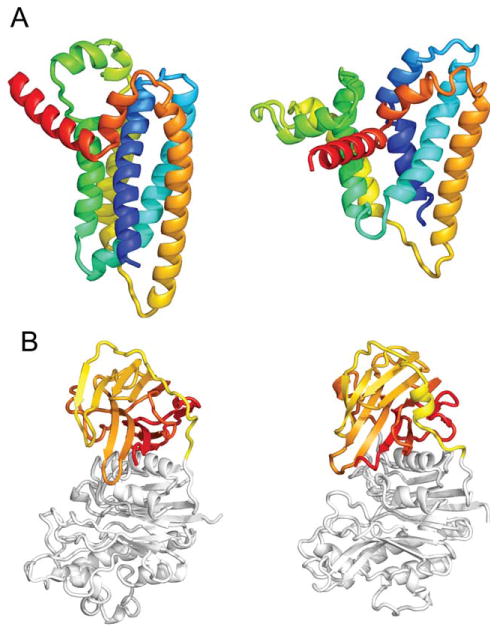
Targets with modeling problems. A: Ts826, biggest failure due to fragment limitation. Best BAKER Ts Prediction (left), proxy for native (right, Tc prediction with GDT_TS 80.47, native structure not deposited yet), (B) Ts794, domain orientation issues. Best BAKER Ts Prediction (left) and native (right).
The remaining targets with relatively low model quality (Ts804, Ts827, Ts794) had domain orientation issues. Although the domain interfaces were roughly correct, the global structures had low GDT-TS values due to lever-arm effects and inaccurate assembly; Ts794 is a good example of this [Fig. 6(B)]. The GDT-TS values for each domain exceed 60 except for Ts794-D1 (57.7). Both Ts804 and Ts827 have small domains that minimally interact with the rest of the protein, and did not converge among our submitted models. The domain orientation was predicted correctly for multi-domain targets Ts777 and Ts814; for these targets, sufficient interdomain contacts were provided for the large interfaces.
Comparison to non-assisted predictions
The extent to which the simulated NMR data improved modeling can be determined by comparing the contact-assisted results with the non-assisted predictions. The comparison between the best non-assisted (TS) predictions among all groups and our assisted (Ts) submissions is displayed in Figure 4. The average GDT-TS for the best predictions improves from 37.5 to 68.6 with the use of contact information [Fig. 4(A)]. Similar results are observed for the Model 1 submissions [Fig. 4(B)]. All targets improve with the exception of T0826, which had fragment quality issues as explained above. Nine out of 19 targets improved significantly (ΔGDT-TS > 30). The improvement of 31.1 GDT-TS units is significantly higher than in the previous CASP contact-assisted experiment, where the improvement was 13.5 GDT-TS over the best non-assisted predictions on average.4 Although there are obvious differences in the targets, contact information, and modeling methods, these results suggest that even with ambiguous data, the increased amount of contact information provided in CASP11 compared to CASP10 led to significant improvements in GDT-TS.
Discussion
Our results show that the amount of information provided in the NMR category was sufficient to generate models with the correct folds. Unambiguous information brought critical clues to the folds of target proteins, in particular for all-β proteins. Ambiguous information helped further refine the structures, yielding significant improvements over the models generated using only the unambiguous information [an example is illustrated in Fig. 1(C)]. The contribution from ambiguous information is evident in the consistent successful selection of one of the best structures as Model 1 (which contrasts with our failure to properly rank models in other prediction categories).
We have several suggestions for future CASP experiments using simulated NMR data. First, the amount of contact information provided was in many cases so large that the prediction problem was not very challenging. Realistically, for proteins >200 residues spin diffusion or other factors3 would likely limit the amount of NOE information for uniformly labeled samples. An optimal amount of information would be somewhere between the very low level (one contact for every 12 residues) provided in CASP10 and the high level (one contact every residue) provided in CASP11. Second, since in any real world NMR structure determination problem chemical shift data and possibly residual dipolar coupling (RDC) data would be available, it would be useful to provide simulated data of these types as well. Chemical shifts and RDC data would largely resolve the fragment quality and domain orientation issues mentioned above. Finally, the simulated NMR data was provided in this experiment with the additional knowledge that there was only a single conformational state and that the targets were monomers, and our modeling methods took advantage of this: we were able to use strong restraints (bounded function) for unambiguous contacts because we could assume the contacts were accurate. Without this assumption, some of the contacts may have been incompatible with each other, and weak restraints (sigmoidal function) would have to have been used which would increase modeling difficulty. It may be useful in subsequent CASP experiments to introduce further ambiguity to better replicate the real world situation. Finally, given the growing importance of protein structure determination using cryo-electron microscopy (cryo-EM),16 prediction challenges with simulated density maps would be useful in future CASP experiments.
Supplementary Material
Acknowledgments
Authors thank Keith Laidig and Darwin Alonso for developing the computational and network infrastructure and Rosetta@home participants for providing the computing resources necessary for this work. Authors would also like to thank the CASP11 organizers, the structural biologists who generously provided targets and Binchen Mao and Brian Koepnick for helping them interpret the simulated NOE data.
Grant sponsor: National Institute for Health Research; Grant number: R01GM092802 (S.O., H.P., Y.L., R.W. and D.B.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raman S, Lange OF, Rossi P, Tyka M, Wang X, Aramini J, Liu G, Ramelot TA, Eletsky A, Szyperski T, Kennedy MA, Prestegard J, Montelione GT, Baker D. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lange OF, Rossi P, Sgourakis NG, Song Y, Lee HW, Aramini JM, Ertekin A, Xiao R, Acton TB, Montelione GT, Baker D. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc Natl Acad Sci USA. 2012;109:10873–10878. doi: 10.1073/pnas.1203013109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim DE, Dimaio F, Yu-Ruei Wang R, Song Y, Baker D. One contact for every twelve residues allows robust and accurate topology-level protein structure modeling. Proteins. 2014;82(Suppl 2):208–218. doi: 10.1002/prot.24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, Kim BH, Das R, Grishin NV, Baker D. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiMaio F, Terwilliger TC, Read RJ, Wlodawer A, Oberdorfer G, Wagner U, Valkov E, Alon A, Fass D, Axelrod HL, Das D, Vorobiev SM, Iwaï H, Pokkuluri PR, Baker D. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature. 2011;473:540–543. doi: 10.1038/nature09964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23:47–55. doi: 10.1002/pro.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simons KT, Ruczinski I, Kooperberg C, Fox BA, Bystroff C, Baker D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins. 1999;34:82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 9.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 10.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson JM, Sgourakis NG, Liu G, Rossi P, Tang Y, Mills JL, Szyperski T, Montelione GT, Baker D. Accurate protein structure modeling using sparse NMR data and homologous structure information. Proc Natl Acad Sci USA. 2012;109:9875–9880. doi: 10.1073/pnas.1202485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 14.J X, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 2010;26:889–895. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 16.Bai XC, McMullan G, Scheres SH. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015;40:49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.