

Abstract
Gravin (AKAP12) is an A-kinase-anchoring-protein that scaffolds protein kinase A (PKA), β2-adrenergic receptor (β2-AR), protein phosphatase 2B and protein kinase C. Gravin facilitates β2-AR-dependent signal transduction through PKA to modulate cardiac excitation-contraction coupling and its removal positively affects cardiac contraction. Trabeculae from the right ventricles of gravin mutant (gravin-t/t) mice were employed for force determination. Simultaneously, corresponding intracellular Ca2+ transient ([Ca2+]i) were measured. Twitch force (Tf)-interval relationship, [Ca2+]i-interval relationship, and the rate of decay of post-extrasysolic potentiation (Rf) were also obtained. Western blot analysis were performed to correlate sarcomeric protein expression with alterations in calcium cycling between the WT and gravin-t/t hearts. Gravin-t/t muscles had similar developed force compared to WT muscles despite having lower [Ca2+]i at any given external Ca2+ concentration ([Ca2+]o). The time to peak force and peak [Ca2+]i were slower and the time to 75% relaxation was significantly prolonged in gravin-t/t muscles. Both Tf-interval and [Ca2+]i-interval relations were depressed in gravin-t/t muscles. Rf, however, did not change. Furthermore, Western blot analysis revealed decreased ryanodine receptor (RyR2) phosphorylation in gravin-t/t hearts. Gravin-t/t cardiac muscle exhibits increased force development in responsiveness to Ca2+. The Ca2+ cycling across the SR appears to be unaltered in gravin-t/t muscle. Our study suggests that gravin is an important component of cardiac contraction regulation via increasing myofilament sensitivity to calcium. Further elucidation of the mechanism can provide insights to role of gravin if any in the pathophysiology of impaired contractility.
Keywords: intracellular calcium, force development, contraction, gravin, cardiac muscle, trabeculae
1. Introduction
A-kinase anchoring proteins (AKAPs) are scaffolding proteins that complex protein kinase A (PKA) along with other signaling molecules and target them to specific subcellular locations thereby increasing the specificity and diversity of cellular signaling (Colledge and Scott, 1999). So far fourteen AKAPs have been identified in the heart and studies have shown that disruption of AKAP/PKA interactions by the synthetic peptide Ht31 resulted in the altered distribution of PKA as well as augmentation of cell shortening in response to β-adrenergic receptor (β-AR) signaling. Interestingly, these changes were associated with a normal rate of Ca2+ cycling, normal intracellular Ca2+ transient amplitude and reduced PKA dependent phosphorylation of cardiac troponin I (cTnI) and cardiac myosin binding protein C (cMyBPC) (Fink et al., 2001; McConnell et al., 2009).
Gravin, also known as AKAP12, AKAP250 or SSeCKS, is highly expressed in the heart and targets a number of signaling molecules including PKA, protein kinase C (PKC), and protein phosphatase 2B (PP2B) to the β2-AR (Fan et al., 2001). Additionally, it has been reported that β-AR agonist treatment strengthens the gravin/β2-AR interaction. Agonist stimulation of β-ARs enhances cardiac function, mainly through PKA-dependent phosphorylation of key components of the excitation-contraction (EC) coupling mechanism (Katz and Lorell, 2000). In addition to facilitating PKA-dependent substrate phosphorylation, gravin also plays an important role in the desensitization/resensitization cycle of the β2-AR (Shih et al., 1999). Moreover knockdown of gravin expression has been shown to abrogate the recruitment of G-protein coupled receptor kinase 2 and β-arrestin, key proteins involved in β-AR desensitization (Lin et al., 2000; Shih et al., 1999). Additionally, we have recently identified that both in the presence and absence of acute β-AR stimulation cardiac function is enhanced in mutant gravin-t/t mice (Guillory et al., 2013). This increased cardiac function in gravin-t/t mice was coupled with reduced β2-AR phosphorylation. Gravin-t/t mice also exhibited increased phosphorylation of cMyBPC at Ser-273 (Guillory et al., 2013), the key site for modulating cardiac function (Guggilam et al., 2013; Jin et al., 2013a) with no change in the phosphorylation levels of phospholamban (PLB), cTnI and two alternative phosphorylation sites of cMyBPC at Ser-282 and Ser-302 compared to the WT (Guillory et al., 2013). These data suggests that that absence of gravin enhances the cardiac contractility via the modulation of the β-AR pathway.
In this study, we measured force development and intracellular Ca2+ ([Ca2+]i) simultaneously in isolated intact cardiac trabeculae from gravin mutant (gravin-t/t) mice. Our objective was to study cardiac contraction and intracellular Ca2+ cycling in gravin-t/t mutant mice myocardium to better understand the functional importance of gravin and gain further insights into the molecular mechanisms for the enhanced cardiac function in gravin-t/t mice. Our results show that gravin-t/t muscles exhibited increased myofilament Ca2+ responsiveness while maintaining their ability to reconstitute in terms of force development and Ca2+ release from the sarcoplasmic reticulum (SR); suggesting that gravin also functions as regulator of myofilament Ca2+ sensitivity in addition to serving as a scaffolding protein and maybe involved in the modulation of the cardiac EC Coupling.
2. Materials and methods
2.1 Gravin Mutant Mice
Gravin mutant mice were produced using gene trap technology to remove the Akap12 (gravin) gene (NM_031185) (Guillory et al., 2013). In brief, embryonic stem (ES) cells containing the gravin trapped vector were obtained from BayGenomics, microinjected into normal blastocysts and surgically implanted into foster mice at the University of Maryland School of Medicine Transgenic / Knockout Core Facility. The mutant gravin allele in these mice contained the gene trap vector that included a splice acceptor site immediately following exon 2, which causes early termination of the transcript. This mutant allele removes the remaining two 3′ exons (exon 3 and exon 4) and is predicted to encode from the amino-terminal to amino acid residue 89 of the 1684 residues from wild-type (WT) gravin, representing less than 6% of the full-length protein. Exons 1 and 2 of the mutant allele are expressed at WT levels. Protein chemical studies have demonstrated that this mutant allele produces less than 10% of the normal amount of gravin. Homozygous mice lacking functional gravin protein (designated gravin-t/t; where t refers to truncation) do not express the critical region required for β2-AR, PKA or PKC binding that are encoded by exon 3. WT mice and homozygous mutant gravin mice (gravin-t/t) are bred on a C57bl/6 background. For experimental studies, male gravin mutant mice aged 10-12 weeks were compared with WT mice.
Research animals were generated and used in compliance with federal, state and local laws and institutional regulations. Specifically, the Institutional Animal Care and Use Committee (IACUC) and ethics committee at the University of Houston (UH) and the Johns Hopkins University (JHU) approved all animal studies. Also, animal care was provided for in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited animal barrier facilities at UH and JHU. Animals were maintained and these experiments were performed in accordance with the guidelines and ethical standards laid down in the 1996 National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and in the 1964 Declaration of Helsinki and its later amendments. All authors of this report gave their informed consent prior to their inclusion in the study.
2.2 Isolation of Intact Trabecular Muscles
Trabeculae from the right ventricle of the hearts were dissected and mounted between a force transducer and a motor arm in a bath with a volume of 300 μl. The muscles were superfused with Krebs-Henseleit solution [K-H, (in mM) NaCl 120, NaHCO3 20, KCl 5, MgCl2 1.2, glucose 10, 0.5 CaCl2 (gassed with 95% O2 and 5% CO2 mixture, pH 7.35–7.45)] at a rate of ∼10 ml/min and stimulated at 0.5Hz at room temperature (21–22°C). Force was measured with a force transducer system (SI, Germany) and expressed in millinewtons per square millimeter (mN/mm2). The muscles were stretched to a length at which unstimulated force reached ∼15% of total twitch force. This resting length corresponded to a sarcomere length of 2.2-2.3 μm (Gao et al., 1995) and was maintained throughout the experiments. Extracellular Ca ([Ca2+]o) was maintained at 0.5 mM during dissection and fura-2 loading (see below) and was increased to 1 mM during the execution of the experimental protocols.
2.3 Measurements of Intracellular Ca2+ ([Ca2+]i) from Trabecular Muscles
[Ca2+]i was measured with the free acid form of fura-2 as described in our previous studies (Gao et al., 1998; Gao et al., 1999; Tan et al., 2009). Fura-2 potassium salt was microinjected iontophoretically into one cell and allowed to spread throughout the whole muscle (via gap junctions). The tip of the electrode (∼0.2μm in diameter) was filled with fura-2 salt (1 mM), and the remainder of the electrode was filled with 150 mM KCl. After a successful impalement into a superficial cell in the unstimulated muscle, a hyperpolarizing current of 5–10 nA was passed continuously for less than 10 min. As previously established, the loading does not affect force development. The epifluorescence of fura-2 was measured by exciting at 380 and 340 nm. The fluorescent light collected at 510 nm by a photomultiplier tube (R1527, Hamamatsu). [Ca2+]i was given by the following equation (after subtraction of the autofluorescence):
| (1) |
where R is the observed ratio of fluorescence (340/380), K'd is the apparent dissociation constant, Rmax is the ratio of 340 nm / 380 nm at saturating [Ca2+], and Rmin is the ratio of 340 nm / 380 nm at zero [Ca2+]i. The values of K'd (=4.0), Rmax (=8.45), and Rmin (0.32) were determined by in vivo calibrations, as described previously (Gao et al., 1998).
2.4 Immunoblotting
Immunoblot analysis was carried out as previously described using antibodies for GAPDH (Cell Signaling), SERCA2A (Cell Signaling), protein phosphatase inhibitor 1 (Santa Cruz Biotechnology), calsequestrin 2 (Pierce antibodies), L-type calcium channel (Santa Cruz Biotechnology) and sodium / calcium exchanger (Thermo Fisher Scientific) (McConnell et al., 2009). Crude heart homogenates or cytosolic fractions were resolved by SDS-PAGE gradient (4–12% Bis-Tris) gels, and transferred to polyvinylidine difluoride membranes. Blots were then incubated overnight at 4°C with primary antibodies. The blots were washed three times with TBS containing 0.1% Tween 20, and then probed with appropriate secondary antibodies (Cell Signaling). Blots were developed by an enhanced chemiluminescence kit (Pierce). NIH Image J was used for densitometric analysis of the immunoblots. In experiments showing the response to acute isoproterenol treatment, measurements by Western blot were obtained with and without isoproterenol infusion via the jugular vein (10 μg/g/min × 3 min), a β-AR agonist, or ascorbic acid (0.002%), a vehicle control.
2.5 Statistical Analysis
These data were analyzed by Student's t-test, one-way ANOVA and repeated two-way multivariate ANOVA (MANOVA) (Systat 10.2 or GraphPad Prism 5). A P-value <0.05 was considered to indicate significant differences between groups. Unless otherwise indicated, data are expressed as mean ± S.E.M.
3. Results
3.1 Altered Response of Gravin-t/t Muscles to [Ca2+]o
We first investigated the effect of changing [Ca2+]o on force development and [Ca2+]i transient in gravin-t/t muscles. Fig. 1 shows raw tracings of force development and [Ca2+]i from representative muscles of WT and gravin-t/t hearts. Within the range of [Ca2+]o tested, force development was similar between gravin-t/t muscles and WT muscles. Both stress and [Ca2+]i amplitude increased in a dose-dependent manner with increased [Ca2+]o in WT and gravin-t/t muscles (Fig. 2A-B). However, [Ca2+]i amplitudes were significantly lower in the gravin-t/t muscles at any given [Ca2+]o, as compared to WT muscles. These findings were consistent with a lower SR Ca2+ content and/or a reduced rate of Ca2+ release from the SR. The dynamics of contractions were studied by measuring the time to peak stress and [Ca2+]i, as well as the time to 50% relaxation of stress and [Ca2+]i. Both the time to peak stress and [Ca2+]i were prolonged in gravin-t/t muscles. Although there were no changes in 50% relaxation, (Fig. 3A-D), the initial relaxation (i.e. from peak to 75% of peak) was significantly slowed gravin-t/t muscles without changes in [Ca2+]i relaxation times (Fig. 3E&F). The slowing of relaxation in force suggests increased myofilament Ca2+ responsiveness in gravin-t/t muscles.
Fig. 1.
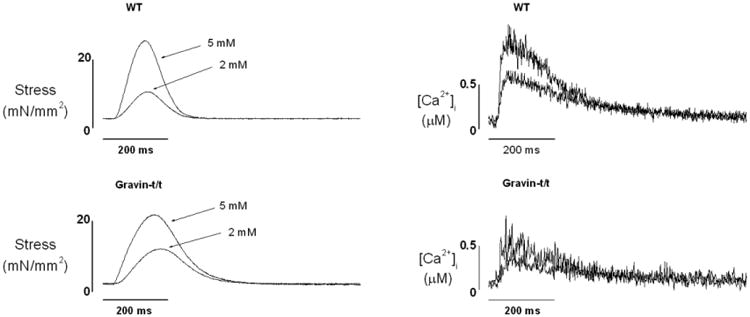
Raw tracings of stress development (left) and [Ca2+]i transient (right) from WT (upper panels) and gravin-t/t (lower panels) trabecular muscles at two [Ca2+]o (2 and 5 mM). Note that [Ca2+]i transients were lower in gravin-t/t muscles while stress remained unchanged.
Fig. 2.
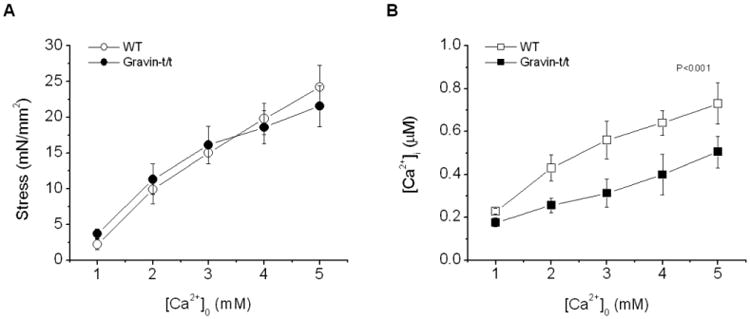
Pooled data of stress (A) and [Ca2+]i transients (B) at varied [Ca2+]o (1-5 mM) from WT and gravin-t/t muscles. Both stress and [Ca2+]i transient increased as [Ca2+]o was raised. Stress was similar in both groups of muscles but [Ca2+]i transients remained significantly lower in gravin-t/t muscles (P<0.001; repeated MANOVA, n=8 in each group).
Fig. 3.
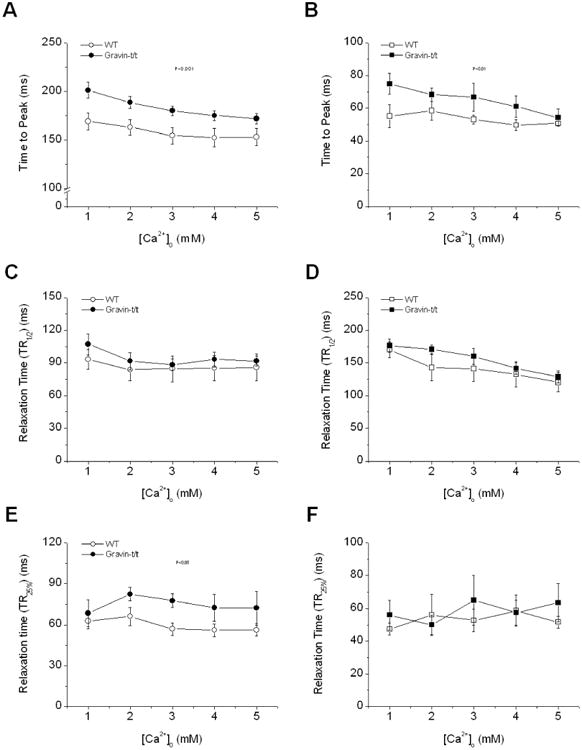
Dynamics of stress and [Ca2+]i transient of trabecular muscles from WT and gravin-t/t mice. The time to peak stress (A) and [Ca2+]i (B) was prolonged in gravin-t/t muscles (as determined by MANOVA) whereas the time to 50% relaxation of stress (C) and [Ca2+]i (D) was not different between WT and gravin-t/t muscles. However, the initial relaxation time (time to 75% of peak) in twitch (E) was significantly slowed in gravin-t/t muscles (P<0.01 repeated MANOVA) whereas the initial relaxation time (time to 75% of peak) in [Ca2+]i (F) was not different between WT and gravin-t/t muscle. (n=8 in each group)
The maintained force development despite low [Ca2+]i indicates that gravin-t/t muscles may have increased response to Ca2+. To further demonstrate this, the peak stress was plotted versus the peak [Ca2+]i (Fig. 4A). If we assume a linear relationship between peak force and peak [Ca2+]i in both groups of muscles, this relationship is shifted leftward with a higher slope in gravin-t/t muscles. To check this point further, we plotted [Ca2+]i vs. force over the entire trajectory of the twitch (i.e. phase-plane plot) (Fig. 4B). As we have pointed out previously (Gao et al., 1995) (Gao et al., 1998), in the phase-plane trajectory, the relaxation phase is determined by the intrinsic properties of the myofilaments and not Ca2+ removal from the cytosol. Thus, a leftward shift of the relaxation trajectory indicates increased myofilament Ca2+ responsiveness. To quantify the shift, we plotted the [Ca2+]i at which 50% relaxation occurred from the phase-plane analysis (Fig. 4C). Gravin-t/t muscles clearly had lower [Ca2+]i levels indicating leftward shift of the relaxation trajectory.
Fig. 4.
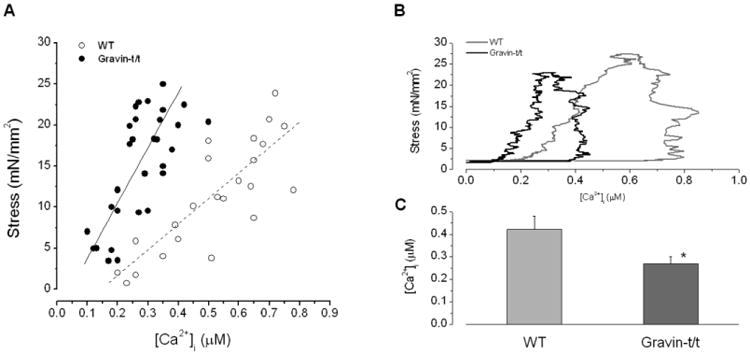
(A) Peak stress and peak [Ca2+]i transient relationships of trabecular muscles from WT and gravin-t/t mice (pooled data, n=8 in each group). In WT, force = 41 [Ca2+]i – 8.22; in gravin-t/t, force = 67 [Ca2+]i – 7.08. The relationship is also shifted leftward in gravin-t/t muscles. These two relationships are significantly different (P<0.001; MANOVA). (B) Representative phase-plane plots for stress vs. [Ca2+]i transient from WT and gravin-t/t muscles. The phase-plane plot is stress vs. [Ca2+]i transient point-to-point over the entire trajectory of the twitch. Note the phase-plane plot of gravin-t/t muscle is shifted to the left. (C) Comparison of [Ca2+]i at which stress relaxed to 50%. *P<0.05 vs. WT; n=6, [Ca2+]o = 1.0 mM.
3.2 Tf- Interval and [Ca2+]i - Interval Relations of Twitches in Gravin-t/t Muscles
The decreased [Ca2+]i transient seen in the gravin-t/t trabeculae (Fig. 1) could be the result of decreased Ca2+ release from the SR as a result of (1) decreased SR Ca2+ content or availability of Ca2+ for release or (2) changes in the kinetics of SR Ca2+ release channels. These possibilities can be tested conveniently in intact electrically stimulated cardiac trabecula based on model of Ca2+ transport as demonstrated previously. (Banijamali et al., 1991; Schouten et al., 1987). In this model, the amplitudes of the test stimulation is plotted against the test intervals (inset of the Fig. 5), yielding a force-test interval (or [Ca2+]i-test interval) relationship. The amplitude of force (and [Ca2+]i) is proportional to Ca2+ released from the SR, which depends on the amount of Ca2+ in the SR, Ca2+ release channels, and Ca2+ extrusion. Fig. 5A and 5B show the results of these experiments. Overall, amplitudes of [Ca2+]i were smaller at all test intervals resulting in lower force. The decreases in force were less dramatic due to increased myofilament Ca2+ sensitivity. The decreases in amplitudes of force and [Ca2+]i, especially the maximums at 60 s test intervals, indicate decreased SR Ca2+ load. It is noticeable that the recovery of force and [Ca2+]i to baseline levels (i.e. at baseline stimulation rate of 0.5 Hz) was rapid in both groups of muscles, suggesting fast rate of recovery of Ca2+ release processes. Together, these results show that, while the amount of Ca2+ load in the SR may be reduced, the rate of recovery of Ca2+ release from the SR is not altered in gravin-t/t muscles.
Fig. 5.
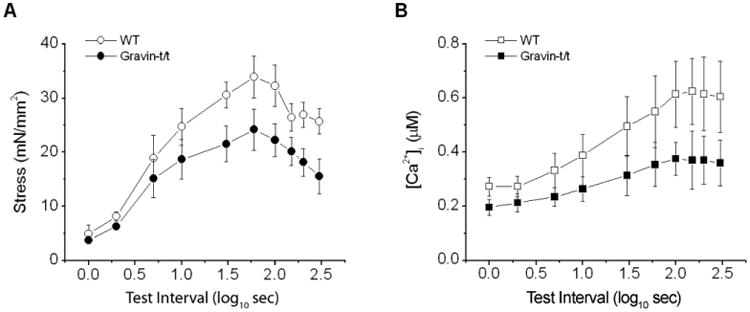
Stress-interval (A) and [Ca2+]i transient-interval (B) relations of trabecular muscles from WT and gravin-t/t mice. The test (rest) intervals are shown on logarithmic scale for the sake of clarity. At short test intervals (<5 s), stress recovered quickly, followed by a slower rise in both groups of muscles. At intervals greater than 60 s, stress started to decline. Gravin-t/t muscle also had lower stress at rest intervals greater than 10 s. [Ca2+]i transients exhibited similar pattern as test interval changed (right), with [Ca2+]i transient being lower in gravin-t/t muscles. [Ca2+]o = 1.0 mM. Note that rate of recovery of [Ca2+]i transient is similar at shorter test intervals in both groups. But the increase (or potentiation) is much slower at test intervals great than 10 s and the total increase is also lower in gravin-t/t muscles. See text for detailed discussion. [Ca2+]o = 1.0 mM.
3.3 Post-extrasystolic Potentiation, or Recirculated Fraction of Ca2+, is not Altered in Gravin-t/t Muscles
In muscles that contracted at a regular rate with stimulation at 0.2 Hz, introduction of a train of extra-stimuli, which causes extrasystoli, results in post-extrasystolic potentiations of the next beats after resuming at 0.2 Hz (inset of Fig. 6). The level of post-extrasystolic potentiation decays to the baseline in an exponential manner and is associated with decay of cytosolic free Ca2+ (Wier and Yue, 1986). The rate of decay, which can be determined from the linear relation between the amplitude of the contractions (n+1) and the amplitude of the preceding contractions (n), is commonly referred to as recirculation fraction (Rf) of Ca2+ (Banijamali et al., 1991; ter Keurs et al., 1990; Ward et al.), which indicates the fraction of Ca2+ taken up by the SR after the preceding release. We determined Rf from both peak stress decay and peak [Ca2+]i transient decay to investigate whether Ca2+ movements across the SR are altered in gravin-t/t muscles (Fig. 6). Rf, determined from either the relationships between the twitches or [Ca2+]i transients, was similar in both groups of muscles, WT and gravin-t/t, suggesting that the fraction of Ca2+ uptake by the SR is the same, thus SR Ca2+ uptake is not altered in the gravin-t/t heart.
Fig. 6.
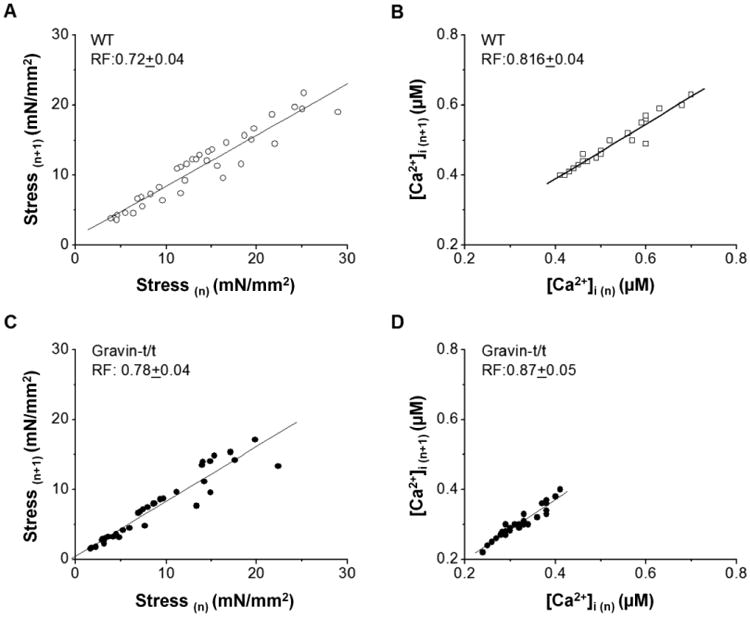
Recirculation fraction (Rf) of Ca2+ in WT (n=5) and gravin-t/t muscles (n=7). Rf was determined from the slopes of the linear relationships between consecutive twitches and their corresponding [Ca2+]i transients at a baseline stimulation rate of 0.5 Hz after potentiation of both stress development and [Ca2+]i transient. Stress (n) and Stress (n+1) are two consecutive twitches, and [Ca2+]i (n) and [Ca2+]i (n+1) refer two consecutive [Ca2+]i transients. [Ca2+]o = 1.0 mM.
3.4 Ca2+ Related Protein Expression
The Rf seen in Fig. 6 indicated that Ca2+ uptake by the SR was unaltered in gravin-t/t muscles. Thus, we measured SERCA and IPP-1 phosphorylation to verify this assumption. SERCA is the main pump responsible for the reuptake of Ca2+ from the cytosol into the sarcoplasmic reticulum, which facilitates relaxation of the cardiac muscle (Kranias and Hajjar, 2012b). Western blot analysis showed that removal of gravin did not affect SERCA expression (Fig. 7A); where it remained equal between WT and gravin-t/t hearts.
Fig. 7.
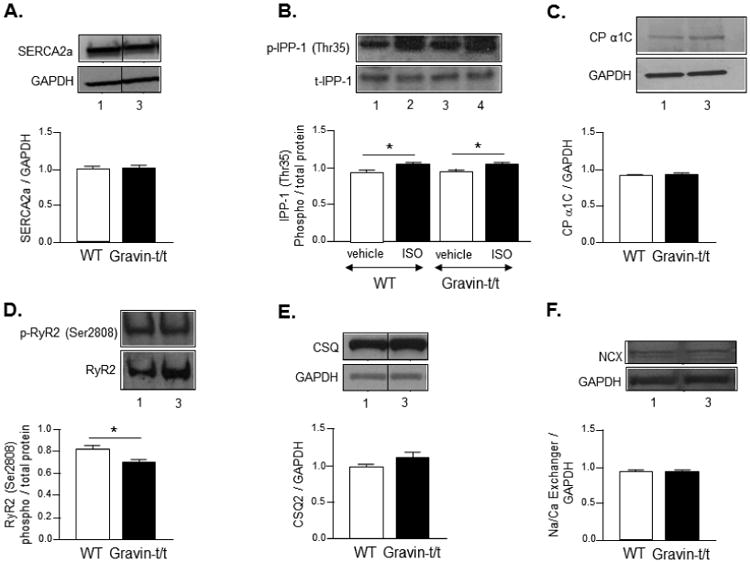
Ca2+-related protein expression. Western blot analysis of (A) sarcoplasmic reticulum Ca2+-ATPase (SERCA), (B) protein phosphatase-1 inhibitor-1 (IPP-1) following acute vehicle (ascorbic acid; 0.002%) or ISO infusion (10 μg/g/min), (C) L-type calcium channel (CP α1C), (D) ryanodine receptor (RyR2), (E) calsequestrin (CSQ2), and (F) sodium / calcium exchanger (NCX) in heart homogenates from WT and gravin-t/t mice. (Lane 1: WT (vehicle); Lane 2: WT (ISO); Lane 3: gravin-t/t (vehicle); Lane 4: gravin-t/t (ISO)). The bar graphs show the ratio of phosphorylated (phospho) to total protein or target protein to GAPDH. All lanes of a specific protein were detected on the same blot and a vertical line indicates where the blots are not contiguous. Data are expressed as the mean ± S.E.M.; n = 4 to 6 samples; *P<0.05 vs. baseline of same phenotype.
Since SERCA expression was unaltered, the next question was whether the activity of the pump was affected by gravin's absence. In order to answer this question, we measured PKA-dependent phosphorylation of IPP-1. IPP-1, when phosphorylated by PKA, inhibits protein phosphatase-1 (PP-1), which is primarily responsible for dephosphorylating key components of the EC coupling mechanism to attenuate contractility. Phospholamban (PLB) is a target of PP-1 and dephosphorylation of PLB results in the inhibition of SERCA to decrease Ca2+ reuptake into the SR (Herzig and Neumann, 2000; Oliver and Shenolikar, 1998). IPP-1 overexpression has been shown to augment cardiac function most likely by maintaining or increasing PLB phosphorylation to disinhibit SERCA (El-Armouche et al., 2003; Oliver and Shenolikar, 1998). We observed that basal IPP-1 phosphorylation and expression were unchanged in the absence of gravin (Fig. 7B). To determine whether β-AR stimulation affected the expression or phosphorylation of proteins involved in Ca2+ cycling, WT and gravin-t/t mice were infused with isoproterenol acutely. Isoproterenol treatment similarly increased IPP-1 phosphorylation in both WT and gravin-t/t hearts (P-value=0.0013) (Fig. 7B). These data corroborates our earlier observations that Ca2+ reuptake into the SR is unchanged in gravin-t/t mice. We further investigated if absence of gravin affects the expression of the key proteins involved in the EC coupling. One of the key proteins, L-type calcium channel plays an important role in modulating EC coupling (Kamp and Hell, 2000). To assess the effect of absence of gravin on L-type calcium channel we evaluated the expression profile of L-type calcium channel and found expression to be the similar between gravin-t/t and WT hearts (Fig. 7C). It is well known that ryanodine receptor (RyR2) is a key component of the calcium regulation in cardiac myocytes and works in tandem with LTCC to regulate the calcium induced calcium release (CICR) to induced contraction. RyR2 when phosphorylated by PKA at serine 2808 increases the sensitivity to calcium leading to CICR. However, hyperphosphorylation of RyR2 by PKA at serine 2808 has also been implicated in leaky RyR2 calcium release leading to impaired contractility and heart failure. In our study, we found that the p-RyR2(S-2808) in the gravin-t/t mice hearts was significantly lower compared to that of the WT mice hearts (P-value=0.0394) (Fig. 7D). To further evaluate whether gravin-t/t mice have altered SR Ca2+ release, we measured cardiac CSQ2 protein expression in WT and gravin-t/t hearts. CSQ2 is a calcium binding protein that has a high capacity for calcium and is a part of the ryanodine receptor Ca2+ release complex (Murphy et al., 2011). Additionally, CSQ2 is thought to be involved in providing a readily releasable pool of Ca2+ to facilitate cardiac contractility as it forms a multi-protein complex with triadin, junctin, and the ryanodine receptor (Scriven et al., 2000) and CSQ2 protein levels appear to be associated with both the level of the SR Ca2+ store as well as the level of SR Ca2+ release (Terentyev et al., 2003). Western blot analysis did not reveal any differences in CSQ2 expression between WT and gravin-t/t hearts (Fig. 7E). These data suggests that SR Ca2+ release would be expected to be similar between untreated or isoproterenol treated WT and gravin-t/t mice. Finally, the NCX is one of the transport mechanisms through which the recycling of the Ca2+ in cardiomyocytes is regulated. No change in NCX expression was observed between the WT and gravin-t/t mice. This suggests that the possible involvement of NCX in Ca2+ cycling, and thus contributing to the decreased Ca2+ transients in gravin-t/t mice in both trabaculae muscles (this study) as well as cardiomyocytes (Guillory et al., 2013), is unlikely (Fig. 7F).
4. Discussion
This study shows that (1) force development was similar in gravin-t/t muscles but the amplitudes of [Ca2+]i transient were lower at almost any given external [Ca2+]s and (2) gravin-t/t muscles had depressed stress-interval and [Ca2+]i-interval relationships as compared to WT muscles, indicating a lower SR Ca2+content. Ca2+ uptake by the SR appeared unaffected since Rf, determined from the relation between consecutive beats after potentiation by extrasystoli remained unchanged. Additionally, (3) SERCA expression, CSQ2 expression, NCX expression and IPP-1 phosphorylation were unaltered in gravin-t/t hearts. This study demonstrates that, at baseline, gravin-t/t muscles have an increased myofilament response to Ca2+. Also in gravin-t/t, the SR Ca2+ load may be reduced but the release process appears intact.
This gravin-t/t mouse model allows us to uniquely test the effect of acute β-AR stimulation on cardiac contractility in vivo in the absence of gravin binding to β2-AR, PKA and other signaling molecules. The absence of functional gravin in these mutant gravin-t/t mice was determined via RT-qPCR, immunohistochemistry (IHC) and Western blot (Guillory et al., 2013). Our observations are also supported by the work of others (Havekes et al., 2012) who independently generated a gravin gene trap mouse model to identify the role of gravin in neuroplasticity. In this article, the authors have reported gravin expression to be less than 10% in their mutant mouse model, wherein this expression has been attributed to the presence of a different gravin isoform (gravin-β) that is not targeted to the plasma membrane (Havekes et al., 2012).
An interesting observation in this study is that gravin-t/t muscles developed normal levels of stress despite having lower [Ca2+]i transient. This finding suggests that the myofilament response to Ca2+ is higher in gravin-t/t muscles, in the absence of receptor stimulation, than in WT muscles. This is a novel observation since, in isolated cardiomyocytes expressing Ht31, an inhibitory peptide that interrupts the binding of PKA to AKAP, none of the parameters for [Ca2+]i transient and cell shortening were changed at baseline (Fink et al., 2001). Also, when Ht31 was expressed in hearts in vivo, there were no changes in baseline LV ejection fraction, stroke volume or LV end-systolic pressure. Correspondingly, there were no differences in PKA-dependent phosphorylation of the measured myofilament proteins between Ht31 and control hearts (McConnell et al., 2009). However, upon stimulation of Ht31 expressing hearts via isoproterenol infusion in vivo, cardiac function, measured by LV ejection fraction and stroke volume, was increased. This increased cardiac function of isoproterenol-stimulated Ht31 expressing hearts, versus isoproterenol-stimulated control hearts, was in part attributed to decreased PKA-dependent phosphorylation of cMyBPC and cTnI, and/or the existence of truncated cTnI (Fink et al., 2001; McConnell et al., 2009). Although further investigation is needed, the mechanism for the increased myofilament Ca2+ response in the gravin-t/t mice is likely due to changes in the phosphorylation states of the myofilament related proteins.
We also observed that [Ca2+]i transients were significantly smaller in gravin-t/t muscles, as we previously reported in isolated cardiomyocytes, in which higher contraction were reported (Guillory et al., 2013). The discrepancy in contraction between the two studies was likely due to differences in magnitudes of intracellular Ca2+ transients under different experimental conditions (such as temperature, stimulation rate, and extracellular Ca2+, etc.), and nevertheless is quantitative in nature. The cellular mechanism for the decreased [Ca2+]i transients may be due to decreased SR release channel function or a lower SR Ca2+ content. Force-interval relationships were often employed to further investigate the relative roles of SR Ca2+ release channel function and SR Ca2+ content in controlling force (Schouten et al., 1987; ter Keurs et al., 1990). In this study, we not only obtained stress-interval relations but also [Ca2+]i-interval relations to get direct evidence of Ca2+ cycling across the SR. Like the force-interval relationship, the [Ca2+]i-interval relation also has three phases: early recovery, rest potentiation, and rest depression. The early recovery of force is dependent on the recovery of SR Ca2+ release channel (Banijamali et al., 1991), and therefore likely reflects the overall function of the SR Ca2+ release channel. The similar rate of recovery of [Ca2+]i transient during shorter intervals <5 s would suggest that the function of SR Ca2+ release channel is not altered in gravin-t/t myocardium.
On the other hand, gravin-t/t muscles showed smaller rest-potentiation (i.e. at test intervals 10-60 s), which is often associated with lower SR Ca2+ accumulation/content. However, this decreased SR Ca2+ content does not appear to be associated with alterations in Ca2+ cycling across the SR as evidenced by unchanged fraction of Ca2+ (re)uptake by the SR (i.e. Rf remained the same as in WT) in gravin-t/t muscles. This indicates that processes involved in the movement of Ca2+ out of the cytosol, such as SERCA and/or sarcolemmal NCX, are independent of gravin modulation under normal conditions. This was further confirmed by Western blot analysis, which showed that the expression of SERCA and NCX was similar in gravin-t/t as well as WT hearts. Of note, the differences in Rf measured from [Ca2+]i decay and that measured from force decay is likely due to the non-linear relationship between Ca2+ and force during contractions.
In the cardiomyocytes, stimulation of the β-adrenergic signaling pathway activates PKA, which phosphorylates a number of substrates involved in cardiac contraction. In Ht31 over-expressed cardiomyocytes, baseline contractility remained unaltered but treatment with isoproterenol resulted in similar magnitudes of changes in WT and Ht31 over-expressed cardiomyocytes (Fink et al., 2001). However, there was greater cell shortening in the Ht31 over-expressed cardiomyocytes. Additionally, LV ejection fraction and stroke volume were significantly increased in Ht31 over-expressed hearts following isoproterenol treatment (McConnell et al., 2009). Furthermore, we have recently shown that the cardiac function is enhanced in gravin-t/t mice in vivo, both in the presence and absence of acute β-AR stimulation (Guillory et al., 2013). In this study, Guillory et al. reports that disruption of gravin's scaffold mediated signaling increases baseline cardiac function and augments contractility in response to acute β-AR stimulation by decreasing β2-AR phosphorylation; where β2-AR phosphorylation has been shown to lead to receptor desensitization. However, we did not observe any differences in PLB or cTnI phosphorylation, either with or without acute β-AR stimulation, between WT and gravin-t/t hearts (Guillory et al., 2013). Instead, we observed significantly increased cMyBPC phosphorylation at position 273 in gravin-t/t hearts in the absence of acute β-AR stimulation (Guillory et al., 2013); where the phosphorylation of cMyBPC at Ser-273 is required for modulating cardiac function (Guggilam et al., 2013; Jin et al., 2013b). Furthermore, this increased phosphorylation of cMyPBC at Ser-273 may be responsible for the increased cardiac function observed in gravin-t/t mice that we reported in Guillory et al. (Guillory et al., 2013); since other studies have shown that increased cMyBPC phosphorylation augments actin-myosin interaction, resulting in enhanced cycling rate of the cross-bridges (Flashman et al., 2004; Stelzer et al., 2006; Tong et al., 2008). Also, we observed significantly increased Hsp20 phosphorylation in gravin-t/t hearts in the presence of acute β-AR stimulation (Guillory et al., 2013); where the phosphorylation of Hsp20 is beneficial to the myocardium (Edwards et al., 2012). Therefore, these results together suggest that the increased cardiac function in gravin-t/t mice may be attributed to the phosphorylation of cMyBPC and Hsp20. Thus, these explanations may account for the observed differences in contractile function in trabeculae muscles versus in vivo, between WT and gravin-t/t.
Calcium induced calcium release (CICR) is one of the most important steps in the initiation of excitation contraction coupling which eventually brings about the contraction of the heart. L-type calcium channel causes an influx of Ca2+ upon membrane depolarization, which further initiates the calcium release from sarcoplasmic reticulum via ryanodine receptors (Bodi et al., 2005). Studies have shown that reduction in the activity of the L-type calcium channel is associated with heart failure (Goonasekera et al., 2012). Thus, there is a strong association between L-type calcium channel, Ca2+ regulation and cardiac contractility. Ablation of gravin did not affect the expression of the L-type calcium channel compared to the WT mice indicating that Ca2+ modulation via the L-type calcium channel remains unaffected in the gravin-t/t mice. The phosphorylation levels of RyR2 at serine 2808 were significantly reduced in the gravin-t/t mice compared to WT mice hearts. RyR2 when phosphorylated by PKA brings about the CICR resulting in contraction. Increased phosphorylation by PKA at this site however is associated with aberrant calcium regulation resulting in impaired contractility and heart failure (Sarma et al., 2010). Though there are contradicting studies about the extent of involvement of phosphorylation of RyR2 at S2802 in heart failure and aberrant contractility, the importance of RyR2 in modulating calcium homeostasis in cardiomyocytes and excitation contraction coupling cannot be refuted (Dobrev and Wehrens, 2014; Houser, 2014). Sarma et al., showed that inhibition of RyR2 phosphorylation by PKA resulted in decreased SR Ca2+ lead in mdx mice which suffer from altered calcium intracellular calcium homeostasis (Sarma et al., 2010). Decreased phosphorylation of RyR2 in gravin t/t mice could be the driving factor for the increased myofilament sensitivity and decreased SR calcium load. The exact mechanism of this however needs to be further explored. The SERCA is a pivotal protein in the modulation of both cardiac contractility and relaxation, as its removal of Ca2+ from the cytosol into the SR facilitates relaxation and its increased SR Ca2+ content, together, results in the augmentation of subsequent contractions (Kranias and Hajjar, 2012a). Alterations in SERCA expression in turn alter Ca2+ homeostasis, which translates into modification of cardiac contractility and relaxation. Additionally, mice with reduced SERCA expression develop heart failure more rapidly compared to WT mice while overexpression of SERCA enhances cardiac function (Baker et al., 1998; He et al., 1997; Schultz Jel et al., 2004). Our study shows that the absence of gravin does not affect SERCA expression. IPP-1 can be phosphorylated by both PKA and PKC but PKA phosphorylation of IPP-1 activates the protein, which then inactivates PP-1. PP-1 dephosphorylates many proteins involved in EC coupling including PLB, the inhibitor of SERCA (Herzig and Neumann, 2000; Oliver and Shenolikar, 1998). IPP-1 null mice have been shown to have decreased cardiac function, increased protein phosphatase-1 activity and decreased PLB phosphorylation (Carr et al., 2002; El-Armouche et al., 2003). Conversely, IPP-1 overexpression results in the augmentation of cardiac function even in the absence of β-adrenergic receptor stimulation (El-Armouche et al., 2003). Thus, PKA mediated phosphorylation of IPP-1 is an important positive modulator of cardiac function. The absence of gravin did not affect IPP-1 expression, in the absence or presence of acute isoproterenol treatment, suggesting that PLB phosphorylation remains unaffected as well, which we have also shown previously (Guillory et al., 2013). This maintenance of PLB phosphorylation enhances the activity of SERCA resulting in the increase of Ca2+ uptake into the SR and the facilitation of cardiac relaxation and contraction. Calsequestrin (CSQ2) is another important component of the EC coupling mechanism. CSQ2 can be found tethered to the ryanodine receptor complex along with junctin and triadin (Scriven et al., 2000) and can have a profound effect upon cardiac function. We observed that, similar to SERCA expression and IPP-1 phosphorylation, basal CSQ2 expression was comparable between WT and gravin-t/t mice. Thus except RyR2, other proteins involved in calcium regulation were unchanged and the exact mechanism of decreased phosphorylation of RyR2 in increased myofilaments sensitivity and decreased calcium load in gravin t/t mice needs to be further explored.
5. Conclusions
In conclusion, gravin-t/t muscle exhibited increased myofilament responsiveness to Ca2+. However, the mechanism for the increased myofilament Ca2+ response is not clear at present. A lower SR Ca2+ load is likely responsible for the decreased [Ca2+]i transient. Though there is decreased RyR2 phosphorylation in gravin-t/t hearts, Ca2+ re-cycling across the SR, however, is not changed as evidenced by unchanged recirculation fraction, unaltered SERCA and CSQ2 expression and IPP-1 phosphorylation. These results not only establish the baseline contraction characteristics of gravin-t/t muscle, but also show that gravin can act as a modulator of cardiac contraction. Our studies support the concept that gravin, in addition to serving as a scaffolding protein, also functions as a regulator of myofilament Ca2+ responsiveness. Further investigation will reveal the physiological and pathophysiological roles of gravin in cardiac excitation-contraction coupling.
Acknowledgments
The material in this paper is based upon a dissertation submitted by Zhitao Li in partial fulfillment of the requirements for a Ph.D. at Harbin Medical University, under the mentorship of Dr. Wei Dong Gao at Johns Hopkins University School of Medicine.
The National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH) under Award Number R01HL085487 (to B.K.M)† and R15HL124458 (to B.K.M.)† supported research reported in this publication. Research was also supported by an American Heart Association (AHA) grant under Award Number 0855439E (to W.D.G)‡.
Footnotes
Author Contributions: Conceived research project: [W.D.G.], [B.K.M.]
Participated in research design: [Z.L.], [S.S.], [W.D.G.], [B.K.M.]
Conducted experiments: [Z.L.], [S.S.], [W.D.], [S.V.S.], [X.S.], [C.S.W.], [W.D.G.]
Contributed new reagents or analytic tools: [W.D.G.], [B.K.M.]
Performed data statistical analysis: [Z.L.], [S.S.], [W.D.], [X.S.], [C.S.W.], [W.D.G.]
Analyzed and interpreted data: [Z.L.], [S.S.], [W.D.], [X.S.], [C.S.W.], [W.D.G.], [B.K.M.]
Handled funding and supervision: [W.D.G.], [B.K.M.]
Wrote or contributed to the writing of the manuscript: [S.S.], [W.D.G.], [B.K.M.]
The content is solely the responsibility of the authors, and does not necessarily represent the official views of the NIH or AHA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, Grupp G, Bhagwhat A, Hoit B, Walsh R, Marban E, Periasamy M. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res. 1998;83:1205–1214. doi: 10.1161/01.res.83.12.1205. [DOI] [PubMed] [Google Scholar]
- Banijamali HS, Gao WD, MacIntosh BR, ter Keurs HE. Force-interval relations of twitches and cold contractures in rat cardiac trabeculae Effect of ryanodine. Circ Res. 1991;69:937–948. doi: 10.1161/01.res.69.4.937. [DOI] [PubMed] [Google Scholar]
- Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114:1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. discussion 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards HV, Scott JD, Baillie GS. PKA phosphorylation of the small heat-shock protein Hsp20 enhances its cardioprotective effects. Biochem Soc Trans. 2012;40:210–214. doi: 10.1042/BST20110673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Rau T, Zolk O, Ditz D, Pamminger T, Zimmermann WH, Jackel E, Harding SE, Boknik P, Neumann J, Eschenhagen T. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:437–439. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- Fan G, Shumay E, Wang H, Malbon CC. The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the beta 2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J Biol Chem. 2001;276:24005–24014. doi: 10.1074/jbc.M011199200. [DOI] [PubMed] [Google Scholar]
- Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, Damron DS, Bond M. AKAP-mediated targeting of protein kinase a regulates contractility in cardiac myocytes. Circ Res. 2001;88:291–297. doi: 10.1161/01.res.88.3.291. [DOI] [PubMed] [Google Scholar]
- Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: its role in physiology and disease. Circ Res. 2004;94:1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- Gao WD, Atar D, Backx PH, Marban E. Relationship between intracellular calcium and contractile force in stunned myocardium. Direct evidence for decreased myofilament Ca2+ responsiveness and altered diastolic function in intact ventricular muscle. Circ Res. 1995;76:1036–1048. doi: 10.1161/01.res.76.6.1036. [DOI] [PubMed] [Google Scholar]
- Gao WD, Perez NG, Marban E. Calcium cycling and contractile activation in intact mouse cardiac muscle. J Physiol. 1998;507(Pt 1):175–184. doi: 10.1111/j.1469-7793.1998.175bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WD, Perez NG, Seidman CE, Seidman JG, Marban E. Altered cardiac excitation-contraction coupling in mutant mice with familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:661–666. doi: 10.1172/JCI5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, Sargent MA, Hofmann F, Moosmang S, Marks AR, Houser SR, Bers DM, Molkentin JD. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–290. doi: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggilam A, Hutchinson KR, West TA, Kelly AP, Galantowicz ML, Davidoff AJ, Sadayappan S, Lucchesi PA. In vivo and in vitro cardiac responses to beta-adrenergic stimulation in volume-overload heart failure. J Mol Cell Cardiol. 2013;57:47–58. doi: 10.1016/j.yjmcc.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillory AN, Yin X, Wijaya CS, Diaz Diaz AC, Rababa'h A, Singh S, Atrooz F, Sadayappan S, McConnell BK. Enhanced Cardiac Function in Gravin Mutant Mice Involves Alterations in the beta-Adrenergic Receptor Signaling Cascade. PLoS One. 2013;8:e74784. doi: 10.1371/journal.pone.0074784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Canton DA, Park AJ, Huang T, Nie T, Day JP, Guercio LA, Grimes Q, Luczak V, Gelman IH, Baillie GS, Scott JD, Abel T. Gravin orchestrates protein kinase A and beta2-adrenergic receptor signaling critical for synaptic plasticity and memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:18137–18149. doi: 10.1523/JNEUROSCI.3612-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–389. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Houser SR. Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ Res. 2014;114:1320–1327. doi: 10.1161/CIRCRESAHA.114.300569. discussion 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin CZ, Jang JH, Kim HJ, Wang Y, Hwang IC, Sadayappan S, Park BM, Kim SH, Jin ZH, Seo EY, Kim KH, Kim YJ, Kim SJ, Zhang YH. Myofilament Ca desensitization mediates positive lusitropic effect of neuronal nitric oxide synthase in left ventricular myocytes from murine hypertensive heart. J Mol Cell Cardiol. 2013a;60C:107–115. doi: 10.1016/j.yjmcc.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Jin CZ, Jang JH, Kim HJ, Wang Y, Hwang IC, Sadayappan S, Park BM, Kim SH, Jin ZH, Seo EY, Kim KH, Kim YJ, Kim SJ, Zhang YH. Myofilament Ca desensitization mediates positive lusitropic effect of neuronal nitric oxide synthase in left ventricular myocytes from murine hypertensive heart. J Mol Cell Cardiol. 2013b doi: 10.1016/j.yjmcc.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- Katz AM, Lorell BH. Regulation of cardiac contraction and relaxation. Circulation. 2000;102:IV69–74. doi: 10.1161/01.cir.102.suppl_4.iv-69. [DOI] [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of Cardiac Contractility by the Phopholamban/SERCA2a Regulatome. Circ Res. 2012a;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012b;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Wang H, Malbon CC. Gravin-mediated formation of signaling complexes in beta 2-adrenergic receptor desensitization and resensitization. J Biol Chem. 2000;275:19025–19034. doi: 10.1074/jbc.275.25.19025. [DOI] [PubMed] [Google Scholar]
- McConnell BK, Popovic Z, Mal N, Lee K, Bautista J, Forudi F, Schwartzman R, Jin JP, Penn M, Bond M. Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J Biol Chem. 2009;284:1583–1592. doi: 10.1074/jbc.M806321200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Mollica JP, Beard NA, Knollmann BC, Lamb GD. Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+-binding protein changes in CSQ2 knockout mice. Am J Physiol Heart Circ Physiol. 2011;300:H595–604. doi: 10.1152/ajpheart.00902.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:D961–972. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:13165–13170. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten VJ, van Deen JK, de Tombe P, Verveen AA. Force-interval relationship in heart muscle of mammals. A calcium compartment model. Biophys J. 1987;51:13–26. doi: 10.1016/S0006-3495(87)83307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz Jel J, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, Shull GE, Kimball TR, Periasamy M. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol. 2004;286:H1146–1153. doi: 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih M, Lin F, Scott JD, Wang HY, Malbon CC. Dynamic complexes of beta2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem. 1999;274:1588–1595. doi: 10.1074/jbc.274.3.1588. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- Tan Z, Dai T, Zhong X, Tian Y, Leppo MK, Gao WD. Preservation of cardiac contractility after long-term therapy with oxypurinol in post-ischemic heart failure in mice. Eur J Pharmacol. 2009;621:71–77. doi: 10.1016/j.ejphar.2009.08.033. [DOI] [PubMed] [Google Scholar]
- ter Keurs HE, Gao WD, Bosker H, Drake-Holland AJ, Noble MI. Characterisation of decay of frequency induced potentiation and post-extrasystolic potentiation. Cardiovasc Res. 1990;24:903–910. doi: 10.1093/cvr/24.11.903. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552:109–118. doi: 10.1113/jphysiol.2003.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ML, Crossman DJ, Loiselle DS, Cannell MB. Non-steady-state calcium handling in failing hearts from the spontaneously hypertensive rat. Pflugers Arch. 2010;460:991–1001. doi: 10.1007/s00424-010-0876-3. [DOI] [PubMed] [Google Scholar]
- Wier WG, Yue DT. Intracellular calcium transients underlying the short-term force-interval relationship in ferret ventricular myocardium. J Physiol. 1986;376:507–530. doi: 10.1113/jphysiol.1986.sp016167. [DOI] [PMC free article] [PubMed] [Google Scholar]