

Abstract
Patient: Female, 48
Final Diagnosis: Inclusion body myositis
Symptoms: Shortness of breath • weakness
Medication: —
Clinical Procedure: Biopsy
Specialty: Neurology
Objective:
Rare disease
Background:
Sporadic inclusion body myositis (IBM) is the most common acquired myopathy seen in adults aged over 50 years, with a prevalence estimated at between 1 and 70 per million. Weakness of the diaphragm with loss of vital capacity is almost universal in IBM. This is almost always asymptomatic. When respiratory complications occur, they are most often due to aspiration. Respiratory failure due to diaphragmatic weakness is exceptionally rare, particularly as the presenting symptom of the disease. It is not currently considered to be a paraneoplastic syndrome.
Case Report:
Our patient presented with hypercarbic respiratory failure. This is the first such reported case without signs of weakness elsewhere of which we are aware. We suspected IBM based on her history of progressive weakness and findings on electromyography. There was a delay of 5 years in obtaining biopsy for confirmation, during which she presented with recurrent episodes of respiratory failure despite using non-invasive ventilation. An autopsy revealed the presence of papillary thyroid carcinoma with spread to local lymph nodes. On the basis that these co-morbidities are unlikely to have occurred by chance (we estimate 1×10−17), we hypothesize that IBM may be a paraneoplastic condition. We acknowledge that proof would require demonstrating a pathogenic antibody.
Conclusions:
IBM should be considered in older patients (age >45) presenting with otherwise unexplained respiratory failure. A workup for possible malignancy in this setting appears reasonable.
MeSH Keywords: Diaphragm, Inclusion Bodies, Myositis, Paraneoplastic Syndromes
Background
Sporadic inclusion body myositis (IBM) is the most frequent acquired myopathy in adults aged over 50 years. Estimates of prevalence (per million population) are 1.0 in Turkey; 4.7 in the Netherlands; 10.7 in Connecticut, USA; 14.9 in western Australia; and 71 in Olmsted County, USA, and are approximately 3-fold higher in those over age 50 [1]. Weakness of the diaphragm is now better recognized as being almost universal in IBM. Progressive loss of vital capacity (VC) is characteristic [2]. Despite this, respiratory failure has only previously been described once as the presenting feature of IBM [3] (the article was published in Spanish and we present a translation in English as Supplementary Material. Furthermore, an association with papillary thyroid carcinoma is novel and we propose a paraneoplastic mechanism for this.
Case Report
Presentation
Our patient was an African-American woman, born in Haiti, previously well, who first presented to our Institution age 48. There was no significant family history. She was found confused at home by her family; this was due to acute-on-chronic hypercarbic respiratory failure, which resolved with the use of bi-level positive airway pressure (BiPAP).
She felt she had been getting weaker over the preceding 3–4 years. She noticed this when walking long distances and when climbing stairs. Power was 5/5, including neck flexors. No deep tendon reflexes were elicited, although she was obese. Blood tests were unremarkable apart from creatinine kinase (CK) of 200 (<170 IU/lt). Acetylcholinesterase receptor antibodies (AChR Abs) were negative. Pulmonary function tests (PFTs) showed a restrictive pattern with a forced vital capacity (FVC) of 30% of expected.
Nerve conduction studies (NCS) were normal apart from 1 absent F-wave in a peroneal nerve. Electromyography (EMG) showed widespread myopathic units with fibrillations at rest. A myositis, particularly IBM, was suspected at this stage due to her history and EMG findings. She was discharged on nocturnal BiPAP. A muscle biopsy was planned; however, we were unable to contact the patient to arrange follow-up in clinic after discharge.
Recurrent respiratory failure
She was admitted again 3 months later with another episode of hypercarbic respiratory failure, which again responded to BiPAP. A muscle biopsy was rescheduled, but she did not attend. The following year she presented elsewhere with respiratory failure requiring intubation and admission to intensive care. She spent 18 months in rehabilitation before returning home. She said she was diagnosed with myasthenia gravis (MG) at that time and had been on pyridostigmine 60 mg q6h and prednisone 5 mg daily since. She needed to use a walker or wheelchair for mobility thereafter, and she required home oxygen during the day with nocturnal BiPAP.
She presented at our Institution again, aged 53, with 3 days of progressive shortness of breath, requiring use of BiPAP during the day. One week prior, she had run out of her home medicines. Power was now 4/5 in the neck flexors and 3/5 in the deltoids; her other muscles remained 5/5. No fatigability was present and there was no ptosis. Reflexes were again absent.
She was treated for suspected community-acquired pneumonia and for possible exacerbation of MG (methylprednisone 1000 mg iv and intravenous immunoglobulin (IVIg) 400 mg/kg/day, both planned for 5 days). AChR R Abs were negative, as were anti-MUSK antibodies. Computerized tomography (CT) of the chest showed only bibasilar atelectasis.
On day 4 of admission, NCS were performed. There was no sign of fatigability or of an electrodecremental response to repeated stimulation, so her IVIg was stopped. EMG showed moderately increased spontaneous activity of the left C5–6 paraspinal muscles, with fibrillation. However, the paraspinal muscles at C6–7 were normal on the same side. The left deltoid showed increased amplitude. All other muscles tested were normal. This was initially interpreted as showing evidence of a C5–6 radiculopathy.
She was unable to complete PFTs. On day 10, a steroid taper was started, decreasing from prednisone 60 mg by 5 mg every 3 days. Pyridostigmine was stopped without signs of deterioration. By day 13, she was stable during the day with intermittent use of BiPAP (3 hours on, 3 hours off). Magnetic resonance imaging (MRI) was not possible due to her use of BiPAP.
Final admission
She was discharged to rehabilitation and readmitted 3 days later with recurrent hypercarbic respiratory failure and acidosis. She was intubated in the Emergency Room. MRI of the brain on day 9 of this admission showed a number of foci of restricted diffusion in the corpus callosum bilaterally, thought to be due to hypoxic-ischemic injury at the time she was intubated. MRI of the neck showed degenerative changes but no signs of radiculopathy or myelopathy. Initially, she was hypotensive and febrile. By day 13, she was sufficiently stable to be extubated to BiPAP.
On day 22 she had a tracheostomy placed due to persistent dependence on BiPAP. She remained ventilator-dependent thereafter. NCS on day 24 showed evidence of superimposed critical-illness neuropathy with significantly lower left median and ulnar motor-evoked response amplitudes. EMG now showed myopathic units in both deltoids and the right bicep muscles (C5–6). The previously documented giant motor units in the left deltoid are were no longer observed.
On day 30, a biopsy of the quadriceps was performed, which confirmed our suspicion of IBM. This is shown in Figures 1 and 2. Variation in myofiber size was noted with atrophic fibers (up to 50% of the specimen) and hypertrophic fibers. Rimmed vacuoles were seen best on Congo red staining. Sparse chronic inflammation was seen, with CD3-positive T cells.
Figure 1.
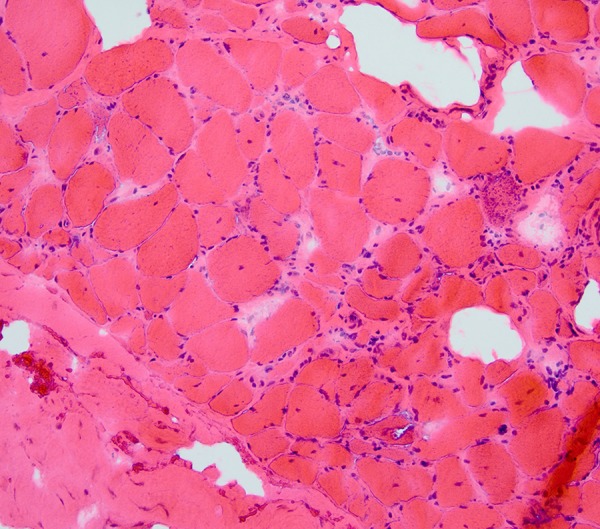
Quadriceps muscle biopsy. Hematoxylin and eosin stain. The muscle fiber size is varied. Many have visible nuclei. Some clusters of atrophied fibers are seen, particularly in the lower right. Endomysial connective tissue is increased in some regions. Inflammation is evident, with lymphocytes. These are most marked in the endomysium. They surround myocytes, which are invaded in some cases (e.g., with splitting of the fiber), as visible in the lower right. No clearly defined inclusion bodies or rimmed vacuoles are noted.
Figure 2.
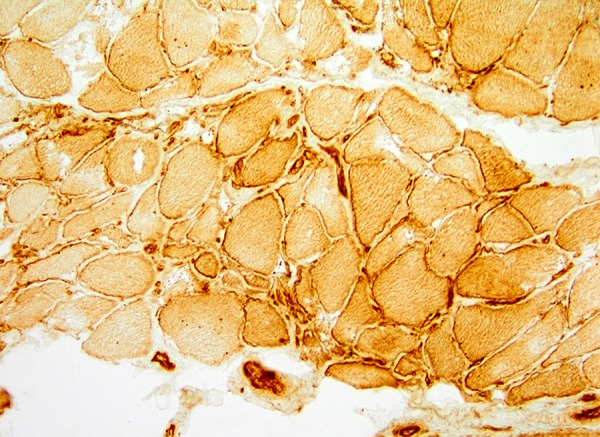
Quadriceps muscle biopsy. Immunohistochemistry for MHC class I. This is up-regulated within the myocytes, particularly on the cell surface. Lymphocytes show dense staining.
Two days later, she became unresponsive and hypotensive, requiring a vasopressor infusion. Throughout the rest of her stay she remained intermittently hypotensive and delirious, responding only to painful stimuli. At 60 days into her admission she developed pulseless electrical activity and died shortly thereafter. An autopsy was performed, which showed severe atrophy and fibrosis of the diaphragm. This was best seen with trichrome staining, showing fibrosis composing up to 80% of the diaphragm, with severely atrophic myofibers coursing through, and separated by, the fibrosis. Incidentally, papillary thyroid carcinoma with metastasis to 2 peri-glandular lymph nodes was also identified.
Discussion
Diagnosis
IBM is a clinical diagnosis, which is confirmed by the findings on NCS, EMG, and biopsy, where possible. Our patient met the first 3 of 4 criteria for clinical diagnosis, given below (criteria from the 2011 European Neuromuscular Centre) [4]:
age at onset >45 years;
duration of weakness >12 months;
CK ≤15× the upper limit of normal (ULN));
finger flexion weakness > shoulder abduction weakness.
In particular, she was lacking the classic pattern of weakness of finger flexors and/or knee extensors.
Our case appears unique in that she had no sign of weakness anywhere else on exam when she first presented. However, her history of progressive weakness and the initial EMG confirmed a myopathic process. The degree of involvement of the diagram was strikingly out of proportion to the degree of involvement elsewhere. Although we were initially concerned about IBM specifically, the delay in performing biopsy to confirm this resulted in significant additional uncertainty. This underscores the need for close follow-up of such patients presenting in crisis.
Similar cases
We were able to find only 5 prior pathologically-confirmed case reports of respiratory failure occurring with IBM [5–7]. One of these occurred in the setting of HTLV, which we did not suspect (or test for) in our patient [8]. The single other case we found presenting primarily with respiratory failure was in a woman aged 74, who, following an episode of pneumonia, developed recurrent hypercarbic respiratory failure requiring BiPAP. After a course of prednisone 1 mg/kg/day for 2 months, she showed no improvement and ultimately required a tracheostomy. It is unclear from the report whether the patient had weakness that was apparent elsewhere on presentation [3].
Pulmonary function in IBM
Asymptomatic impairment of respiratory function is common in patients with IBM, with forced vital capacity (FVC) <80% of predicted in 44% (7/16) of patients in one series. Only one case of hypercarbia amongst this group was noted [9]. PFTs (and polysomnography) have therefore been recommended routinely in IBM. The importance of respiratory involvement as a late/terminal event in the course of IBM was highlighted in a series of 64 patients from 2011 [10]. Recurrent aspiration pneumonia appears to be the principal problem. Primary respiratory failure was not identified as a late complication in the series.
Treatment
Earlier diagnosis may have changed our approach to treatment. Steroids and other immune suppressants are typically used empirically in patients with IBM. In our case, the fact that she presented again with respiratory failure on two occasions when her steroid dose was reduced or stopped suggests that she was deriving some benefit from these. While experience suggests that there are individuals who respond to such measures, this is typically not sustained. Although one series of 8 patients showed probable improvement on biopsy following use of prednisone for up to 12 months, this was not accompanied by an improvement in muscle strength [11]. Results with other immune suppressants have been disappointing to date, although clinical trials with more novel agents are ongoing.
There may have been some empiric advantage in the use of agents been shown to improve contractility of the diaphragm. Neostigmine and aminophylline have been shown experimentally to counteract the impairment of contractility caused by hypercapnia. Thus, her use of pyridostigmine may have been of some benefit, albeit inadvertently as it was prescribed for MG [12].
BiPAP was beneficial for our patient. Whether this should be routinely employed in those with IBM and involvement of the diaphragm has not been investigated. Such a strategy appears rational and has been shown to improve quality of life in patients with amyotrophic lateral sclerosis. Diaphragmatic stimulation of dysfunctional muscle would be unlikely to produce additional gains in respiratory function.
A paraneoplastic disease?
Without an autopsy, the possibility of this being a paraneoplastic disorder would not have been considered further. As PTC was not diagnosed ante-mortem, we were unable to analyze serum to look for a potentially causative antibody. IBM is not currently recognized to be a paraneoplastic phenomenon, although cases associated with bladder and renal cell carcinoma have been identified [13,14]. Our patient’s IBM qualifies as a “possible paraneoplastic syndrome” based on expert panel criteria from 2004: “A non-classical syndrome, no onconeural antibodies and cancer present within 2 years of diagnosis” [15].
Could the co-occurrence of IBM and PTC be simply a matter of chance? For an estimate, we begin with the age-adjusted incidence of thyroid cancer (TC), which is 12.4/100 000/year in African-American women in the USA [16]. We then reason with the following approximate assumptions:
TC with metastases to local lymph nodes developed in our patient age 45–49;
The age group 45–50 accounts for 7.2% of the standardized population (used in age adjustment) [17]; however
Newly diagnosed TC may be more common in this age group, accounting for approximately 12% of cases (vs. other age groups) [14];
80% of cases of TC are PTC [18];
12–80% of cases of PTC have nodal metastases at presentation [19].
Thus, her annual risk of developing PTC, in this age range, may be given as:
The age-and-sex-adjusted incidence of sporadic IBM in adults age 30 or older has been estimated at 0.41/100,000/year [20]. We assume:
Sex is approximately evenly matched in the standardized population, i.e. male: female ≈1: 1.
20% of cases of IBM are diagnosed in those age <60; therefore, the incidence in the age group 30–60 is ≈20% of that in those >60 [21].
The age group 30–59 accounts for 41.7% of the standardized population.
We then calculated her annual risk of IBM, based on her age, as follows:
Thus, the risk of both conditions developing within a year, as is typical for paraneoplastic disorders, is of the order of 1×10−17, which we believe is sufficiently unlikely to make chance a remote possibility.
Alternatively, we may view the PTC as having been longstanding and asymptomatic. PTC, large enough to be detectable by ultrasound, has been estimated to occur in up to 5% of adults. The natural history of such asymptomatic lesions remains unclear. Although surgery is usually performed, the utility of extensive resection (beyond thyroid lobectomy) has been questioned; the estimated survival rate at 20 years is thought be 99% with current approaches [22].
A paraneoplastic etiology appears more plausible if we consider IBM as part of the spectrum of T cell-mediated autoimmune processes directed at intracellular antigens, particularly dermatomyositis (DMM) and polymyositis (PM). DMM, PM, and, to a lesser extent, IBM are associated with cancer at rates which exceed that expected due to surveillance bias (with a standardized incidence-ratio for IBM of 2.4 (95% CI 1.2–4.9) [23]. This is particularly true for DMM, and in some patients a paraneoplastic disease course for DMM has been described [24].
PTC is a rare cause of paraneoplastic syndromes. Spread to lymph nodes, as in our patient, is thought to be a prerequisite for such syndromes. Reported paraneoplastic conditions in patients with PTC are limited to case reports and include DMM (5 patients in total), PM, propriospinal myoclonus, cerebellar syndrome, acquired reactive perforating collagenosis (a hyperkeratotic skin disorder), and a condition resembling adult-onset Still’s disease [25–30]. As in our patient, no pathognomonic antibodies were identified in these cases.
A possible association between IBM and PTC should be falsifiable, such as with the use of the standard ‘One-Sample Proportion Test’ (with a significance, as is conventional, of 5%). As above, we assume that thyroid cancer, of sufficient size to be detectable by ultrasound, has a prevalence of around 5% in adults [20]. With any sample size (i.e., patients with IBM) and an observed prevalence of asymptomatic PTC of 5%, we can conclude there is no association with a power of 95%. We can accept such an association with a power of ≈95% with, for example, a sample size of 60 and an observed prevalence of 15% or a sample size of 20 and a prevalence of 22.5% [31]. Such sample sizes have already been achieved in case series of patients with IBM attending specialized centers. Thus, to settle the question, an empiric policy of screening for PTC at the time of diagnosis of IBM could be instituted at a tertiary center for a period of 5–10 years.
Conclusions
This case highlights the importance of early recognition of respiratory failure as a possible presenting feature of IBM. As this is a potentially life-threatening manifestation, we suggest that IBM should be considered as a possibility in any patient aged over 45 with new-onset of otherwise unexplained respiratory failure. A workup for malignancy appears reasonable on an individual basis. To this end, a CT with contrast, followed by PET/CT, appears reasonable, as is suggested in the workup of suspected paraneoplastic disorders. A thyroid-focused ultrasound may be a reasonable alternative.
Supplementary Materials
Diaz PJ, Garcia SF, Sanchez LJ, Saborido FJ.
Inclusion body myositis with respiratory onset.
Revista Clinica Espanola 2003; 203(3): 164
Inclusion body myositis (IBM) is a spontaneous, inflammatory, idiopathic myopathy which is occasionally hereditary. It affects more females than males (3: 1) and is the most common myopathy in the elderly population i.e. over 50 years old.
Weakness and muscle atrophy are painless and can be generalized or limited just to the limbs. In 20% of cases weakness starts with the quadriceps or the finger flexors – mainly flexor pollicis longus.
Deep tendon reflexes are normal at the beginning but they decrease as the disease progresses. Dysphagia is frequent. CPK is normal or can be elevated. EMG shows a myopathic pattern (i.e. mixed potentials), fibrillation or rarely a neuropathic pattern. Muscle biopsy shows structural changes and inflammatory basophilic and eosinophilic intracytoplasmic vacuoles, which stain for β-amyloid protein. Electronic microscopy (EM) identifies an accumulation of β-amyloid with abnormal tubular filaments in the nucleus and cytoplasm. Respiratory compromise is rare.
The following case is that of a 74 year-old woman with a history of hyperlipidemia and depression; she was not taking any treatment for these. She presented with three days of fever, cough and yellow sputum. She had severe respiratory distress with a PO2 of 42 mmHg and a lobar consolidation in the right lung. Mechanical ventilation and cefotaxime with clarithromycin were started with clinical and radiological improvement. After extubation, she had episodes when her PO2 dropped and PCO2 rose.
EMG showed a myopathic pattern with mixed potentials and short and prolonged motor unit potentials. A quadriceps muscle biopsy showed small, angled, striated fibers and small, basophilic nuclear vacuoles. Trichrome, Gomori and PAS stains were negative; β-amyloid was positive. EM showed intranuclear and intracytoplasmic bodies with disruption of the myofibers and the sarcolemma. She was started on prednisone 1 mg/kg/day po for 2 months, with no response. Breathing was maintained by mechanical ventilation (a tracheostomy was performed).
Respiratory compromise is rare in IBM; aspiration pneumonia and interstitial pneumonia have been described. The pathophysiology is unknown but an immunologic mechanism has been postulated such as toxicity from cytotoxic T cells, T cell cytokines, macrophages and adhesion molecules. This proposed immunologic mechanism is supported by the association [of IBM] with SLE and sarcoidosis. Some inducers of autoimmunity have been proposed, such as HTLV-1 and HCV, but no clear association has been shown.
Other factors which may be important for an immunologic etiology are the deposition of β-amyloid, mitochondrial anomalies and deletions of mitochondrial DNA in the muscle fibers. The vacuoles of the muscle fiber contain β2-secretases with proteolytic activity (BACE-1 and BACE-2), suggesting its participation in processing of the abnormal β-amyloid protein.
Treatment in general is discouraging. Steroids, ivIg, cyclosporine, cyclophosphamide and interferon β-1a have been used with variable responses but poor success overall. Respiratory compromise is infrequent. Therefore, we recommend IBM should be considered in any older patient (aged >50) with respiratory failure.
Translated with permission of the Publisher. Original source: Díaz Peromingo JA, García Suárez F, Sánchez Leira J, Saborido Froján J. Inclusion body myositis with respiratory onset. Rev Clin Esp, 2003; 203(3): 164.
Copyright © 2003 Elsevier España, S.L.U. y Sociedad Española de Medicina Interna. Published by Elsevier. All rights reserved.
Acknowledgments
The authors would like to acknowledge Jenny Libien MD PhD, who provided the autopsy report, as well as Florence Shum MD and Charles Abrams MD PhD, who performed and interpreted the NCS and EMGs.
Footnotes
Conflict of interests
None.
References:
- 1.Greenberg SA. Inclusion body myositis. Curr Opin Rheumatol. 2011;23(6):574–78. doi: 10.1097/BOR.0b013e32834b53cc. [DOI] [PubMed] [Google Scholar]
- 2.Teixeira A, Cherin P, Demoule A, et al. Diaphragmatic dysfunction in patients with idiopathic inflammatory myopathies. Neuromuscul Disord. 2005;15(1):32–39. doi: 10.1016/j.nmd.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Diaz PJ, Garcia SF, Sanchez LJ, Saborido FJ. [Inclusion body myositis with respiratory onset] Revista Clinica Espanola. 2003;203(3):164. doi: 10.1157/13044932. [in Spanish] [DOI] [PubMed] [Google Scholar]
- 4.Brady S, Squier W, Hilton-Jones D. Clinical assessment determines the diagnosis of inclusion body myositis independently of pathological features. J Neurol Neurosurg Psychiatry. 2013;84(11):12400–46. doi: 10.1136/jnnp-2013-305690. [DOI] [PubMed] [Google Scholar]
- 5.Martin SE, Gondim DD, Hattab EM, et al. Inclusion body myositis involving the diaphragm: Report of a pathologically confirmed case. Neurol India. 2014;62(1):66–67. doi: 10.4103/0028-3886.128313. [DOI] [PubMed] [Google Scholar]
- 6.Voermans N, Vaneker M, Hengstman G, et al. Primary respiratory failure in inclusion body myositis. Neurology. 2004;63(11):2191–92. doi: 10.1212/01.wnl.0000145834.17020.86. [DOI] [PubMed] [Google Scholar]
- 7.Cohen R, Lipper S, Dantzker D. Inclusion body myositis as a cause of respiratory failure. Chest. 1993;104(3):975–77. doi: 10.1378/chest.104.3.975. [DOI] [PubMed] [Google Scholar]
- 8.Littleton E, Man W, Holton J, et al. Human T cell leukaemia virus type I associated neuromuscular disease causing respiratory failure. J Neurol Neurosurg Psychiatry. 2002;72(5):650–52. doi: 10.1136/jnnp.72.5.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruz PMR, Needham M, Hollingsworth P, et al. Sleep disordered breathing and subclinical impairment of respiratory function are common in sporadic inclusion body myositis. Neuromuscul Disord. 2014;24(12):1036–41. doi: 10.1016/j.nmd.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Cox FM, Titulaer MJ, Sont JK, et al. A 12-year follow-up in sporadic inclusion body myositis: An end stage with major disabilities. Brain. 2011;134(11):3167–75. doi: 10.1093/brain/awr217. [DOI] [PubMed] [Google Scholar]
- 11.Breithaupt M, Schmidt J. Update on treatment of inclusion body myositis. Curr Rheumatol Rep. 2013;15(5):1–6. doi: 10.1007/s11926-013-0329-z. [DOI] [PubMed] [Google Scholar]
- 12.Howell S, Fitzgerald R, Roussos C. Effects of aminophylline, isoproterenol, and neostigmine on hypercapnic depression of diaphragmatic contractility. Am Rev Respir Dis. 1985;132(2):241–47. doi: 10.1164/arrd.1985.132.2.241. [DOI] [PubMed] [Google Scholar]
- 13.Jensen ML, Wieting JM, Andary MT, et al. Inclusion body myositis and transitional cell carcinoma of the bladder: Significant resolution of symptoms after tumor excision. Arch Phys Med Rehabil. 1997;78(3):327–29. doi: 10.1016/s0003-9993(97)90043-5. [DOI] [PubMed] [Google Scholar]
- 14.Ytterberg SR, Roelofs RI, Mahowald ML. Inclusion body myositis and renal cell carcinoma. Report of 2 cases and review of the literature. Arthritis Rheum. 1993;36(3):416–21. doi: 10.1002/art.1780360319. [DOI] [PubMed] [Google Scholar]
- 15.Graus F, Delattre J, Antoine J, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135–40. doi: 10.1136/jnnp.2003.034447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.SEER Cancer Statistics Factsheets . Thyroid Cancer. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/statfacts/html/thyro.html [Accessed 8/20/16] [Google Scholar]
- 17.Klein RJ, Schoenborn CA. Age adjustment using the 2000 projected U.S. population. Hyattsville, Maryland: National Center for Health Statistics; Jan, 2001. Healthy People Statistical Notes, no. 20. [PubMed] [Google Scholar]
- 18.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995. Cancer. 1998;83:2638–48. doi: 10.1002/(sici)1097-0142(19981215)83:12<2638::aid-cncr31>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 19.Randolph GW, Duh QY, Heller KS, et al. The prognostic significance of nodal metastases from papillary thyroid carcinoma can be stratified based on the size and number of metastatic lymph nodes, as well as the presence of extranodal extension. Thyroid. 2012;22(11):1144–52. doi: 10.1089/thy.2012.0043. [DOI] [PubMed] [Google Scholar]
- 20.Wilson FC, Ytterberg SR, St Sauver JL, Reed AM. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35(3):445–47. [PubMed] [Google Scholar]
- 21.Dimachkie MM, Barohn RJ. Inclusion body myositis. Semin Neurol. 2012;32(3):237–45. doi: 10.1055/s-0032-1329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brito JP, Hay ID, Morris JC, et al. Low risk papillary thyroid cancer. BMJ. 2014;348:g3045. doi: 10.1136/bmj.g3045. [DOI] [PubMed] [Google Scholar]
- 23.Buchbinder R, Hill CL. Malignancy in patients with inflammatory myopathy. Curr Rheum Rep. 2002;4(5):415–26. doi: 10.1007/s11926-002-0087-9. [DOI] [PubMed] [Google Scholar]
- 24.Callen JP. Relationship of cancer to inflammatory muscle diseases. dermatomyositis, polymyositis, and inclusion body myositis. Rheum Dis Clin North Am. 1994;20(4):943–53. [PubMed] [Google Scholar]
- 25.Shah M, Shah NB, Moder KG, Dean D. Three cases of dermatomyositis associated with papillary thyroid cancer. Endocr Pract. 2013;19(6):154–57. doi: 10.4158/EP13145.CR. [DOI] [PubMed] [Google Scholar]
- 26.Kalliabakos D, Pappas A, Lagoudianakis E, et al. A case of polymyositis associated with papillary thyroid cancer: A case report. Cases J. 2008;1(1):289. doi: 10.1186/1757-1626-1-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attarian H, Applebee G, von Lepel A. Paraneoplastic myoclonus with papillary thyroid carcinoma. Eur Neurol. 2007;58(3):182–83. doi: 10.1159/000104721. [DOI] [PubMed] [Google Scholar]
- 28.Gratwicke JP, Alli A, Rollin M, et al. Paraneoplastic cerebellar syndrome and sensory ganglionopathy with papillary thyroid carcinoma. J Neurol Sci. 2014;341(1):183–84. doi: 10.1016/j.jns.2014.03.042. [DOI] [PubMed] [Google Scholar]
- 29.Yazdi S, Saadat P, Young S, et al. Acquired reactive perforating collagenosis associated with papillary thyroid carcinoma: A paraneoplastic phenomenon? Clin Exp Dermatol. 2010;35(2):152–55. doi: 10.1111/j.1365-2230.2009.03211.x. [DOI] [PubMed] [Google Scholar]
- 30.Inoue R, Kato T, Kim F, et al. A case of adult-onset Still’s disease (aosd)-like manifestations abruptly developing during confirmation of a diagnosis of metastatic papillary thyroid carcinoma. Mod Rheumatol. 2012;22(5):796–800. doi: 10.1007/s10165-011-0588-3. [DOI] [PubMed] [Google Scholar]
- 31.Chow S, Shao J, Wang H. Sample size calculations in clinical research. 2nd ed. Chapman & Hall/CRC; Biostatistics Series: 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Diaz PJ, Garcia SF, Sanchez LJ, Saborido FJ.
Inclusion body myositis with respiratory onset.
Revista Clinica Espanola 2003; 203(3): 164
Inclusion body myositis (IBM) is a spontaneous, inflammatory, idiopathic myopathy which is occasionally hereditary. It affects more females than males (3: 1) and is the most common myopathy in the elderly population i.e. over 50 years old.
Weakness and muscle atrophy are painless and can be generalized or limited just to the limbs. In 20% of cases weakness starts with the quadriceps or the finger flexors – mainly flexor pollicis longus.
Deep tendon reflexes are normal at the beginning but they decrease as the disease progresses. Dysphagia is frequent. CPK is normal or can be elevated. EMG shows a myopathic pattern (i.e. mixed potentials), fibrillation or rarely a neuropathic pattern. Muscle biopsy shows structural changes and inflammatory basophilic and eosinophilic intracytoplasmic vacuoles, which stain for β-amyloid protein. Electronic microscopy (EM) identifies an accumulation of β-amyloid with abnormal tubular filaments in the nucleus and cytoplasm. Respiratory compromise is rare.
The following case is that of a 74 year-old woman with a history of hyperlipidemia and depression; she was not taking any treatment for these. She presented with three days of fever, cough and yellow sputum. She had severe respiratory distress with a PO2 of 42 mmHg and a lobar consolidation in the right lung. Mechanical ventilation and cefotaxime with clarithromycin were started with clinical and radiological improvement. After extubation, she had episodes when her PO2 dropped and PCO2 rose.
EMG showed a myopathic pattern with mixed potentials and short and prolonged motor unit potentials. A quadriceps muscle biopsy showed small, angled, striated fibers and small, basophilic nuclear vacuoles. Trichrome, Gomori and PAS stains were negative; β-amyloid was positive. EM showed intranuclear and intracytoplasmic bodies with disruption of the myofibers and the sarcolemma. She was started on prednisone 1 mg/kg/day po for 2 months, with no response. Breathing was maintained by mechanical ventilation (a tracheostomy was performed).
Respiratory compromise is rare in IBM; aspiration pneumonia and interstitial pneumonia have been described. The pathophysiology is unknown but an immunologic mechanism has been postulated such as toxicity from cytotoxic T cells, T cell cytokines, macrophages and adhesion molecules. This proposed immunologic mechanism is supported by the association [of IBM] with SLE and sarcoidosis. Some inducers of autoimmunity have been proposed, such as HTLV-1 and HCV, but no clear association has been shown.
Other factors which may be important for an immunologic etiology are the deposition of β-amyloid, mitochondrial anomalies and deletions of mitochondrial DNA in the muscle fibers. The vacuoles of the muscle fiber contain β2-secretases with proteolytic activity (BACE-1 and BACE-2), suggesting its participation in processing of the abnormal β-amyloid protein.
Treatment in general is discouraging. Steroids, ivIg, cyclosporine, cyclophosphamide and interferon β-1a have been used with variable responses but poor success overall. Respiratory compromise is infrequent. Therefore, we recommend IBM should be considered in any older patient (aged >50) with respiratory failure.