

Abstract
Endogenous danger signals, or damage-associated molecular patterns (DAMPs), are generated in response to cell stress and activate innate immunity to provide a pivotal mechanism by which an organism can respond to damaged self. Accumulating experimental and clinical data have established the importance of DAMPs, which signal through innate pattern recognition receptors (PRRs) or DAMP-specific receptors, in regulating the alloresponse to solid organ transplantation (SOT). Moreover, DAMPs may incite distinct downstream cellular responses that could specifically contribute to the development of allograft fibrosis and chronic graft dysfunction. A growing understanding of the role of DAMPs in directing the immune response to transplantation has suggested novel avenues for the treatment or prevention of allograft rejection that complement contemporary immunosuppression and could lead to improved outcomes for solid organ recipients.
Introduction
Solid organ transplantation (SOT) provides life-extending treatment for patients with advanced kidney, heart, liver, or lung failure. Despite growth in transplant volumes and improved short-term post-transplant outcomes, the success of each of these commonly transplanted solid organs is limited by the development of acute rejection and chronic graft dysfunction. The frequent occurrence of acute rejection despite contemporary T cell–based immunosuppression highlights the shortcomings of available therapies. Furthermore, the immune mechanisms leading to the development of chronic graft dysfunction remain poorly understood. A greater understanding of the host immunological response to SOT is required to develop novel approaches to improve patient outcomes.
While allograft rejection was first understood to occur as a result of the host adaptive immune response to donor MHC antigens, an increasingly complex picture is emerging involving many more facets of host immunity. Recent studies have established innate immunity as a critical mediator of solid organ transplant rejection (1–3). Innate immunity relies on recognition of highly conserved molecular patterns by germline-encoded pattern recognition receptors (PRRs). Although such innate mechanisms provide host defense against invading pathogens through recognition of pathogen-associated molecular patterns (PAMPs), endogenous “danger signals” generated in response to cell stress (in the absence of infection) can similarly activate innate PRRs, providing a pivotal mechanism by which an organism can sense and respond to damaged self (4).
In the 20 years following the description of this “danger hypothesis” by Polly Matzinger (4), numerous endogenous molecules, collectively referred to as damage-associated molecular patterns (DAMPs), have been identified that activate innate immunity. DAMPs are of particular relevance in the context of organ transplantation because of the injury inherent in the removal of an organ from one individual and later implantation into another. A body of literature has emerged describing release of DAMPs upon SOT, and studies have begun to elucidate a role for specific DAMPs and their relevant receptors in regulating the host immune response to the allograft. This Review will discuss experimental and clinical evidence supporting the importance of DAMPs in solid organ transplant rejection, emphasize mechanisms by which DAMP-mediated innate activation influences the development of alloimmunity, and consider how emerging knowledge of the immune response to DAMPs could be exploited to derive novel therapeutics to improve solid organ transplant outcomes.
DAMPs as endogenous danger signals in SOT
Broadly, DAMPs can be divided into two categories: (a) intracellular constituents such as nucleic acids or HSPs that remain sequestered from the immune system in homeostatic conditions, but are released into the extracellular milieu or exposed on the cell surface following cell damage, and (b) extracellular constituents, such as extracellular matrix (ECM) components, that are modified in the context of cell stress or injury. There is now compelling evidence that clinical events occurring across all commonly transplanted solid organs can generate DAMPs (Figure 1). For instance, all transplanted organs necessarily sustain some degree of ischemia/reperfusion injury (IRI) in the process of procurement and reimplantation, and IRI has been shown to directly generate innate DAMPs (5). In the case of cadaveric organs, donor brain death results in accumulation of DAMPs in tissues subsequently procured for transplantation (1, 6, 7). The clinical relevance of these observations is supported by evidence that despite HLA mismatch, recipients of live, unrelated donor kidneys that undergo only a brief duration of warm ischemia have improved long-term outcomes as compared with recipients of highly HLA-matched cadaveric organs that necessarily experience donor brain death and longer periods of cold ischemia (8).
Figure 1. Danger signals in solid organ transplantation (SOT).
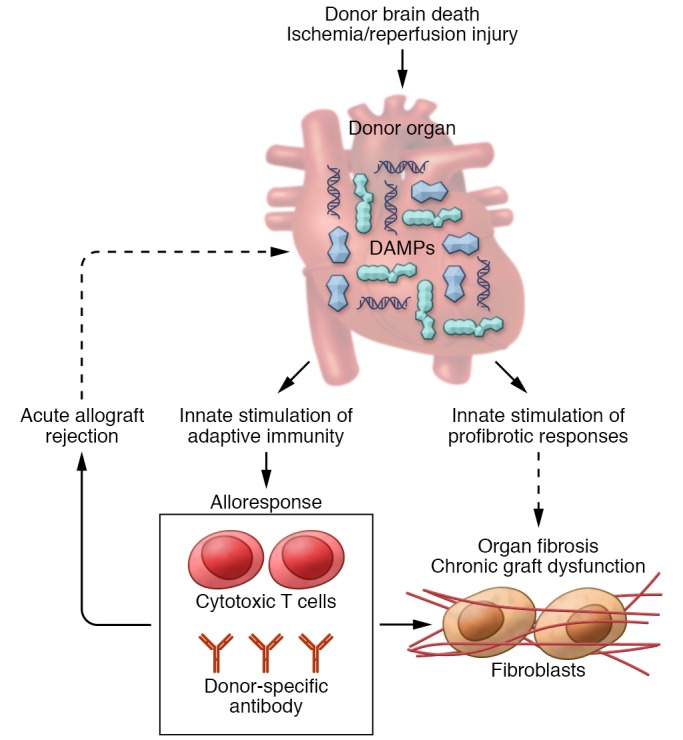
In the context of SOT, DAMPs can accumulate in the donor organ during the process of donor brain death or as a result of IRI. DAMPs act through TLRs or DAMP-specific receptors to activate innate immunity and enhance the alloresponse by upregulating donor alloantigen presentation, providing costimulation to recipient T cells and driving effector T cell recruitment and differentiation. Cytotoxic T cells and donor-specific antibody generated from the alloresponse can cause acute allograft rejection, which may in turn cause sustained graft injury, thereby potentiating DAMP release. Evolving data suggest DAMPs may also directly promote fibrotic responses, thereby potentially contributing, along with alloimmunity, to the development of organ fibrosis and chronic graft dysfunction.
In addition to perioperative injury, there are likely to be organ transplant–specific mechanisms of DAMP generation. Specifically, cytotoxic T cells and alloantibody generated as part of host alloimmunity can result in significant noninfectious injury to structural graft cells, leading to DAMP release. In keeping with this idea, acute rejection has been associated with intragraft DAMP accumulation (9–11). Thus, it is possible that the ongoing alloresponse to the graft can generate DAMPs, which in turn drive further immune activation toward the allograft (Figure 1). The persistent generation of DAMPs over the lifetime of the allograft, even after IRI has resolved, could represent a critical mechanism by which alloreactivity is perpetuated after organ transplantation.
A majority of DAMPs signal through TLRs, a family of prototypic innate PRRs (12). A variety of cell types express TLRs, including most immune cells as well as some structural cells. TLR ligation initiates a complex intracellular kinase cascade, ultimately activating the transcription factor NF-κB and altering expression of inflammatory response genes. Depending on the stimuli and adaptor proteins involved, TLR signaling can lead to inflammatory cytokine secretion, neutrophil recruitment, antigen-presenting cell maturation, and upregulation of costimulatory molecules and MHC. In the context of organ transplantation, TLR signaling promotes alloimmunity and potentiates organ immunogenicity by upregulating donor alloantigen presentation, providing requisite costimulation to recipient T cells, and creating a cytokine environment that drives effector T cell recruitment and differentiation.
Early experimental evidence implicating TLR signaling in allograft rejection was provided by studies of minor mismatch skin transplantation, in which the downstream adaptor protein MyD88 was demonstrated to be necessary for the development of graft rejection (13). Further experimental data confirmed the importance of MyD88 in kidney and heart rejection (14, 15). Interestingly, while MyD88 deficiency in either the donor or recipient improved graft outcomes in these experimental models, the greatest attenuation of rejection occurred when TLR signaling was impaired in both donor and recipient. The first human data corroborating the relevance of TLR signaling to solid organ transplant outcomes demonstrated reduced acute rejection and improved long-term outcomes in lung transplant recipients with a loss-of-function polymorphism in TLR4 (16). Subsequently, similar results were reported with this TLR4 polymorphism in kidney and liver transplant donors (6, 17).
Although TLRs are the receptors most commonly employed by DAMPs, some DAMPs also mediate effects through non-PRRs. For example, ligation of the transmembrane receptor for advanced glycation end products (RAGE) can activate kinases independent of TLR signaling and act in cooperation with TLRs to modulate the inflammatory response to DAMPs. The DAMPs and their corresponding receptors with supporting evidence in SOT are outlined in Table 1 and further detailed below.
Table 1. DAMPs with supporting evidence in solid organ transplantation.

DAMPs derived from intracellular constituents
High-mobility group box 1.
High-mobility group box 1 (HMGB1) is a nuclear protein that maintains chromosomal architecture but, when released into the extracellular space by damaged cells, can also act as a DAMP. Extracellular HMGB1 induces several responses through ligation of TLR4 and RAGE, including stimulating proinflammatory cytokines and chemokines and inducing cell proliferation and migration. HMGB1 release occurs in the context of IRI of each commonly transplanted solid organ, including the kidney (5), heart (18), liver (19), and lung (7). The clinical significance and consequences of HMGB1 release on allograft function were illustrated in a translational study of kidney transplant donors. HMGB1 and TLR4 were upregulated in the renal tubules of cadaveric, as compared with live, kidney donors. In vitro experiments demonstrated that HMGB1 induces inflammatory responses in human renal tubular cells in a TLR-dependent manner. Finally, the organs from kidney donors with loss-of-function TLR4 genetic variants were protected against the development of early allograft dysfunction and exhibited diminished intragraft inflammatory cytokine transcript expression (6). In a similar translational study of lung transplantation, elevated serum HMGB1 in cadaveric donors correlated with impaired oxygenation of the donor lung before and early after lung transplant. Consistent with these clinical observations, experimental data from murine lung transplant models demonstrated brain death occurring after traumatic brain injury (a common mode of death among lung donors) induced pulmonary dysfunction and increased RAGE levels. Moreover, both RAGE-deficient and WT mice treated with HMGB1 antibody were protected against pulmonary dysfunction and had improved lung allograft outcomes. Interestingly, increased IL-10 was observed in the RAGE-deficient mice, suggesting that activation of the HMGB1/RAGE axis contributes to inferior graft outcomes by inhibiting the production of antiinflammatory cytokines (7). Studies in heart transplant models suggest additional mechanisms linking HMGB1 to graft rejection, including promotion of dendritic cell (DC) maturation, stimulation of Th1 immunity, and activation of IL-17–producing invariant NKT cells (20–22).
While these and other studies have established the importance of HMGB1 in acute allograft rejection, recent experimental and clinical studies have extended the role of HMGB1 to chronic graft dysfunction in heart and lung transplantation. In an experimental model of chronic cardiac graft dysfunction, HMGB1 inhibition reduced graft inflammation and organ fibrosis (23). Similarly, in a murine experimental model of chronic lung allograft dysfunction, HMGB1 induced latent TGF-β release and enhanced lung fibrosis through a RAGE-dependent mechanism (24). Further support for HMGB1 in chronic graft dysfunction after lung transplantation was provided in a clinical study demonstrating elevated HMGB1 in the lung fluid of patients with restrictive allograft syndrome, a recently described phenotype of chronic lung allograft dysfunction hallmarked by parenchymal pulmonary fibrosis (25).
ATP.
In contrast to the role of intracellular ATP as a universal energy source, extracellular ATP functions as a DAMP. Unlike other DAMPs that signal through common innate PRRs, extracellular ATP is recognized by specific cell surface nucleoside receptors in the P2 receptor family, including P2X and P2Y receptors. P2X receptors consist of ion channels that regulate cellular cation transport, while P2Y receptors are GPCRs. Binding of extracellular ATP to P2X or P2Y receptors has a wide range of immune and inflammatory effects on macrophages, DCs, mast cells, and lymphocytes (26).
The direct relevance of extracellular ATP as a DAMP in the context of SOT was highlighted by a study demonstrating that expression of P2X7 was elevated in graft-infiltrating lymphocytes in patients and mice after heart transplantation. Targeting P2X7 with the irreversible antagonist periodate-oxidized ATP for 14 days after fully mismatched murine heart transplant promoted long-term cardiac transplant survival, which was associated with reduced donor-specific T cell activation, reduced Th1 and Th17 differentiation, and inhibited STAT3 phosphorylation in T cells. Finally, in a separate murine model of chronic heart transplant dysfunction, P2X7 inhibition prevented the development of chronic heart allograft vasculopathy (27).
Extracellular ATP is hydrolyzed by nucleotidases including NTPDase 1 (CD39), which converts ATP to AMP to maintain homeostatic ATP levels and shut down purinergic signaling. Further evidence implicating ATP as a mediator of solid organ transplant rejection was provided by a study demonstrating that fully MHC-mismatched liver transplants performed in CD39-deficient mice have evidence of increased allograft dysfunction, increased IFN-γ production by liver-infiltrating CD8+ T cells, and greater proliferative responses to donor alloantigens in comparison with mice receiving WT grafts. Moreover, Treg frequency and overall graft survival were significantly reduced in CD39-deficient livers (28).
Nucleic acids and mitochondrial DAMPs.
Release of nuclear DNA fragments, mitochondrial DNA (mtDNA) fragments, and other mitochondrial contents can occur upon tissue injury, activating innate immunity to promote downstream adaptive immune responses through ligation of TLR9 or NALP3 (cryopyrin). Such nucleic acid or mitochondrial products accumulate in the extracellular space and enter into the circulation as a consequence of IRI or in the context of acute or chronic allograft rejection (29–34). Consistent with this idea, recent work in heart, lung, and kidney transplantation has identified accumulation of donor-derived cell-free DNA in the blood of rejecting organ recipients (34). Defining the influence of these cell-free DNA fragments on intragraft immune responses is an important area for further study. In particular, there is interest in understanding whether the cell-free fraction includes mtDNA, which contains unmethylated CpG motifs similar to bacterial DNA and has been associated with proinflammatory responses in non–transplant-related experimental or human studies (35, 36).
HSPs.
The highly conserved heat shock response, characterized by upregulation of HSPs, is triggered in a range of potentially damaging situations, including hypoxia, oxidative stress, inflammation, or infection. While intracellular HSPs mediate important homeostatic functions, HSPs released or secreted into the extracellular space during tissue injury can act as DAMPs and amplify the alloresponse in the context of SOT. In keeping with this idea, graft-infiltrating lymphocytes from a rodent heterotopic heart transplant model showed a marked increase in proliferation when exposed to donor MHC in the presence of HSP65 or HSP70 as compared with donor MHC in isolation (37). Early clinical studies in heart and renal allograft recipients corroborated these findings and suggested that human allograft-derived T cells demonstrate HSP reactivity (38, 39).
HSP expression has since been demonstrated to be increased in the context of kidney (40–42), heart (43–46), and lung (47, 48) transplant rejection; however, some of these studies have yielded conflicting results. For example, while HSP27 has been shown to be significantly increased in rejecting as compared with nonrejecting cardiac allografts (45), loss of vascular HSP27 has also been associated with the development of chronic cardiac allograft vasculopathy (49). Additional findings in clinical liver transplantation point to a role for increased HSP70 expression in protecting against early graft rejection (50). These inconsistent findings potentially relate to context-dependent actions of HSPs. While some studies have demonstrated immune-stimulating properties of HSPs, other experimental evidence supports an immune-inhibitory role. HSPs have been proposed as resolution-associated molecular patterns that induce the production of immunoregulatory cytokines such as IL-10 to mitigate further tissue damage (51). Consistent with this idea, studies in skin transplant models have demonstrated that elevated HSP expression increases IL-10 levels and Treg recruitment, and prolongs allograft survival (52–54). These studies emphasize that the impact of HSP expression on the subsequent alloimmune response is likely to be highly context-specific and possibly even organ-specific.
DAMPs derived from extracellular constituents
Haptoglobin.
Haptoglobin is an acute-phase serum protein that, in addition to binding free hemoglobin, influences free radical formation and angiogenesis. More recently, haptoglobin has been described as a novel mediator of innate immune activation after heart transplantation (55, 56). Haptoglobin expression is increased in the context of both human and experimental cardiac transplant rejection, and targeted depletion of haptoglobin led to sustained costimulatory blockade–induced tolerance in an experimental heart transplant model, suggesting that its presence impairs maintenance of tolerance. In vitro studies demonstrated that haptoglobin exerts its immune effects through increased expression of the proinflammatory cytokines IL-6 and macrophage inflammatory protein-2 (MIP-2), while reducing levels of the antiinflammatory cytokine IL-10. Haptoglobin also enhanced DC graft recruitment and augmented antidonor T cell responses (56). Previous studies using a murine skin transplant model demonstrated that haptoglobin activates donor DCs in a MyD88-dependent manner, implying a role for TLR signaling in mediating the effects of haptoglobin in transplantation (55). The relevance of haptoglobin in driving the alloresponse to other solid organs remains unknown and represents a promising area for further exploration.
Hyaluronan.
Hyaluronan (HA) is an essential component of the ECM that exists as a high–molecular weight polymer (HMWHA) under homeostatic conditions. HA has been shown to mediate a wide array of biological functions owing to the breadth of HA receptors (including alternatively spliced and variant forms) in addition to its modification to lower–molecular weight HA (LMWHA) fragments or cross-linked leukocyte-adhesive HA matrices (57, 58). While HMWHA has been demonstrated to attenuate inflammation and protect from inflammatory damage (57, 59–61), LMWHA fragments generated in response to tissue injury or cell stress can act as potent DAMPs. HA has been demonstrated to signal through CD44 to activate MAP kinase cascades or through interaction with TLR4 to coordinate the innate response and influence outcomes after SOT.
In a rat model of fully MHC-mismatched heart transplant, IRI upregulated tissue expression of HA and transcript levels of HA synthases 1–3 (HAS1–HAS3) in the early post-transplant period. This early increase in HA was followed by an exaggerated T cell inflammatory infiltrate, increased Th17-related cytokines, and worse graft fibrosis (62). In another study using a rodent model of chronic heart allograft dysfunction, chronically rejecting hearts had extensive graft accumulation of extracellular HA, and fibroblasts isolated from affected hearts demonstrated increased CD44 mRNA when compared with fibroblasts isolated from nonrejecting heart allografts. Furthermore, HA promoted in vitro fibroblast adhesion and migration in a CD44-dependent manner and potentiated ex vivo fibroblast survival (63). These studies suggest that the CD44/HA axis augments alloreactivity and contributes to the pathobiology of allograft fibrosis in chronic graft dysfunction.
In a translational study of human and murine lung transplantation, Todd and colleagues demonstrated that human lung allografts with chronic graft dysfunction have extensive tissue accumulation of HA in regions of airway fibrosis, in addition to increased lung fluid HA, and significantly upregulated tissue transcripts for HAS1–3. Parallel studies in a murine orthotopic lung transplant model revealed that administration of LMWHA (but not intact HA) can overcome costimulatory blockade–induced tolerance and promote graft alloimmunity through a TLR2/4-dependent mechanism (64). While previous studies suggested that PAMP exposure could break established tolerance after SOT (2, 3), this study demonstrated that even in the absence of infectious stimuli, DAMPs can overcome tolerance to promote alloimmunity and graft rejection. Cui and colleagues extended these findings to demonstrate increased HA expression in acute lung rejection in humans (65). Further experiments in a murine lung transplant model showed that LMWHA accumulates in acute lung rejection as a result of diminished clearance through the lymphatic vessel endothelial HA receptor LYVE-1. This finding is particularly notable in light of the prior study by Todd and colleagues that demonstrated that LMWHA, at least when given exogenously, can break established tolerance. Taken together, these studies suggest that LMWHA accumulation in the setting of acute rejection may represent a mechanism by which the alloresponse to the graft is perpetuated and ultimately contributes to graft fibrosis and failure. Notably, in the experimental model, therapeutic lymphangiogenesis improved HA clearance and attenuated acute rejection histology (65), indicating a potentially novel approach to mitigate HA-induced endogenous innate immune activation in the context of transplantation. Further studies are needed to determine whether this approach would be specific to lung transplantation or whether similar mechanisms of impaired lymphatic LMWHA clearance exist and contribute to graft rejection in other commonly transplanted solid organs.
Heparan sulfate.
Heparan sulfate (HS) is a proteoglycan ubiquitously expressed on cell surfaces and in the ECM of normal human tissues. HS can interact with a wide array of proteins to regulate functions including endothelial cell integrity, blood coagulation, and complement activation. Both surface-bound and soluble HS can act as DAMPs to initiate the immune response in the context of cell injury. Moreover, cell surface HS can enable the response to other DAMPs, specifically HMGB1, by facilitating the oligomerization of RAGE, an essential step in RAGE signal transduction, while soluble HS can interact with TLR4 to release proinflammatory cytokines and promote DC maturation (66–68).
In the context of SOT, expression of the HS proteoglycan perlecan, an important component of the vascular matrix, is elevated in arteries affected by transplant vasculopathy in a rodent model of chronic renal allograft dysfunction (69). Furthermore, perlecan has been shown to interact with FGF2 to contribute to fibroblast activation in the development of chronic graft impairment (70). Studies in human renal transplant recipients have confirmed the clinical relevance of perlecan expression in kidney rejection, as higher serum or urinary levels of the perlecan C-terminal fragment associate with acute vascular rejection or advanced chronic allograft nephropathy, respectively (71, 72). A recent translational study by Adepu and colleagues also implicated the HS syndecan-1 as important in contributing to graft outcomes in experimental and clinical kidney transplantation (73).
Tenascin C.
Tenascin C (TNC) is a large matrix glycoprotein expressed at low levels in normal ECM, but is upregulated in response to tissue injury, where it can function as a DAMP. Elevated tissue TNC expression has been reported in rodent models of chronic allograft dysfunction involving kidney (74), lung (75), and heart (76) transplantation. The mechanistic role for TNC in organ transplantation remains to be fully determined; however, a role for TNC as a DAMP in allograft rejection and fibrosis is suggested by studies of cardiac remodeling, wherein TNC-deficient mice are protected against post–myocardial infarction fibrosis (77). Consistent with this observation, TNC augmented expression of proinflammatory cytokines and proteins important to tissue remodeling in cardiac myofibroblasts in a TLR4-dependent manner (78). Still other nontransplant models point to a potential role for TNC in mediating DC maturation and promoting Th17 immunity (79).
Fibronectin.
Similar to TNC, fibronectin is a ubiquitously expressed ECM glycoprotein. In the setting of tissue damage, cellular fibronectin containing an alternatively spliced exon encoding type III extra domain A (EDA) is generated. Such EDA-containing fibronectin fragments (EDA-fibronectin) activate innate immunity through TLR4 (80). Fibronectin expression was increased in the context of chronic vascular rejection in a nonhuman primate kidney transplant model, and plasma EDA-fibronectin levels correlated with acute rejection in human renal transplant recipients (81, 82). Other experimental and clinical studies have examined the role of EDA-containing fibronectin in the pathogenesis of cardiac allograft vasculopathy and cardiac fibrosis after heart transplantation (83–87). Booth and colleagues used a murine heterotopic cardiac transplant model to demonstrate that although heart allografts in EDA-fibronectin–deficient recipients experienced acute rejection similarly to those in WT recipients, they were protected from the development of allograft fibrosis and chronic graft dysfunction (85). Importantly, a clinical study by Franz and colleagues noted tissue expression of EDA-fibronectin associated with inflammation and histological signs of chronic rejection in human heart transplant biopsies (87). These data provide a strong rationale for further studies examining the exact signaling pathways stimulated by EDA-fibronectin in organ transplant rejection and indicate that strategies targeting fibronectin may hold promise to aid in diagnosing, preventing, or treating cardiac rejection in particular.
Beyond these DAMPs of known importance in solid organ transplant, many additional endogenous danger molecules have been recognized to exert effects through innate immunity (88–90). A critical area of future research will be to elucidate the potential role for these DAMPs in the transplant setting. Other ECM proteoglycans, such as biglycan or decorin, are likely to be modified in the setting of allograft injury similarly to HA or fibronectin. In particular, soluble decorin has been demonstrated to exert antifibrotic effects and confer protection from apoptosis in nontransplant models of renal disease (91), making it an attractive area for further study in chronic allograft nephropathy after kidney transplantation. Intracellular DAMPs beyond those currently implicated in SOT could be released as a consequence of IRI. For example, studies in nontransplant models of cardiac IRI point to the importance of extracellular RNA signaling through a TLR3/TIR domain–containing adaptor–inducing interferon-β (TRIF) pathway (92). Understanding the full range of DAMP expression in SOT will be essential to provide a more comprehensive view of the role of DAMPs in directing the immune response to the allograft and potentially identify promising strategies to improve graft outcomes.
Evolving paradigms for DAMPs in SOT
Collectively, this body of basic and clinical work across multiple transplanted solid organs indicates that DAMPs act through TLRs or DAMP-specific receptors to mediate acute rejection and chronic graft dysfunction. Much remains to be understood with respect to the role of DAMPs in regulating adaptive alloimmunity. Furthermore, significant gaps remain in understanding the exact mechanisms by which DAMPs contribute to chronic graft dysfunction and the extent to which certain DAMPs are unique to individual organs or potentially drive organ-specific differences in the frequency or manifestations of rejection.
To this point, a particularly exciting area is the development of a more nuanced understanding of the manner in which DAMPs may drive fibrosis-biased tissue remodeling within an allograft. Subtle yet important distinctions in downstream signaling induced by DAMPs versus PAMPs have now been identified, even when sharing a common receptor. For example, Piccinini and colleagues recently demonstrated divergent cellular response in macrophages exposed to the bacterial PAMP LPS or the endogenous DAMP TNC, despite shared signaling through TLR4. While both stimuli activated NF-κB and MAP kinase, each initiated different downstream signaling pathways leading to distinct patterns of cytokine and MMP expression and unique functional cellular responses. Specifically, in response to TNC, macrophages increased synthesis and phosphorylation of matrix, while in response to LPS, the macrophage capacity for matrix degradation was increased. While the mechanisms underlying these observations are not fully understood, the authors demonstrated that macrophages exposed to TNC were shifted toward an IL-4–induced activation phenotype and produced less TNF-α and IL-10 than LPS-exposed macrophages (93). These data provide clues to the way in which DAMPs may induce distinct signals in the context of transplant that differ from those initiated in response to exogenous infections/PAMPs.
These data, in addition to the preponderance of matrix-related DAMPs described in chronic graft dysfunction after SOT, support the hypothesis that DAMPs, in contrast to PAMPs, may be directly involved in promoting allograft fibrosis. Further supporting this idea, deposition of EDA-fibronectin precedes fibroblast α-smooth muscle actin expression (commonly associated with transition to a myofibroblast phenotype) in vivo in a rodent model of wound repair and in vitro during fibroblast stimulation with TGF-β1 (94). Similarly, others have demonstrated that the fibroblast response to TGF-β1 can be regulated by modification of the level of fibroblast-generated HA either through overexpression of HAS2 or by blockade of HA synthesis (95). Collectively, these observations extend the existing conceptual model in which DAMPs activate TLRs to promote alloimmunity, which in turn drives acute rejection and chronic graft dysfunction, and implicate novel DAMP-driven innate mechanisms that could drive graft fibrosis.
Another gap in our understanding of clinical outcomes after SOT relates to organ-specific differences in the rates of acute rejection and manifestations of chronic graft dysfunction among the transplanted solid organs. For example, despite the use of similar immunosuppression, acute rejection occurs in less than 10% of kidney transplant recipients but in more than 35% of lung recipients (96). Furthermore, a key feature of chronic allograft dysfunction in heart transplantation is intraluminal obliteration of the coronary vasculature, while in lung transplantation histological manifestations of chronic graft dysfunction generally include intraluminal obliteration of the epithelialized airways. It is plausible that these differences could be, at least in part, explained by the location and type of DAMPs expressed in response to early transplant IRI or alloimmune-mediated injury, consistent with the organ-specific differences in significant DAMPs identified across the commonly transplanted organs (Table 1). This line of investigation represents an important area of future study with potential to impact the development of organ-specific treatments.
Implications for DAMPs in SOT therapies
A growing understanding of the role of DAMPs in initiating and directing the immune response to SOT offers the opportunity to reconsider therapeutic approaches to prevent or treat allograft rejection. At the same time, increasing application of ex vivo organ preservation systems presents a unique opportunity to manipulate donor organs before implantation so as to reduce IRI, thereby diminishing innate activation and potentially promoting immune tolerance. For example, in lung transplantation organs procured through ex vivo perfusion have been used successfully for clinical transplantation and have been successfully manipulated in experimental models to reduce IRI through adenovirus-mediated IL-10 production (97–99).
To this point, interestingly, laboratory studies have suggested that low-level DAMP exposure prior to transplantation, or preconditioning, can reduce the impact of IRI. Ischemic preconditioning with HMGB1 protected against renal or hepatic IRI, an effect shown to be dependent on TLR4 signaling (100, 101). Similarly, genetic overexpression of HSPs in the donor organ can alter alloimmunity in the context of experimental heart transplantation. Specifically, hearts transplanted from transgenic mice overexpressing HSP27 experienced a delayed onset of acute rejection and reduced IRI-induced apoptosis as compared with HSP27-negative hearts from littermate controls (102). A potential mechanism to explain these observations is innate immune-driven immunological memory occurring as a result of epigenetic reprogramming after stimulation. While immune memory was previously thought to be driven exclusively by adaptive immunity, new evidence suggests that tissue-derived signals can induce epigenetic changes to regulate the magnitude and type of future immune responses (103). Such epigenetic modifications can diminish subsequent responses, as is the case with LPS tolerance. Further understanding the mechanisms of innate memory could provide avenues to dampen the response to endogenous DAMPs induced in the transplant setting.
Decreasing DAMP expression, enhancing DAMP clearance, or blocking DAMP signaling may also represent novel approaches to mitigate allograft rejection. A recent study investigated the impact of recombinant soluble thrombomodulin, which can bind HMGB1 and prevent HMGB1-mediated proinflammatory responses, on IRI in liver grafts. Thrombomodulin treatment attenuated levels of HMGB1 and inflammatory cytokines, decreased markers of liver graft injury, and improved organ function (104). Other studies suggest that HA production may be an actionable target. The HAS inhibitor 4-methylumbelliferone, a drug approved for use in Europe in patients with colon cancer, has been shown to reduce expression of HA and the severity of IRI in the kidney, suggesting it could be used to treat donor allografts to reduce early graft dysfunction (105). Alternatively, in experimental lung transplantation, impaired lymphangiogenesis leads to LMWHA accumulation, a process shown to be ameliorated by specific pharmacotherapy with VEGF-C156S treatment (9).
Beyond these therapeutics directed at modulating DAMP accumulation, several TLR antagonists are under development for treatment of inflammatory diseases or cancer (106). Progress with such agents has moved slowly and proved ineffective in the treatment of conditions such as sepsis. Additionally, while an abundance of experimental data suggests that treatments targeting TLRs have potential to attenuate the alloresponse in the context of organ transplantation, substantial concerns exist regarding the increased risk for infectious complications with these agents. Ultimately, strategies targeting specific DAMPs or precise points in innate pathways may represent more promising avenues to effectively prevent acute rejection and chronic graft dysfunction after SOT.
Conclusion
Accumulating data support the paradigm that, independent of donor MHC, the response to SOT entails activation of innate immunity through endogenous DAMPs. DAMPs, such as HMGB1 and HA, have been identified as modulators of graft outcomes across multiple solid organs and act through TLRs as well as DAMP-specific receptors to create a proinflammatory environment that promotes alloresponsiveness. Recent evidence suggests that allograft-induced persistent immune stimuli could promote DAMP release. Furthermore, DAMPs may activate distinct downstream signals that could promote tissue remodeling and fibrosis. Given structural differences among the cells and matrix constituents of the commonly transplanted solid organs, it is likely that evidence will emerge to suggest that distinct DAMPs contribute to organ-specific differences in acute rejection rates and manifestations of chronic allograft dysfunction. The emerging understanding of DAMPs and their role in innate/adaptive crosstalk in SOT has suggested novel therapeutic approaches that could complement contemporary immunosuppression. These approaches should be aggressively pursued in clinical studies to test the hypothesis that inhibition of innate, as well as adaptive, immunity is necessary to achieve successful long-term allograft outcomes.
Footnotes
Conflict of interest: J.L. Todd discloses research support granted to the institution from Boehringer Ingelheim, GlaxoSmithKline, and Theravance Biopharma Inc. J.L. Todd has served in an advisory role for Therabron Therapeutics Inc. S.M. Palmer discloses research support granted to the institution from Boehringer Ingelheim and Gilead Sciences. S.M. Palmer has served in an advisory role for Boehringer Ingelheim, Chimerix, Renovion, Sofinnova Partners, and Therabron Therapeutics Inc. S.M. Palmer’s spouse is an employee of Lexicon.
Reference information:J Clin Invest. 2017;127(7):2464–2472.https://doi.org/10.1172/JCI90594.
References
- 1.Kim IK, Bedi DS, Denecke C, Ge X, Tullius SG. Impact of innate and adaptive immunity on rejection and tolerance. Transplantation. 2008;86(7):889–894. doi: 10.1097/TP.0b013e318186ac4a. [DOI] [PubMed] [Google Scholar]
- 2.Thornley TB, et al. TLR agonists abrogate costimulation blockade-induced prolongation of skin allografts. J Immunol. 2006;176(3):1561–1570. doi: 10.4049/jimmunol.176.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, et al. TLR engagement prevents transplantation tolerance. Am J Transplant. 2006;6(10):2282–2291. doi: 10.1111/j.1600-6143.2006.01489.x. [DOI] [PubMed] [Google Scholar]
- 4.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 5.Wu H, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krüger B, et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A. 2009;106(9):3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber DJ, et al. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med. 2014;6(252):252ra124. doi: 10.1126/scitranslmed.3009443. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Terasaki PI, Cecka JM, Gjertson DW, Takemoto S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med. 1995;333(6):333–336. doi: 10.1056/NEJM199508103330601. [DOI] [PubMed] [Google Scholar]
- 9.Cui Y, et al. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J Clin Invest. 2015;125(11):4255–4268. doi: 10.1172/JCI79693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnsson C, Tufveson G. Serum hyaluronan — a potential marker of cardiac allograft rejection? J Heart Lung Transplant. 2006;25(5):544–549. doi: 10.1016/j.healun.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 11.Rao PN, et al. Monitoring of acute lung rejection and infection by bronchoalveolar lavage and plasma levels of hyaluronic acid in clinical lung transplantation. J Heart Lung Transplant. 1994;13(6):958–962. [PubMed] [Google Scholar]
- 12.O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors — redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111(10):1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, et al. Absence of MyD88 signaling induces donor-specific kidney allograft tolerance. J Am Soc Nephrol. 2012;23(10):1701–1716. doi: 10.1681/ASN.2012010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tesar BM, Zhang J, Li Q, Goldstein DR. TH1 immune responses to fully MHC mismatched allografts are diminished in the absence of MyD88, a toll-like receptor signal adaptor protein. Am J Transplant. 2004;4(9):1429–1439. doi: 10.1111/j.1600-6143.2004.00544.x. [DOI] [PubMed] [Google Scholar]
- 16.Palmer SM, et al. The role of innate immunity in acute allograft rejection after lung transplantation. Am J Respir Crit Care Med. 2003;168(6):628–632. doi: 10.1164/rccm.200303-447OC. [DOI] [PubMed] [Google Scholar]
- 17.Oetting WS, Guan W, Schladt DP, et al. Donor polymorphisms of TLR4 associated with graft failure in liver transplant recipients. Liver Transpl. 2012;18(12):1399–1405. doi: 10.1002/lt.23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaczorowski DJ, et al. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87(10):1455–1463. doi: 10.1097/TP.0b013e3181a36e5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z, et al. TLR4 as receptor for HMGB1-mediated acute lung injury after liver ischemia/reperfusion injury. Lab Invest. 2013;93(7):792–800. doi: 10.1038/labinvest.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, Laubach VE. Receptor for advanced glycation end products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant. 2013;13(9):2255–2267. doi: 10.1111/ajt.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, et al. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7(4):799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]
- 22.Duan L, et al. High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab Invest. 2011;91(1):43–53. doi: 10.1038/labinvest.2010.141. [DOI] [PubMed] [Google Scholar]
- 23.Zou H, et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory-like mDCs. Am J Transplant. 2014;14(8):1765–1777. doi: 10.1111/ajt.12781. [DOI] [PubMed] [Google Scholar]
- 24.He L, et al. HMGB1 exacerbates bronchiolitis obliterans syndrome via RAGE/NF-κB/HPSE signaling to enhance latent TGF-β release from ECM. Am J Transl Res. 2016;8(5):1971–1984. [PMC free article] [PubMed] [Google Scholar]
- 25.Saito T, et al. Distinct expression patterns of alveolar “alarmins” in subtypes of chronic lung allograft dysfunction. Am J Transplant. 2014;14(6):1425–1432. doi: 10.1111/ajt.12718. [DOI] [PubMed] [Google Scholar]
- 26.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–317. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vergani A, et al. Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation. 2013;127(4):463–475. doi: 10.1161/CIRCULATIONAHA.112.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida O, et al. CD39 deficiency in murine liver allografts promotes inflammatory injury and immune-mediated rejection. Transpl Immunol. 2015;32(2):76–83. doi: 10.1016/j.trim.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Interewicz B, Olszewski WL, Stanislawska J, Sikora M, Durlik M, Maksymowicz M. DNA released from rejecting organs is an indicator of the degree of graft cellular damage. Transplant Proc. 2005;37(1):98–101. doi: 10.1016/j.transproceed.2005.01.064. [DOI] [PubMed] [Google Scholar]
- 30.Ott U, Aschoff A, Fünfstück R, Jirikowski G, Wolf G. DNA fragmentation in acute and chronic rejection after renal transplantation. Transplant Proc. 2007;39(1):73–77. doi: 10.1016/j.transproceed.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 31.Hu Q, Wood CR, Cimen S, Venkatachalam AB, Alwayn IP. Mitochondrial damage-associated molecular patterns (MTDs) are released during hepatic ischemia reperfusion and induce inflammatory responses. PLoS One. 2015;10(10):e0140105. doi: 10.1371/journal.pone.0140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Vlaminck I, et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med. 2014;6(241):241ra77. doi: 10.1126/scitranslmed.3007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Vlaminck I, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci U S A. 2015;112(43):13336–13341. doi: 10.1073/pnas.1517494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gielis EM, et al. Cell-Free DNA: An Upcoming Biomarker in Transplantation. Am J Transplant. 2015;15(10):2541–2551. doi: 10.1111/ajt.13387. [DOI] [PubMed] [Google Scholar]
- 35.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao H, et al. Circulatory mitochondrial DNA is a pro-inflammatory agent in maintenance hemodialysis patients. PLoS One. 2014;9(12):e113179. doi: 10.1371/journal.pone.0113179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moliterno R, Valdivia L, Pan F, Duquesnoy RJ. Heat shock protein reactivity of lymphocytes isolated from heterotopic rat cardiac allografts. Transplantation. 1995;59(4):598–604. doi: 10.1097/00007890-199502270-00027. [DOI] [PubMed] [Google Scholar]
- 38.Moliterno R, et al. Heat shock protein-induced T-lymphocyte propagation from endomyocardial biopsies in heart transplantation. J Heart Lung Transplant. 1995;14(2):329–337. [PubMed] [Google Scholar]
- 39.Trieb K, Grubeck-Loebenstein B, Eberl T, Margreiter R. T cells from rejected human kidney allografts respond to heat shock protein 72. Transpl Immunol. 1996;4(1):43–45. doi: 10.1016/S0966-3274(96)80032-3. [DOI] [PubMed] [Google Scholar]
- 40.O’Neill S, Ingman TG, Wigmore SJ, Harrison EM, Bellamy CO. Differential expression of heat shock proteins in healthy and diseased human renal allografts. Ann Transplant. 2013;18:550–557. doi: 10.12659/AOT.889599. [DOI] [PubMed] [Google Scholar]
- 41.Maehana T, et al. Heat shock protein 90α is a potential serological biomarker of acute rejection after renal transplantation. PLoS One. 2016;11(9):e0162942. doi: 10.1371/journal.pone.0162942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe K, et al. Interstitial expression of heat shock protein 47 and alpha-smooth muscle actin in renal allograft failure. Nephrol Dial Transplant. 2000;15(4):529–535. doi: 10.1093/ndt/15.4.529. [DOI] [PubMed] [Google Scholar]
- 43.Shi J, Yang D, Cong X, Li Y, Yang X, Liu Y. Expression of HSPA12B in acute cardiac allograft rejection in rats. Pathol Res Pract. 2015;211(1):20–26. doi: 10.1016/j.prp.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 44.Sarri S, Shaw SM, Gieschen-Krische MA, Archer L, Yonan N, Fildes JE. Myocardial heat shock protein 60 expression is upregulated following acute cardiac rejection. Transpl Immunol. 2009;21(3):140–142. doi: 10.1016/j.trim.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Schimke I, et al. Increased level of HSP27 but not of HSP72 in human heart allografts in relation to acute rejection. Transplantation. 2000;70(12):1694–1697. doi: 10.1097/00007890-200012270-00005. [DOI] [PubMed] [Google Scholar]
- 46.Baba HA, et al. Possible relationship between heat shock protein 70, cardiac hemodynamics, and survival in the early period after heart transplantation. Transplantation. 1998;65(6):799–804. doi: 10.1097/00007890-199803270-00006. [DOI] [PubMed] [Google Scholar]
- 47.Wood KL, et al. The role of heat shock protein 27 in bronchiolitis obliterans syndrome after lung transplantation. J Heart Lung Transplant. 2010;29(7):786–791. doi: 10.1016/j.healun.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizzo M, et al. Increased expression of HDJ-2 (heat shock protein 40) and heat shock protein 70 in biopsy specimens of transplanted human lungs. J Heart Lung Transplant. 1998;17(3):241–249. [PubMed] [Google Scholar]
- 49.De Souza AI, Wait R, Mitchell AG, Banner NR, Dunn MJ, Rose ML. Heat shock protein 27 is associated with freedom from graft vasculopathy after human cardiac transplantation. Circ Res. 2005;97(2):192–198. doi: 10.1161/01.RES.0000174815.10996.08. [DOI] [PubMed] [Google Scholar]
- 50.Flohé S, Speidel N, Flach R, Lange R, Erhard J, Schade FU. Expression of HSP 70 as a potential prognostic marker for acute rejection in human liver transplantation. Transpl Int. 1998;11(2):89–94. doi: 10.1007/s001470050110. [DOI] [PubMed] [Google Scholar]
- 51.Shields AM, Panayi GS, Corrigall VM. Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin Exp Immunol. 2011;165(3):292–300. doi: 10.1111/j.1365-2249.2011.04433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borges TJ, et al. Prolonged survival of allografts induced by mycobacterial Hsp70 is dependent on CD4+CD25+ regulatory T cells. PLoS One. 2010;5(12):e14264. doi: 10.1371/journal.pone.0014264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luna E, et al. Treatment with encapsulated Hsp60 peptide (p277) prolongs skin graft survival in a murine model of minor antigen disparity. Scand J Immunol. 2007;66(1):62–70. doi: 10.1111/j.1365-3083.2007.01951.x. [DOI] [PubMed] [Google Scholar]
- 54.Kovalchin JT, Mendonca C, Wagh MS, Wang R, Chandawarkar RY. In vivo treatment of mice with heat shock protein, gp 96, improves survival of skin grafts with minor and major antigenic disparity. Transpl Immunol. 2006;15(3):179–185. doi: 10.1016/j.trim.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Shen H, et al. Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J Clin Invest. 2012;122(1):383–387. doi: 10.1172/JCI58344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen H, et al. Haptoglobin enhances cardiac transplant rejection. Circ Res. 2015;116(10):1670–1679. doi: 10.1161/CIRCRESAHA.116.305406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91(1):221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang A, de la Motte C, Lauer M, Hascall V. Hyaluronan matrices in pathobiological processes. FEBS J. 2011;278(9):1412–1418. doi: 10.1111/j.1742-4658.2011.08069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang D, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 60.Campo GM, et al. The inhibition of hyaluronan degradation reduced pro-inflammatory cytokines in mouse synovial fibroblasts subjected to collagen-induced arthritis. J Cell Biochem. 2012;113(6):1852–1867. doi: 10.1002/jcb.24054. [DOI] [PubMed] [Google Scholar]
- 61.Shimizu M, et al. Hyaluronan inhibits matrix metalloproteinase-1 production by rheumatoid synovial fibroblasts stimulated by proinflammatory cytokines. J Rheumatol. 2003;30(6):1164–1172. [PubMed] [Google Scholar]
- 62.Syrjälä SO, et al. Increased Th17 rather than Th1 alloimmune response is associated with cardiac allograft vasculopathy after hypothermic preservation in the rat. J Heart Lung Transplant. 2010;29(9):1047–1057. doi: 10.1016/j.healun.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Wu GD, Wang H, Zhu H, He Y, Barr ML, Klein AS. Genetic modulation of CD44 expression by intragraft fibroblasts. J Biochem. 2008;144(5):571–580. doi: 10.1093/jb/mvn106. [DOI] [PubMed] [Google Scholar]
- 64.Todd JL, et al. Hyaluronan contributes to bronchiolitis obliterans syndrome and stimulates lung allograft rejection through activation of innate immunity. Am J Respir Crit Care Med. 2014;189(5):556–566. doi: 10.1164/rccm.201308-1481OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cui Y, et al. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J Clin Invest. 2015;125(11):4255–4268. doi: 10.1172/JCI79693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu D, et al. Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem Biol. 2013;8(7):1611–1620. doi: 10.1021/cb4001553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Platt JL, Wrenshall LE, Johnson GB, Cascalho M. Heparan Sulfate Proteoglycan Metabolism and the Fate of Grafted Tissues. Adv Exp Med Biol. 2015;865:123–140. doi: 10.1007/978-3-319-18603-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168(10):5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 69.Rienstra H, et al. Differential expression of proteoglycans in tissue remodeling and lymphangiogenesis after experimental renal transplantation in rats. PLoS One. 2010;5(2):e9095. doi: 10.1371/journal.pone.0009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katta K, et al. Renal heparan sulfate proteoglycans modulate fibroblast growth factor 2 signaling in experimental chronic transplant dysfunction. Am J Pathol. 2013;183(5):1571–1584. doi: 10.1016/j.ajpath.2013.07.030. [DOI] [PubMed] [Google Scholar]
- 71.Soulez M, et al. The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ Res. 2012;110(1):94–104. doi: 10.1161/CIRCRESAHA.111.250431. [DOI] [PubMed] [Google Scholar]
- 72.O’Riordan E, et al. Urinary proteomic analysis of chronic allograft nephropathy. Proteomics Clin Appl. 2008;2(7-8):1025–1035. doi: 10.1002/prca.200780137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adepu S, et al. Incipient renal transplant dysfunction associates with tubular syndecan-1 expression and shedding. Am J Physiol Renal Physiol. 2015;309(2):F137–F145. doi: 10.1152/ajprenal.00127.2015. [DOI] [PubMed] [Google Scholar]
- 74.Doege C, Koch M, Heratizadeh A, Sótonyi P, Mengel M, Nashan B. Chronic allograft nephropathy in athymic nude rats after adoptive transfer of primed T lymphocytes. Transpl Int. 2005;18(8):981–991. doi: 10.1111/j.1432-2277.2005.00153.x. [DOI] [PubMed] [Google Scholar]
- 75.Päiväniemi OE, Maasilta PK, Alho HS, Vainikka TL, Salminen US. Epithelial tenascin predicts obliterative airway disease. J Heart Lung Transplant. 2008;27(4):400–407. doi: 10.1016/j.healun.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 76.Franz M, et al. Extra cellular matrix remodelling after heterotopic rat heart transplantation: gene expression profiling and involvement of ED-A+ fibronectin, alpha-smooth muscle actin and B+ tenascin-C in chronic cardiac allograft rejection. Histochem Cell Biol. 2010;134(5):503–517. doi: 10.1007/s00418-010-0750-6. [DOI] [PubMed] [Google Scholar]
- 77.Nishioka T, et al. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2010;298(3):H1072–H1078. doi: 10.1152/ajpheart.00255.2009. [DOI] [PubMed] [Google Scholar]
- 78.Maqbool A, et al. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J Cardiol. 2016;8(5):340–350. doi: 10.4330/wjc.v8.i5.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruhmann M, Piccinini AM, Kong PL, Midwood KS. Endogenous activation of adaptive immunity: tenascin-C drives interleukin-17 synthesis in murine arthritic joint disease. Arthritis Rheum. 2012;64(7):2179–2190. doi: 10.1002/art.34401. [DOI] [PubMed] [Google Scholar]
- 80.Okamura Y, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276(13):10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 81.Tanaka M, et al. A novel monitoring of kidney allograft rejection with plasma EDA(+) fibronectin. Transplant Proc. 1999;31(1–2):310–311. doi: 10.1016/s0041-1345(98)01640-6. [DOI] [PubMed] [Google Scholar]
- 82.Wieczorek G, et al. Acute and chronic vascular rejection in nonhuman primate kidney transplantation. Am J Transplant. 2006;6(6):1285–1296. doi: 10.1111/j.1600-6143.2006.01307.x. [DOI] [PubMed] [Google Scholar]
- 83.Franz M, et al. Expression of extra domain A containing fibronectin in chronic cardiac allograft rejection. J Heart Lung Transplant. 2011;30(1):86–94. doi: 10.1016/j.healun.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 84.Coito AJ, Kato H, Azimi R, Kupiec-Weglinski JW. Chronic allograft rejection versus tolerance: a critical role for EIIIA(+) fibronectin. Transplant Proc. 2001;33(1-2):526–527. doi: 10.1016/S0041-1345(00)02124-2. [DOI] [PubMed] [Google Scholar]
- 85.Booth AJ, et al. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol. 2012;226(4):609–618. doi: 10.1002/path.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Franz M, et al. Extra cellular matrix remodelling after heterotopic rat heart transplantation: gene expression profiling and involvement of ED-A+ fibronectin, alpha-smooth muscle actin and B+ tenascin-C in chronic cardiac allograft rejection. Histochem Cell Biol. 2010;134(5):503–517. doi: 10.1007/s00418-010-0750-6. [DOI] [PubMed] [Google Scholar]
- 87.Franz M, et al. De novo expression of fetal ED-A(+) fibronectin and B (+) tenascin-C splicing variants in human cardiac allografts: potential impact for targeted therapy of rejection. J Mol Histol. 2014;45(5):519–532. doi: 10.1007/s10735-014-9573-4. [DOI] [PubMed] [Google Scholar]
- 88.Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289(51):35237–35245. doi: 10.1074/jbc.R114.619304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Land WG. The role of damage-associated molecular patterns (DAMPs) in human diseases: part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ Med J. 2015;15(2):e157–e170. [PMC free article] [PubMed] [Google Scholar]
- 90.Land WG. The role of damage-associated molecular patterns in human diseases: part I — promoting inflammation and immunity. Sultan Qaboos Univ Med J. 2015;15(1):e9–e21. [PMC free article] [PubMed] [Google Scholar]
- 91.Schaefer L. Small leucine-rich proteoglycans in kidney disease. J Am Soc Nephrol. 2011;22(7):1200–1207. doi: 10.1681/ASN.2010050570. [DOI] [PubMed] [Google Scholar]
- 92.Chen C, et al. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J Am Heart Assoc. 2014;3(1):e000683. doi: 10.1161/JAHA.113.000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Piccinini AM, Zuliani-Alvarez L, Lim JM, Midwood KS. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci Signal. 2016;9(443):ra86. doi: 10.1126/scisignal.aaf3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Serini G, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142(3):873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Webber J, Meran S, Steadman R, Phillips A. Hyaluronan orchestrates transforming growth factor-β1-dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284(14):9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yusen RD, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Lung and Heart-Lung Transplant Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant. 2016;35(10):1170–1184. doi: 10.1016/j.healun.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 97.Cypel M, et al. Normothermic ex vivo lung perfusion in clinical lung transplantation. N Engl J Med. 2011;364(15):1431–1440. doi: 10.1056/NEJMoa1014597. [DOI] [PubMed] [Google Scholar]
- 98.Machuca TN, Cypel M, Bonato R, et al. Safety efficacy of ex vivo donor lung adenoviral IL-10 gene therapy in a large animal lung transplant survival model. Hum Gene Ther. doi: 10.1089/hum.2016. [published online ahead of print January 4, 2017]. https://doi.org/10.1089/hum.2016.070. [DOI] [PubMed] [Google Scholar]
- 99.Yeung JC, et al. Ex vivo adenoviral vector gene delivery results in decreased vector-associated inflammation pre- and post-lung transplantation in the pig. Mol Ther. 2012;20(6):1204–1211. doi: 10.1038/mt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu H, et al. Preconditioning with recombinant high-mobility group box 1 protein protects the kidney against ischemia-reperfusion injury in mice. Kidney Int. 2014;85(4):824–832. doi: 10.1038/ki.2013.475. [DOI] [PubMed] [Google Scholar]
- 101.Izuishi K, et al. Cutting edge: high-mobility group box 1 preconditioning protects against liver ischemia-reperfusion injury. J Immunol. 2006;176(12):7154–7158. doi: 10.4049/jimmunol.176.12.7154. [DOI] [PubMed] [Google Scholar]
- 102.Seemampillai B, Germack R, Felkin LE, McCormack A, Rose ML. Heat shock protein-27 delays acute rejection after cardiac transplantation: an experimental model. Transplantation. 2014;98(1):29–38. doi: 10.1097/TP.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Netea MG, Latz E, Mills KH, O’Neill LA. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol. 2015;16(7):675–679. doi: 10.1038/ni.3178. [DOI] [PubMed] [Google Scholar]
- 104.Kashiwadate T, et al. Soluble thrombomodulin ameliorates ischemia-reperfusion injury of liver grafts by modulating the proinflammatory role of high-mobility group box 1. Tohoku J Exp Med. 2016;239(4):315–323. doi: 10.1620/tjem.239.315. [DOI] [PubMed] [Google Scholar]
- 105.Colombaro V, et al. Inhibition of hyaluronan is protective against renal ischaemia-reperfusion injury. Nephrol Dial Transplant. 2013;28(10):2484–2493. doi: 10.1093/ndt/gft314. [DOI] [PubMed] [Google Scholar]
- 106.Chong AS, Alegre ML. The impact of infection and tissue damage in solid-organ transplantation. Nat Rev Immunol. 2012;12(6):459–471. doi: 10.1038/nri3215. [DOI] [PMC free article] [PubMed] [Google Scholar]