

Abstract
Aldosterone-producing adenomas (APAs) are benign tumors of the adrenal gland that constitutively produce the salt-retaining steroid hormone aldosterone and cause millions of cases of severe hypertension worldwide. Either of 2 somatic mutations in the potassium channel KCNJ5 (G151R and L168R, hereafter referred to as KCNJ5MUT) in adrenocortical cells account for half of APAs worldwide. These mutations alter channel selectivity to allow abnormal Na+ conductance, resulting in membrane depolarization, calcium influx, aldosterone production, and cell proliferation. Because APA diagnosis requires a difficult invasive procedure, patients often remain undiagnosed and inadequately treated. Inhibitors of KCNJ5MUT could allow noninvasive diagnosis and therapy of APAs carrying KCNJ5 mutations. Here, we developed a high-throughput screen for rescue of KCNJ5MUT-induced lethality and identified a series of macrolide antibiotics, including roxithromycin, that potently inhibit KCNJ5MUT, but not KCNJ5WT. Electrophysiology demonstrated direct KCNJ5MUT inhibition. In human aldosterone-producing adrenocortical cancer cell lines, roxithromycin inhibited KCNJ5MUT-induced induction of CYP11B2 (encoding aldosterone synthase) expression and aldosterone production. Further exploration of macrolides showed that KCNJ5MUT was similarly selectively inhibited by idremcinal, a macrolide motilin receptor agonist, and by synthesized macrolide derivatives lacking antibiotic or motilide activity. Macrolide-derived selective KCNJ5MUT inhibitors thus have the potential to advance the diagnosis and treatment of APAs harboring KCNJ5MUT.
Keywords: Endocrinology, Nephrology
Introduction
Hypertension affects more than 1.1 billion people (1) and is a major risk factor for heart attack, stroke, and congestive heart failure, contributing to more than nine million deaths worldwide each year (2). In patients referred to hypertension clinics, adrenal gland tumors that constitutively secrete the steroid hormone aldosterone are found in about 5% (3); they affect one to ten million people worldwide. These tumors are virtually always benign, and their surgical removal is commonly curative, motivating efforts to make a timely and accurate diagnosis. Aldosterone-producing adenoma (APA) diagnosis is typically based upon the findings of hypertension, elevated plasma aldosterone levels with suppressed plasma renin activity (indicating autonomous adrenal aldosterone production), imaging evidence of an adrenal tumor, and invasive bilateral adrenal vein sampling (AVS) showing increased aldosterone levels in the adrenal vein ipsilateral to the tumor (4). Tumor removal is typically performed laparoscopically or retroperitoneoscopically (5).
Unfortunately, the screening procedure for APAs is expensive and time-consuming. In addition, AVS is an invasive and technically challenging procedure that is only available in a limited number of tertiary care centers. As a result, many patients are not diagnosed and do not receive optimal treatment. (11)C-metomidate PET-CT has been proposed as a noninvasive alternative (6). However, lower sensitivity and specificity, lack of availability in a typical outpatient setting, and high cost are potential concerns, and this procedure is not generally recommended by current guidelines (4). With rising levels of hypertension in low- and middle-income countries in particular (1), there is a need for simple and inexpensive new screening approaches for potentially curable forms of secondary hypertension. New pharmacologic strategies for the diagnosis and/or treatment of APAs have the potential to improve patient outcome.
By exome sequencing of APAs and matched normal tissue, either of 2 somatic missense mutations in the potassium channel KCNJ5 were shown to commonly cause APAs (7). Large multicenter studies have confirmed these findings and have shown that these mutations (G151R and L168R) account for 34%–47% of APAs in people of European ancestry (8–10) and 60%–77% of people of Asian ancestry (11–14), with differences likely attributable to differences in disease definition. There is also a striking sex dimorphism in European and some Asian cohorts (8, 10, 11, 15, 16); European cohorts consistently show that these mutations account for 50%–60% of women with APAs, but only 20% of men, and KCNJ5 mutations are more prevalent in younger patients (8, 10). A small number of additional KCNJ5 mutations are very rare causes of APAs (8, 10, 11). KCNJ5G151R and KCNJ5L168R both modify the channel’s selectivity filter, which normally confers high selectivity of the channel for K+ conductance; the mutant channel loses selectivity and shows similar conduction of K+ and Na+ (7). This results in Na+ influx, leading to membrane depolarization, activation of voltage-gated Ca2+ channels, and Ca2+ influx (7). Increased intracellular Ca2+ is the signal for both cellular proliferation and aldosterone production in adrenal glomerulosa cells (17), thereby accounting for the cardinal features of APAs. Evidence that these single mutations are sufficient for the formation of a hormone-producing tumor comes from the finding of identical or related germline KCNJ5 mutations in a rare Mendelian form of early onset and severe hypertension due to primary aldosteronism with massive adrenal hyperplasia (7, 18) as well as a striking case of germline mosaicism with hyperplastic lesions restricted to areas carrying a KCNJ5 mutation (19). Further, APAs harboring KCNJ5 mutations have very few additional protein-altering somatic mutations (only about 3 per tumor), which virtually never alter other genes involved in cell proliferation or aldosterone biosynthetic pathways (20).
Inhibition of KCNJ5WT has the potential to cause cardiac side effects due to KCNJ5 expression in the atrium, where it contributes to slowing of the heart rate in response to acetylcholine (21). However, loss of K+ selectivity in KCNJ5G151R and KCNJ5L168R channels as well as loss of sensitivity to the K+ channel blockers barium (7) and tertiapin-Q (22) suggest that the pore of mutant channels is sufficiently altered to enable small molecules to block ion passage through the mutant, but not the WT, channel.
These observations motivated efforts to identify specific inhibitors of both KCNJ5G151R and KCNJ5L168R (both mutations are hereafter referred to as KCNJ5MUT) channels that do not inhibit KCNJ5WT and might be used as diagnostic or therapeutic agents for APAs harboring these mutations.
Results
Development of screening assays for inhibitors of KCNJ5MUT and KCNJ5WT.
To screen for inhibitors of mutant and WT KCNJ5 channels, we generated stable HEK293 cell lines using the commercially available Flp-In T-REx System, in which single copies of desired genes are integrated into a common site in the genome under control of a tetracycline-inducible CMV promoter. Cell lines with inducible expression of KCNJ5WT, KCNJ5G151R, or KCNJ5L168R were produced (see Methods). While induction of KCNJ5WT expression had no effect on cell viability, cells expressing KCNJ5G151R or KCNJ5L168 died rapidly, an effect we previously demonstrated to be Na+ dependent (18).
We measured cellular ATP levels as a proxy for cell viability, using a commercially available luciferase assay (23) (see Methods). Induction of KCNJ5WT had no effect on ATP levels. In contrast, induction of KCNJ5G151R or KCNJ5L168R led to drastically reduced ATP levels 72 hours after induction (Figure 1A). This allowed high-throughput screening for inhibitors that prevented reduction in ATP levels upon induction of KCNJ5G151R or KCNJ5L168R.
Figure 1. Establishment of screening assays for KCNJ5MUT and KCNJ5WT inhibition.

(A) Reduced ATP levels following induction of KCNJ5G151R or KCNJ5L168R, but not KCNJ5WT, in HEK293 cells. Cell survival was measured using an ATP-based viability assay with luminescence as a read-out in stable inducible cell lines in the uninduced and induced states. Whereas induction of KCNJ5WT expression had no significant effect on ATP levels, induction of KCNJ5G151R and KCNJ5L168R led to a significant decrease in ATP levels. P values represent 2-tailed unpaired Student’s t test comparing uninduced vs. induced for each line. n = 8 for each condition, 2,000 cells per well (WT and G151R) or 4,000 cells per well (L168R). Bars represent mean, and error bars represent SD. (B) Evaluation of the KCNJ5WT membrane potential assay using BaCl2 using a standard curve of BaCl2 concentrations. Depolarization caused by increasing BaCl2 concentrations leads to an increase in fluorescence. Relative fluorescence at 2 mM BaCl2 was defined as 100%. Data were fitted with a 4-parameter nonlinear regression of log-dose versus response. n = 6. Dots represent mean, error bars represent SD.
Conditions were optimized to maximize reproducibility and the signal-to-background ratios (S/B) (24) comparing ATP levels in uninduced and induced cells. Screening was performed in 384-well plates; compounds were added 24 hours after plating, and channel expression was induced immediately after compound addition. ATP levels were measured 72 hours after induction.
Because expression of KCNJ5WT in HEK293 cells does not cause lethality (Figure 1A), a different assay for inhibition of this channel was required. Overexpression of KCNJ5WT in HEK293 cells leads to K+ efflux and membrane hyperpolarization (7), an effect that is reversible upon inhibition of the channel with the potassium channel blocker barium. Changes in membrane potential were detected using a well-established fluorescence-based measure of membrane potential, and the efficacy of the assay was demonstrated with inhibition by barium (Figure 1B, see Methods). This assay may also detect depolarization by compounds that inhibit endogenous K+ channels, which would not be desired due to the resulting toxicity.
High-throughput screen for KCNJ5MUT inhibitors.
KCNJ5G151R, the most frequent mutation in APAs (8), was used for the primary screen. Levels of ATP in uninduced and induced cells without added test compound defined 100% and 0% effect, respectively, and served as a proxy for lethality. We screened 73,001 compounds from 19 libraries for rescue of KCNJ5G151R-induced lethality, measured as increased levels of ATP, at a concentration of 10 μM (see Methods). Of all compounds, 94.6% showed less than a 10% effect, and 398 compounds (0.5%) showed at least a 33.3% effect. Upon repeat testing, 92 distinct compounds (0.1% of all compounds from the initial screen) produced a 33.3% or greater increase in ATP (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI91733DS1).
Among these 92 hits, 31 produced a more than 66.6% mean increase in ATP. These included 6 series of structurally related compounds. One of these comprised 2 macrolide antibiotics — roxithromycin and erythromycin (see Figure 2 for structures). The others were a structural series of compounds from the ChemDiv library (http://www.chemdiv.com/) (4 compounds from D089, 3 compounds from 3132, 2 each from G281, F471, and E612; Supplemental Figure 1). Notably, compounds from ChemDivD089 and ChemDivE612 share an aniline or benzylamine derivative connected to a benzofuran (in D089) or isoindoline (in E612) derivative, respectively.
Figure 2. Structure activity relationship of macrolide inhibition of KCNJ5G151R.

Select compounds are shown. The locations of desosamine and cladinose moieties are indicated on the roxithromycin structure. Values of IC50 against KCNJ5G151R are given for active compounds. See Table 1 for detailed screen results.
We next screened these 92 compounds in dose-response assays along with 6 additional compounds sharing structural similarity to active macrolide antibiotics. Each compound was tested at concentrations of 0.12 μM, 0.37 μM, 1.1 μM, 3.3 μM, and 10 μM. Of these 98 compounds, 91 showed a dose response on KCNJ5G151R. The results revealed roxithromycin as the most potent KCNJ5G151R inhibitor in this assay (IC50 0.37 μM; Supplemental Table 2). Remarkably, virtually complete rescue of ATP level was achieved, and microscopy revealed vital cells.
All 98 compounds were also tested for inhibition of the other common mutation in APAs, KCNJ5L168R. Thirty-six compounds showed a dose response for this channel as well; these included the macrolide compounds roxithromycin, roxithromycin-D7, N-demethyl roxithromycin, erythromycin A oxime, clarithromycin, erythromycin estolate, and erythromycin (Supplemental Table 2). Some other potent inhibitors of KCNJ5G151R channels, e.g., NSC18883 and KC-159, demonstrated either high IC50 values or low maximum inhibition of KCNJ5L168R channels.
Screen for KCNJ5WT inhibition.
To determine the effect of these compounds on KCNJ5WT channels, the compounds were also tested for inhibition of KCNJ5WT by measurement of membrane potential after induction in the presence and absence of drug. Eight macrolides or macrolide derivatives tested had no significant effect on KCNJ5WT channels; several of these, including roxithromycin and clarithromycin, showed robust inhibition of both KCNJ5G151R and KCNJ5L168R (Supplemental Table 2 and Figure 2). Nonmacrolide compounds typically were not selective for mutant channels or did not robustly inhibit both KCNJ5G151R and KCNJ5L168R (Supplemental Table 2 and Supplemental Figure 1).
Macrolides are potent inhibitors of KCNJ5MUT, but not KCNJ5WT.
Macrolides feature a many-membered lactone ring with one or more deoxy sugars attached (25). Those with antibiotic activity inhibit protein synthesis by binding of the desosamine sugar and the lactone ring to bacterial 23S ribosomal RNA (26). Binding to other targets produces antiinflammatory activity (27) and stimulation of gastrointestinal motility (28), which can have clinical utility.
To further characterize the structure-activity relationship and inhibitory characteristics of the macrolide series, we screened 14 macrolides and their derivatives in 10-point dilution curves (Table 1 and Figure 3). This analysis confirmed potent effects of some macrolide antibiotics, including roxithromycin and clarithromycin, but demonstrated negligible activity of others, such as flurithromycin. The inhibitory activity of roxithromycin and clarithromycin was greater than that of erythromycin. Clarithromycin differs from erythromycin only by methylation of the hydroxyl group at the 6 position of the lactone ring; roxithromycin differs only by an N-oxime side chain on the lactone ring (Figure 2). These groups may stabilize the interaction between the macrolides and the mutant channel. Other modifications of the lactone ring in azithromycin and flurithromycin diminish or eliminate inhibition of KCNJ5MUT, supporting interaction of ring constituents with mutant channels. In addition, the cladinose sugar of roxithromycin appears to be critical, since its removal in decladinose roxithromycin and telithromycin results in loss of inhibition of KCNJ5MUT (Figure 2). Erythromycin B showed greater activity than erythromycin C and A, other components of erythromycin, suggesting that hydroxylation at the 12 position, which is present in most macrolides, may decrease anti-KCNJ5MUT activity.
Table 1. Inhibition of mutant and WT KCNJ5 by macrolides and their derivatives.

Figure 3. Dose-response curves of lead macrolide compounds on WT and mutant KCNJ5.

Dose-response curves of roxithromycin, clarithromycin, and idremcinal on activity of WT and mutant (G151R and L168R) KCNJ5 are shown. For mutant KCNJ5, percentage of inhibition denotes increase in ATP, measured by luminescence, in the presence of drug. For WT KCNJ5, percentage of inhibition denotes change in membrane potential in the presence of drug (see Methods). Compounds were tested in technical triplicates at 20, 10, 5, 2.5, 1.25, 0.625, 0.3125, 0.1563, 0.0781, and 0.0391 μM concentrations. Data were fit with a 4-parameter nonlinear regression of log-dose versus response. While inhibition of mutant channels showed a strong dose-response relationship with all 3 compounds, there was no significant inhibition of WT channels. See Table 1 for inhibition data.
In addition to macrolide antibiotics, nonantibiotic macrolide derivatives, such as the motilin receptor agonist idremcinal (EM 574) (28, 29), were also potent and selective inhibitors of KCNJ5MUT (idremcinal IC50 0.60 μM for KCNJ5G151R and 1.99 μM for KCNJ5L168R, no dose response for KCNJ5WT; Figure 2 and Table 1). Several other compounds with macrolide structures, including troleandomycin, josamycin, kitasamycin, oleandomycin, oligomycin C, rapamycin, spiramycin, dirithromycin, rondomycin, and natamycin, had little or no activity against KCNJ5G151R in the primary screen (see Figure 2 for selected structures).
Synthesis of macrolide derivatives without antibiotic or motilide activity.
The use of macrolide antibiotics is associated with a comparatively low risk of Clostridium difficile infection (30), and maintenance treatment with macrolides has been suggested for patients with frequent exacerbations of chronic obstructive pulmonary disease (31), cystic fibrosis (32), and non–cystic fibrosis bronchiectasis (33). Yet for the treatment of patients with primary aldosteronism, the potential induction of pathogen resistance through antibiotic treatment or increased gastrointestinal side effects through motilide activity would not be desired. We thus sought to identify compounds that retain selective channel inhibition without antibiotic or motilide activity, as these would have the most potential for clinical use as KCNJ5MUT inhibitors. In the development of idremcinal, compounds were identified that lacked both antibiotic and motilide activity (28). We synthesized several of these compounds (see Methods), confirmed their lack of antibiotic activity using the Kirby-Bauer disk diffusion assay (Supplemental Table 3, see Methods), and tested intermediates and target compounds for their effects on KCNJ5MUT. PLUX38, -36, -37, and -33 all showed significant inhibition of both mutant channels with no inhibition of KCNJ5WT (Figure 4 and Supplemental Table 3). These findings suggest the ability to develop drugs that retain selective inhibition of mutant KCNJ5 channels without antibiotic or gastrointestinal motility activities.
Figure 4. Structures of synthesized compounds with and without inhibition of mutant KCNJ5.

Values of IC50 for KCNJ5G151R are given for active compounds. See Supplemental Table 3 for detailed screen results.
Electrophysiology demonstrates direct inhibition of KCNJ5MUT by roxithromycin and synthesized compounds.
While KCNJ5 channels exist as heterotetramers with KCNJ3 and as homotetramers in heart (34), their subunit composition in the adrenal gland is currently unknown. To directly demonstrate that macrolides are inhibiting mutant KCNJ5 channels, we performed electrophysiology of HEK293T cells expressing KCNJ5 homotetramers or KCNJ5/KCNJ3 heterotetramers (7). Currents were measured by the whole-cell patch-clamp technique (7, 18). Figure 5A shows cation currents of heterotetramers recorded in physiological solutions (high extracellular Na+, high intracellular K+) (7). Expression of KCNJ3/KCNJ5WT channels resulted in inwardly rectifying K+ currents and a negative reversal potential as a result of high K+ conductance. Consistent with published results (7), expression of KCNJ3/KCNJ5G151R channels caused marked depolarization as a result of Na+ conductance. Addition of 20 μM roxithromycin led to almost complete inhibition of KCNJ3/KCNJ5G151R currents across all voltages tested. Of note, a shift of the reversal potential to more negative voltages was observed after addition of roxithromycin, consistent with inhibition of the depolarizing Na+ conductance. Similar results were obtained for KCNJ3/KCNJ5L168R. The remaining current for KCNJ3/KCNJ5L168R may be a potassium current because it was blocked by the known potassium channel pore-blocker barium. In contrast, roxithromycin showed no inhibition of potassium currents in KCNJ3/KCNJ5WT channels, and currents showed normal sensitivity to barium in the presence of roxithromycin. Parallel studies of KCNJ5G151R and KCNJ5L168R homotetramers confirmed these results, and no effect on KCNJ5WT homotetramers was seen. KCNJ5G151R homotetramers demonstrated 80% inhibition with 20 μM roxithromycin and 53% inhibition with 10 μM roxithromycin (Supplemental Figure 2), suggesting that IC50s for current inhibition are higher than those for rescue of cellular lethality. These results demonstrate direct inhibition by roxithromycin of channels containing KCNJ5 mutations. Similarly, PLUX37 and PLUX38, which were tested as representative synthesized inhibitors without antibiotic or motilide activity, demonstrated inhibition of KCNJ5MUT without effect on KCNJ5WT in electrophysiological studies at a 10 μM concentration (Figure 6).
Figure 5. Roxithromycin inhibits KCNJ3/KCNJ5MUT-induced Na+ current and KCNJ5MUT-induced aldosterone production.

(A) Cation currents were measured with the perforated whole cell recording (see Methods). Current-voltage plots of KCNJ3/KCNJ5G151R, KCNJ3/KCNJ5L168R, and KCNJ3/KCNJ5WT heterotetramers before and 10 minutes after addition of 20 μM roxithromycin and 1 mM BaCl2 are shown. Shown are 3 (G151R, WT) or 4 (L168R) biological replicates. (B) HAC15 cells stably transfected with KCNJ5WT, KCNJ5G151R, KCNJ5L168R, or empty vector were treated with the indicated concentrations of roxithromycin or with vehicle control for 6 hours. Aldosterone in the supernatant was quantified by ELISA and normalized to cell count (see Methods). Four independent experiments were performed for each condition (each measured in duplicate of 2 dilutions). (C) H295R cells transiently transfected with KCNJ5WT, KCNJ5G151R, KCNJ5L168R, or empty vector were treated with roxithromycin or vehicle control for 18 hours. CYP11B2 expression (with GAPDH as control) was determined by qPCR; 6 independent experiments were performed. Values were normalized to the respective values of empty vector. Error bars represent SEM. NS denotes P > 0.05. *P ≤ 0.05; **P ≤ 0.01; #P ≤ 0.001, Mann-Whitney U test versus 0 μM roxithromycin.
Figure 6. Inhibition of KCNJ5MUT by PLUX37 and PLUX38.

Cation currents of heterotetramers of KCNJ3 and mutant or WT KCNJ5 were measured with the perforated patch-clamp technique without (blue) or with (red) 10 μM inhibitor (A) PLUX37 and (B) PLUX38. Extracellular solution: 140 mM NaCl/5 mM KCl; intracellular solution: 140 mM KCl. For both compounds, n = 5 biological replicates for KCNJ3/KCNJ5G151R and KCNJ3/KCNJ5L168R; n = 4 for KCNJ3/KCNJ5WT (mean and SEM). (C) H295R cells transiently transfected with KCNJ5WT, KCNJ5G151R, KCNJ5L168R, or empty vector were treated with PLUX38 or vehicle control for 18 hours. CYP11B2 expression (with GAPDH as control) was determined by qPCR; 5 independent experiments were performed. Values were normalized to the respective values of empty vector. Error bars represent SEM. NS denotes P > 0.05. **P ≤ 0.01, Mann-Whitney U test versus 0 μM roxithromycin.
Roxithromycin inhibits KCNJ5MUT-induced aldosterone production in human adrenal-derived H295R cells.
To assess effects of KCNJ5 inhibition on aldosterone production, we used the HAC15 cell line (a subclone of NCI-H295R, derived from human adrenocortical carcinoma and a commonly used in vitro model of zona glomerulosa aldosterone production; refs. 35, 36). We stably transduced cells with KCNJ5WT, KCNJ5G151R, and KCNJ5L168R or empty vector control and measured aldosterone production from supernatants. Aldosterone production was about 5-fold increased in cells expressing KCNJ5MUT channels. Importantly, roxithromycin inhibited aldosterone production in a dose-dependent fashion in cells expressing either KCNJ5G151R or KCNJ5L168R (Figure 5B), whereas levels showed virtually no change in cells expressing KCNJ5WT. Aldosterone levels in cells expressing KCNJ5MUT did not decrease to the levels of control cells, possibly because aldosterone accumulates in the supernatant and inhibition is not instantaneous. Membrane depolarization resulting from Na+ conductance of KCNJ5MUT is known to induce expression of CYP11B2, encoding aldosterone synthase. This enzyme catalyzes the 3 terminal steps in aldosterone biosynthesis (hydroxylation at positions C11 and C18 of the steroid nucleus, followed by oxidation at position C18). Accordingly, we measured expression of CYP11B2 by quantitative PCR (qPCR) in NCI-H295R cells transiently transfected with KCNJ5WT, KCNJ5G151R, and KCNJ5L168R. Consistent with published results (37), cells transfected with KCNJ5G151R or KCNJ5L168R showed an approximately 7-fold increase in CYP11B2 expression compared with cells transfected with KCNJ5WT. Roxithromycin inhibited expression of CYP11B2 in a dose-dependent fashion in cells expressing either KCNJ5G151R or KCNJ5L168R, whereas levels showed virtually no changes in cells expressing KCNJ5WT. Similar results were obtained for the synthesized inhibitor PLUX38 (Figure 6C).
Discussion
These results demonstrate that specific macrolides selectively and potently inhibit channels harboring mutant forms of KCNJ5. The doses required to rescue overexpression-induced lethality are lower than those required to achieve near-complete channel inhibition, in line with the observation that moderate Na+ influx at physiological expression levels is associated with hyperplasia and tumor formation, rather than lethality, in vivo (7).
Prior exploration of candidate molecules (22) included inhibitors of Na+/H+ exchangers, Na+/Ca2+ exchangers, calcium channels, and sodium channels. While several were shown to have some potency to inhibit KCNJ5L168R, the effect on KCNJ5G151R was reported for verapamil only (31% inhibition at high therapeutic levels, ref. 22; the compound did not pass the threshold for KCNJ5G151R inhibition in our primary screen), and effects on KCNJ5WT were not studied. Our study is the first we are aware of to perform an unbiased screen for compounds that specifically inhibit both KCNJ5G151R and KCNJ5L168R and to have screened more than a handful of compounds. This demonstrates the value of using a robust high-throughput screen. Our findings suggest that macrolides or related compounds could be used for the clinical diagnosis of APAs, eliminating the need for cost-intensive and invasive screening by adrenal venous sampling in a substantial number of cases. Because aldosterone synthase and aldosterone both have short half-lives (38), short-term inhibition of expression of aldosterone synthase is sufficient to produce rapid reductions in plasma and urinary aldosterone levels, as occurs with dexamethasone inhibition of aldosterone synthase expression in the Mendelian disease glucocorticoid-remediable aldosteronism (39). Thus, in patients in whom an APA is suspected from the finding of hypertension due to primary aldosteronism, short-term administration of an inhibitor of mutant KCNJ5 would be expected to acutely reduce serum and urinary aldosterone levels in patients in whom somatic KCNJ5 mutations are the cause of APAs. Tumors with KCNJ5 mutations are typically large (40), with characteristic morphology upon imaging (41). Radiological evidence of such a unilateral adrenal tumor, in conjunction with the reduction in aldosterone production after pharmacological inhibition, would likely be sufficient to proceed to surgical intervention, obviating the need for invasive AVS. The large fraction of APAs caused by these mutations suggests that such a test would have substantial clinical impact. This approach to diagnosis could improve the identification of patients whose hypertension could be cured by surgical intervention.
Moreover, the long history of pharmacologic use of macrolides, with their established safety and oral bioavailability, suggests a useful path forward for developing drugs similar to the tool compounds PLUX37 and PLUX38 that could be optimized for channel inhibition, lack of antibiotic and gastrointestinal effects, and pharmacokinetics. The results thus far demonstrate structure activity relationships that can likely be built upon to achieve these goals, and a recent report on the fully synthetic assembly of 300 new macrolide candidates suggests further candidates for screening (42).
Even though spironolactone, a mineralocorticoid receptor antagonist, is available for the medical treatment of patients with APAs who elect not to undergo surgery, its use is associated with marked side effects, including gynecomastia, which often limits compliance. Eplerenone, a more selective mineralocorticoid receptor antagonist, is more expensive and not approved for the therapy of hypertension in Europe (4). In addition to a role in the diagnosis of APAs, it is also possible that long-term treatment with inhibitors of KCNJ5MUT could be efficacious in the treatment of hypertension in patients with APAs due to these mutations, potentially eliminating the need for surgery. This may be particularly relevant for patients who are poor surgical candidates. By inhibiting the proliferative signal as well as aldosterone production, it is also possible that the tumor might shrink with treatment, potentially eventually allowing withdrawal of therapy. In addition, inhibitors of KCNJ5MUT could be beneficial in individuals with rare germline KCNJ5 mutations who do not respond to therapy with mineralocorticoid antagonists and would otherwise require bilateral adrenalectomy (18). For long-term use, antibiotic and gastrointestinal effects would need to be eliminated from such a drug. Our studies indicate that channel inhibition can be separated from these other effects, affording promise that such inhibitors can be developed.
Finally, there is precedent for the pharmacologic treatment of hormone-producing tumors. Dopamine agonists have dramatically affected the treatment of prolactinomas, hormone-producing tumors of the pituitary gland. Dopamine agonists typically restore normal prolactin levels and reduce tumor mass, with recent data demonstrating that therapy can be withdrawn in a subset of patients after normalization of hormone levels and tumor disappearance (43). These observations provide optimism that an analogous approach could have similar efficacy for APAs.
KCNJ5 channels are not expressed in rodent zona glomerulosa (44), and a mouse model of KCNJ5MUT largely does not replicate the human phenotype (45), hindering the testing of macrolide compounds in animal models. The effect of macrolides on aldosterone production of primary cultures from APAs carrying KCNJ5MUT could be tested ex vivo (46), with limited informative value beyond our studies in H295R cells. The most promising path forward, however, involves clinical trials in humans. The potential of macrolides as diagnostic tools could be tested immediately with roxithromycin, which is marketed as an antibiotic in the European Union and whose safety has been established. After a single dose of 300 mg to healthy adults, maximum serum concentrations are 16.4 mg/l (~20 μM; half-life ~11 hours) (47), concentrations that strongly inhibit KCNJ3/KCNJ5MUT heterotetramers and KCNJ5MUT homotetramers in vitro and reduce KCNJ5MUT-induced CYP11B2 expression by about 70% and KCNJ5MUT-induced aldosterone production by more than 50%. A decrease in blood pressure and/or aldosterone levels upon short-term roxithromycin treatment that specifically occurs in KCNJ5MUT carriers (for clinical trials, mutation testing can be performed after routine surgery to correlate response to mutation) would therefore not only demonstrate diagnostic potential, but also provide additional support for the initiation of safety and efficacy studies of nonantibiotic macrolide compounds in primary aldosteronism.
Methods
Generation of stable inducible HEK293 cell lines.
WT or mutant human KCNJ5 cDNAs (7) were subcloned into the pcDNA5/FRT/TO vector using BamHI and HindIII. Single copies of WT or mutant human KCNJ5 cDNAs expressed under the control of a tetracycline-inducible promoter were introduced into HEK293 cells using the Flp-In T-REx System (Invitrogen) according to the manufacturer’s instructions. Cells were grown in selective media containing 10% Tet System Approved FBS (Clontech) until induction.
Generation of HAC15 cell lines stably expressing KCNJ5.
HAC15 cells (a gift of William Rainey, University of Michigan, Ann Arbor, Michigan, USA) authenticated by short tandem repeat (STR) analysis (ATCC Cell Line Authentication Service) and tested for mycoplasma (Lonza MycoAlert Mycoplasma Detection Kit) were cultured at 37°C and 5% CO2 in DMEM:F12 (1:1, Gibco, Thermo Fisher Scientific) supplemented with 5% Cosmic Calf Serum (CCS) (Hyclone Laboratories), 1% penicillin/streptomycin, 1% insulin-transferrin-selenium, 1% nonessential amino acids, and 0.1% lipid mixture (all Gibco, Thermo Fisher Scientific). HEK293T cells were cultured in DMEM (Biochrom) with 10% FBS (Biochrom) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific) in 5% CO2 at 37°C.
pLX303 KCNJ5G151R, pCMV-VSV-G, and psPAX2 were gifts of C. Gomez-Sanchez (University of Mississippi Medical Center, Jackson, Mississippi, USA) (48). pLX303 KCNJ5WT was generated from plx303 KCNJ5G151R using a PCR-based strategy (QuikChange, Stratagene) according to the manufacturer’s instructions, and pLX303 KCNJ5L168R was similarly generated from plx303 KCNJ5WT. pLX303 empty vector was generated by digest of pLX303 KCNJ5G151R with BsrGI (NEB) and self-ligation. Plasmids for transduction were prepared using the QIAGEN Maxi Plus Kit. For lentiviral production, HEK293T cells were seeded in T75 flasks and transfected with pLX303 constructs, pCMV-VSV-G, and psPAX2 in a molar ratio of 12:8:14 (total DNA amount 33.5 μg, filled up to 610 μl with PBS) plus 96 μg polyethylenimine ([PEI]87) (a gift of C. Gomez-Sanchez) in 610 μl PBS. After 20 minutes incubation, 12.4 ml medium containing roxithromycin to a final concentration of 20 μM was added. After overnight incubation, the medium was changed. After 48 hours, the supernatant containing the lentivirus was harvested, centrifuged at 1,690 g and 4°C for 30 minutes, filtered through a 0.45-μM particle filter, and frozen at –80°C. For spinoculation (49), HAC15 cells were seeded in 24-well plates at 150,000 cells per well. The next day, 0.9 ml supernatant containing lentivirus and 0.8 μg/ml polybrene (Hexadimetrinebromide, Sigma-Aldrich) were added to each well. The plates were centrifuged for 2 hours at 1,200 g and 15°C and incubated at 37°C, 5% CO2 for 4 hours. The medium was changed to HAC15 medium containing 20 μM roxithromycin. The following day, the spinoculation was repeated. Selection was performed with 5 μg/ml blasticidin in the presence of 20 μM roxithromycin. Correct genotypes and transgene expression were verified by RNA isolation and Sanger sequencing of KCNJ5 cDNA.
Compound libraries.
All compound libraries were from the Yale Center for Molecular Discovery Research Collection. Compound libraries are formatted as 10 mM stocks in DMSO. For dilution curves, 10 mM stocks of compounds in DMSO were prepared and diluted 2-fold serially in DMSO. In the primary screen, the following libraries were screened: Gen-Plus, Pure Natural Products, Pharmakon 1600 (all 3 Microsource), NIH Clinical Collection, Oncology Set 2, Diversity Set 2, Mechanistic Diversity Set, Natural Products Set (all 4 NCI), Epigenetics Library, Kinase Inhibitor Library, Phosphatase Inhibitor Set, Ion Channel Ligand Library, Bioactive Lipid Library, Metabotropic Glutamatergic Ligand Library, 640 FDA-approved drugs, Nuclear Receptor Ligand Library, Protease Inhibitor Library (all 9 Enzo), and ChemDiv Library. Suppliers for compounds are indicated in Supplemental Tables 1 and 2. Additional compounds and suppliers (Table 1) were as follows: roxithromycin (Sigma-Aldrich, R4393), idremcinal (Enzo, ALX-380-264), pseudo erythromycin enol ether (Toronto Research, P839500), clarithromycin (Sigma-Aldrich, A3487), erythromycin B (Santa Cruz Biotechnology Inc., SC-362735), azithromycin (Sigma-Aldrich, 75199), anhydroerythromycin A (Toronto Research, A638950), erythromycin C (Santa Cruz Biotechnology Inc., SC-362736), erythromycin (Sigma-Aldrich, E6376), dirithromycin (Sigma-Aldrich, D4065), and flurithromycin (Waterstone Tech, 40589).
Cell viability assay.
20 μl of G151R and L168R cells was plated into sterile white with clear bottom tissue culture–treated 384-well plates (Corning) at a density of 2,000 cells/well (G151R) or 4,000 cells/well (L168R) using a Multidrop Combi Reagent Dispenser (Thermo Scientific). Assay plates were centrifuged and incubated overnight at 37°C, 5% CO2. 20 nl was transferred from the compound source plate to the cell assay plate using an Aquarius (Tecan) with a 384-well pin tool (V&P Scientific). The final concentration of compound for screening was 10 μM, and the final DMSO concentration was 0.1%. 1 μl of tetracycline (final concentration 1 μg/ml) was added to compound-containing and negative control wells by Multidrop Combi to induce expression. 1 μl of media was added to positive control wells. Assay plates were centrifuged and incubated for 72 hours at 37°C, 5% CO2. CellTiter-Glo (Promega) was used to measure cell viability in the assay wells according to the manufacturer’s instructions. 10 μl/well of CellTiter-Glo reagent was added to the assay plates using a MultiDrop Combi. The plates were shaken on a Thermomixer R (Eppendorf) at 1,100 rpm for 1 minute and incubated in the dark for 10 minutes at room temperature. Luminescence was read on an Envision plate reader (PerkinElmer) with 0.3 second sampling time per well. Raw data (luminescence counts per second) were normalized to percentage of effect by the following formula: 100 – (((sample – negative control mean)/(positive control mean – negative control mean)) × 100).
Membrane potential assay.
20 μl of WT cells were plated into sterile black with clear bottom tissue culture-treated 384-well plates (Corning) at a density of 10,000 cells/well using a Multidrop Combi. Assay plates were centrifuged at 46 g for 10 seconds and incubated overnight at 37°C in a humidified 5% CO2 incubator. After the overnight incubation, 1 μl of tetracycline (final concentration, 1 μg/ml) was added to all wells by Multidrop Combi to induce KCNJ5. Assay plates were centrifuged at 46 g for 10 seconds and incubated overnight at 37°C in a humidified 5% CO2 incubator. The next day, 10 μl of medium was removed from each well using a PlateMate Plus (Thermo Scientific). 10 μl of Blue Formula FLIPR Membrane Potential Dye (Molecular Devices) was added to cells using a multichannel pipettor. Assay plates were incubated for 30 minutes at 37°C. After incubation, 20 nl was transferred twice from the compound source plate to the cell assay plate using an Aquarius with a 384-well pin tool for a total transfer volume of 40 nl and a final DMSO concentration of 0.2%. 1 μl of 20 mM BaCl2 (1 mM final concentration) was added to positive control wells using a multichannel pipettor, and results were defined as 100% inhibition. The assay plate was incubated 20 minutes at room temperature in the dark. Fluorescence was read in a FlexStation II 384 plate reader (Molecular Devices) at 530/565 nm ex/em, bottom read.
Synthesis of macrolide derivatives.
The synthesis of PLUX30B, -31A, -32, -33, -35A, -37 and -38, -40 (50), and PLUX36 (28) was performed as previously described. The acylation reaction needed to synthesize PLUX34, -39, and -42 has also been described in the literature (51). The synthesis of PLUX33 or N-demethyl-9-deoxo-9-hydroxy-N-butyl-(9S)-erythromycin was made in a manner identical to that of PLUX32, only 1-iodobutane was used as the alkylating agent. Crude product was purified by silica gel chromatography, eluting with 6% (8:1, ethanol/ammonium hydroxide) in ethyl acetate to give 142 mg, 52% product as a colorless solid. 1H NMR peaks (400 MHz, DMSO-d6) were as follows: 1H NMR (400 MHz, DMSO-d6) δ 5.56 (d, J = 5.7 Hz, 1H), 4.92 (d, J = 9.8 Hz, 1H), 4.79 (d, J = 4.7 Hz, 1H), 4.53 (s, 1H), 4.39 (d, J = 2.1 Hz, 1H), 4.28 (d, J = 7.4 Hz, 1H), 3.86 (m, 5H), 3.70 (s, 1H), 3.59 (d, J = 5.1 Hz, 1H), 3.55(2, 1H), 3.20 (s, 3H), 3.04 (d, J = 8.5 Hz, 2H), 2.88 (dd, J = 9.4, 7.4 Hz, 1H), 2.67(m,1H), 2.54 (m, 1H), 2.48(m, 1H), 2.32 (m, 1H), 2.27 (d, J = 15.2 Hz, 1H), 2.15 (s, 3H), 1.89 (s, 1H), 1.73 (m, 3H), 1.56(d, J = 10.9 Hz, 1H) 1.46 (m, 2H), 1.40-1.19 (m, 5H), 1.15- 0.92 (m, 29H), 0.85 (t, J = 7.2 Hz, 3H), 0.74 (t, J = 7.4 Hz, 3H). m/z: 778 [M+H]+ (found [M+H]+, 778.5310, requires C40H75NO13 [M+H]+ 778.5317). PLUX36 or N-demethyl-8,9-anhydroerythromycin A 6,9-hemiacetal and PLUX37 or N-demethyl- N-isobutyl-8,9-anhydroerythromycin A 6,9-hemiacetal were synthesized as previously described (28). PLUX40 or N-demethyl-N-(2-methylpropyl)-erythromycin was produced by alkylating known N-demethylerythromycin in a manner identical to that done with PLUX32, using 1-iodo-2-methylpropane as the alkylating agent. Crude product was purified by silica gel chromatography eluting with 4% (8:1, ethanol/ammonium hydroxide) in dichloromethane to give product 85 mg, 52% yield. 1H NMR peaks (400 MHz, DMSO-d6) were as follows: 1H NMR (400 MHz, DMSO-d6) δ 5.13 – 5.04 (m, 1H), 4.72 (d, J = 4.8 Hz, 1H), 4.50 (s, 1H), 4.36 (d, J = 7.6 Hz, 1H), 4.30 – 4.23 (m, 2H), 4.08 – 3.94 (m, 2H), 3.90 – 3.82 (m, 2H), 3.75 (dd, J = 5.9, 2.0 Hz, 1H), 3.60 (s, 1H), 3.46 (d, J = 7.2 Hz, 1H), 3.19 (s, 3H), 3.03 (td, J = 8.0, 7.5, 3.8 Hz, 1H), 2.93 – 2.71 (m, 4H), 2.65 (m, 1H), 2.46 – 2.38 (m, 1H), 2.33 – 2.08 (m, 6H), 1.92 – 1.60 (m, 5H), 1.60 – 1.45 (m, 3H), 1.43-1.32 (m, 2H), 1.26 (s, 4H), 1.15 (d, J = 6.1 Hz, 3H), 1.11 (s, 3H), 1.09 (d, J = 7.3 Hz, 3H), 1.07-1.01 (m, 6H), 1.02-0.96 (m, 6H), 0.82 (dd, J = 6.5, 2.6 Hz, 6H), 0.74 (t, J = 7.4 Hz, 3H) m/z: 776 [M+H]+ (found [M+H]+, 776.5184, requires C40H73NO13 [M+H]+ 776.5160).
Kirby-Bauer disk diffusion assay.
Staphylococcus aureus (ATCC 25923) was streaked on Kirby-Bauer plates. 18 nmol compound was added to a sterile filter paper disk and placed on the plate. After incubation at 37°C, the diameter of the inhibitory zone was recorded.
Electrophysiology.
HEK293T cells (ATCC) were used for transient expression of GFP-tagged KCNJ5 or KCNJ3. The cells were grown in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) in 5% CO2 and 95% air at 37°C. Cells were grown to 50%–70% confluence for transfection, and the corresponding cDNAs (7) were simultaneously applied to the cells using TurboFect transfection reagent according to the manufacturer’s protocol (Fermentas). Briefly, a cDNA cocktail (0.5 μg GFP-tagged KCNJ5 and 0.5 μg KCNJ3) was diluted with 200 μl serum-free DMEM and further mixed with 4 μl TurboFect transfection reagent for the transfection of cells cultured in 35-mm Petri dishes. For studying the K+/cation channel activity of homotetramer KCNJ5, we used 1 μg cDNA for transfection. Cells transfected with vector alone were used as a control, and their background currents were subtracted from the experimental groups. After 15 minutes incubation at room temperature, the mixture of the transfection agents was applied to the cells followed by an additional 24 hours of incubation before use. Roxithromycin (Sigma-Aldrich) was dissolved in the bath solution to reach the designated concentrations. To determine the Ba2+-sensitive K+ currents, we used 1 mM BaCl2 at the end of experiments. For the examination of the effect of roxithromycin on cation currents in cells transfected with KCNJ5 mutants (G151R or L168R), 1 mM BaCl2 was present in the bath solution throughout the experiments.
Within 24 hours after transfection, cells were treated with trypsin-containing medium (TrypLE Express) (Gibco, Thermo Fisher Scientific) for 10 minutes to detach the cells. We carried out the perforated whole-cell patch-clamp experiments at room temperature. The cells were incubated with a KCl bath solution containing 140 mM KCl, 1.8 mM MgCl2, 1.8 mM CaCl2, and 10 mM HEPES (pH 7.4) or with a Na Ringer bath solution (140 mM NaCl, 5 mM KCl, 1.8 mM MgCl2, 1.8 mM CaCl2, and 10 mM HEPES). Fluorescence signal (an indication of positive transfection) was detected with an intensified video imaging system including a SIT 68 camera (Long Island Industries). Borosilicate glass (1.7-mm OD) was used to make the patch-clamp pipettes that were pulled with a Narishege electrode puller. The pipette had a resistance of 2–4 MΩ when filled with 140 mM KCl. The tip of the pipette was filled with pipette solution containing 140 mM KCl, 2 mM MgCl2, 1 mM EGTA, and 5 mM HEPES (pH 7.4). The pipette was then back-filled with amphotericin B (20 μg/0.1 ml) containing pipette solution. After forming a high-resistance seal (>2 GΩ), the membrane capacitance was monitored until the whole-cell patch configuration was formed. The cell membrane capacitance was measured and compensated. The K/cation currents were measured from –80 mV to 20 mV with 20 mV steps by an Axon 200A patch-clamp amplifier. The currents were low-pass filtered at 1 KHz and digitized by an Axon interface (Digidata 1200) and were analyzed using the pClamp Software System 9 (Axon). Data are presented as mean ± SEM.
Real-time PCR.
H295R cells (a gift of Matthias Haase), authenticated by STR analysis (ATCC Cell Line Authentication Service) and tested for mycoplasma (Lonza MycoAlert Mycoplasma Detection Kit), were cultured in DMEM/F12, HEPES (Gibco, Thermo Fisher Scientific) supplemented with 2.5% Ultroser G (Pall Biosepra), 1% ITS+ Premix (Corning), and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific) at 37°C and 5% CO2. Plasmids were purified using the EndoFree Maxi Kit (QIAGEN). Roxithromycin stock solutions were 100 mM in DMSO (both Sigma-Aldrich). PLUX38 stock concentration was 50 mM in DMSO. 3 × 106 cells were transfected with 3 μg pIRES2 EGFP containing no insert and WT or mutant KCNJ5 using an Amaxa Nucleofector device (program P-20) and the Nucleofector Kit R (Lonza). Cells were allowed to recover briefly in RPMI 1640 medium (Gibco, Thermo Fisher Scientific). Transfected cells were resuspended in medium containing 20 μM roxithromycin and plated on a 12-well plate. Transfection was confirmed via fluorescence microscopy. After 25 hours, cells were washed with PBS, and medium containing 0.1% Ultroser G and DMSO (vehicle) and 5, 20, or 50 μM compound was added. After an additional 18 hours, RNA was harvested using the RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. 300 ng RNA was transcribed using the Quantitect Reverse Transcription Kit (QIAGEN). Ct levels of CYP11B2 and GAPDH were determined in triplicate in a 7300 Real-Time PCR System (Applied Biosystems) using TaqMan Gene Expression Master Mix and assays Hs01597732_m1 (CYP11B2) or HS02758991_g1 (GAPDH) (all Applied Biosystems). ΔCt values were calculated by subtracting GAPDH Ct from CYP11B2 Ct, and ΔΔCt values were calculated by subtracting the average ΔCt of vehicle-treated empty vector–transfected cells from each ΔCt value. Fold changes in expression levels are shown as 2–ΔΔCt, normalized to the value of vector-transfected cells at the respective inhibitor concentration.
Aldosterone ELISA.
250,000 stably transfected HAC15 cells/well were seeded on a white clear-bottom 96-well plate (Corning). After 24 hours, cells were washed with PBS, and medium was changed to 0.1% CCS, 1% penicillin/streptomycin, 1% nonessential amino acids, 1% ITS, and 0.1% lipid concentrate (all Gibco, Thermo Fisher Scientific), with 20 μM roxithromycin, but no blasticidin. After an additional 24 hours, cells were again washed with PBS, and medium containing 0, 5, 20, or 50 μM roxithromycin was added. After 6 hours, the plate was centrifuged, the supernatant removed, and 100 μl of medium containing the same roxithromycin concentration as before was added. After 45 minutes incubation at room temperature, a CellTiter-Glo assay (Promega) was performed according to the manufacturer’s instructions. Luminescence was analyzed on an Enspire 2300 Multilabel Reader (PerkinElmer) with a measurement time of 0.25 seconds. Cell supernatants were centrifuged again, transferred to a fresh tube, and frozen at –20°C. They were analyzed in duplicate at 2 dilutions using the Cayman Aldosterone EIA Kit (catalog 501090) following the manufacturer’s instructions. For values outside the assay range, both replicates of the respective dilution were removed from the analysis. Cell counts were interpolated in GraphPad Prism using a nonlinear fit of the standard curve of 1,000,000, 750,000, 500,000, 250,000, 125,000, 62,500, 31,250, and 15,625 cells seeded in triplicate. Aldosterone per well was normalized to cell count.
Statistics.
Data were analyzed using the programs Excel (Microsoft) and Prism (GraphPad). Error bars are shown as SD unless otherwise indicated in the legend. Dose-response curves were fitted with the following 4-parameter logistic equation:
(Equation 1)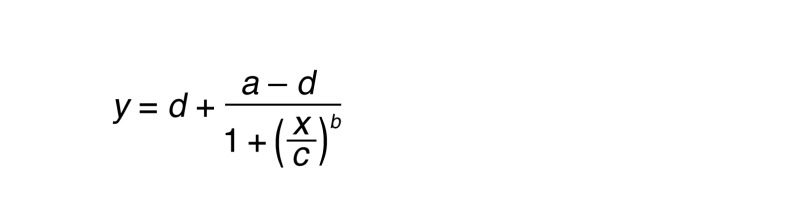
where y = dependent variable, a = minimum inhibition, d = maximum inhibition, x = independent variable, c = inflection point, and b = Hill’s slope. For Student’s t test, data passed normality tests in Prism. P ≤ 0.05 was considered statistically significant.
Author contributions
UIS and RPL conceived the project. UIS generated plasmids, stable inducible HEK293, and stable HAC15 cell lines. JZ contributed to cell maintenance. LA, UIS, and JSM designed and performed high-throughput screening assays. MP synthesized compounds. CZ and WW performed and analyzed electrophysiology. ENR performed qPCR analysis and ELISAs. UIS and BIK performed Kirby-Bauer disk-diffusion assays. UIS prepared figures and tables. UIS and RPL wrote and edited the main text and supplemental data. UIS, DH, JSM, WW, and RPL oversaw parts of the project or had advisory roles.
Supplementary Material
Acknowledgments
We thank the staff of the Yale Center for Molecular Discovery, Vincent Piscitelli (Yale University), the staff of the Düsseldorf Department of Hematology, Oncology, and Clinical Immunology, and Eric Seidel (Heinrich Heine University Düsseldorf) for their assistance and helpful discussions and Matthias Haase (Heinrich Heine University Düsseldorf) for providing the H295R cell line. The HAC15 cell line was a gift of William E. Rainey (University of Michigan), and plasmids and reagents used for the generation of stable HAC15 cell lines were gifts of Celso E. Gomez-Sanchez (The University of Mississippi Medical Center). These studies were supported in part by grants from the Yale Center for Molecular Discovery Center Pilot Program (to UIS and RPL), the Leducq Transantlantic Network on Hypertension (to RPL), the NIH (DK54983 to WW), the Howard Hughes Medical Institute (to RPL), and the Ministry of Innovation, Science, and Research of the State of North Rhine-Westphalia, Germany (to UIS). UIS is a member of the young college of the North Rhine-Westphalia Academy for Sciences and Arts.
Footnotes
CZ’s present address is: Department of Physiology, Xuzhou Medical College, Xuzhou, Jiangsu, China.
RPL’s present address is: Laboratory of Human Genetics and Genomics, The Rockefeller University, New York, New York, USA.
Conflict of interest: Yale University has filed a provisional patent application: US 61/949,577, Compositions and Methods for Diagnosing and Treating Diseases and Disorders Associated with Mutant KCNJ5. R.P. Lifton is a nonexecutive director of Genentech and F. Hoffmann–La Roche AG. U.I. Scholl is a part-time resident at University Hospital Düsseldorf.
Reference information:J Clin Invest. 2017;127(7):2739–2750.https://doi.org/10.1172/JCI91733.
Contributor Information
Laura Abriola, Email: laura.abriola@yale.edu.
Chengbiao Zhang, Email: zhangcbs@sina.com.
Mark Plummer, Email: mark.plummer@yale.edu.
Barbara I. Kazmierczak, Email: barbara.kazmierczak@yale.edu.
Junhui Zhang, Email: junhui.zhang@yale.edu.
Denton Hoyer, Email: denton.hoyer@yale.edu.
Wenhui Wang, Email: wenhui_wang@nymc.edu.
Richard P. Lifton, Email: richard.lifton@yale.edu.
References
- 1.NCD Risk Factor Collaboration (NCD-RisC) Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet. 2017;389(10064):37–55. doi: 10.1016/S0140-6736(16)31919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim SS, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi GP, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol. 2006;48(11):2293–2300. doi: 10.1016/j.jacc.2006.07.059. [DOI] [PubMed] [Google Scholar]
- 4.Funder JW, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2016;101(5):1889–1916. doi: 10.1210/jc.2015-4061. [DOI] [PubMed] [Google Scholar]
- 5.Walz MK, et al. Retroperitoneoscopic adrenalectomy in Conn’s syndrome caused by adrenal adenomas or nodular hyperplasia. World J Surg. 2008;32(5):847–853. doi: 10.1007/s00268-008-9513-0. [DOI] [PubMed] [Google Scholar]
- 6.Powlson AS, Gurnell M, Brown MJ. Nuclear imaging in the diagnosis of primary aldosteronism. Curr Opin Endocrinol Diabetes Obes. 2015;22(3):150–156. doi: 10.1097/MED.0000000000000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi M, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331(6018):768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Åkerström T, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One. 2012;7(7):e41926. doi: 10.1371/journal.pone.0041926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandes-Rosa FL, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension. 2014;64(2):354–361. doi: 10.1161/HYPERTENSIONAHA.114.03419. [DOI] [PubMed] [Google Scholar]
- 10.Boulkroun S, et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension. 2012;59(3):592–598. doi: 10.1161/HYPERTENSIONAHA.111.186478. [DOI] [PubMed] [Google Scholar]
- 11.Zheng FF, et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension. 2015;65(3):622–628. doi: 10.1161/HYPERTENSIONAHA.114.03346. [DOI] [PubMed] [Google Scholar]
- 12.Wang B, et al. Prevalence and characterization of somatic mutations in Chinese aldosterone-producing adenoma patients. Medicine (Baltimore) 2015;94(16):e708. doi: 10.1097/MD.0000000000000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu VC, et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci Rep. 2015;5:11396. doi: 10.1038/srep11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamoto T, et al. Clinical and steroidogenic characteristics of aldosterone-producing adenomas with ATPase or CACNA1D gene mutations. J Clin Endocrinol Metab. 2016;101(2):494–503. doi: 10.1210/jc.2015-3284. [DOI] [PubMed] [Google Scholar]
- 15.Hong AR, et al. Genetics of aldosterone-producing adenoma in Korean patients. PLoS One. 2016;11(1):e0147590. doi: 10.1371/journal.pone.0147590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J Atheroscler Thromb. 2015;22(2):191–200. doi: 10.5551/jat.24455. [DOI] [PubMed] [Google Scholar]
- 17.Spät A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev. 2004;84(2):489–539. doi: 10.1152/physrev.00030.2003. [DOI] [PubMed] [Google Scholar]
- 18.Scholl UI, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A. 2012;109(7):2533–2538. doi: 10.1073/pnas.1121407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura A, et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol Cell Endocrinol. 2017;441:134–139. doi: 10.1016/j.mce.2016.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scholl UI, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. 2013;45(9):1050–1054. doi: 10.1038/ng.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krapivinsky G, Gordon EA, Wickman K, Velimirović B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature. 1995;374(6518):135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 22.Tauber P, et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology. 2014;155(4):1353–1362. doi: 10.1210/en.2013-1944. [DOI] [PubMed] [Google Scholar]
- 23.Crouch SP, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160(1):81–88. doi: 10.1016/0022-1759(93)90011-U. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25. MacDougall C, Chambers HF. Aminoglycosides. In: Brunton LL, Chabner BA, Knollmann BC eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 2011:1505–1520. [Google Scholar]
- 26.Schlünzen F, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413(6858):814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 27.Parnham MJ. Immunomodulatory effects of antimicrobials in the therapy of respiratory tract infections. Curr Opin Infect Dis. 2005;18(2):125–131. doi: 10.1097/01.qco.0000160901.71813.fe. [DOI] [PubMed] [Google Scholar]
- 28.Tsuzuki K, et al. Motilides, macrolides with gastrointestinal motor stimulating activity. I. O-substituted and tertiary N-substituted derivatives of 8,9-anhydroerythromycin A 6,9-hemiacetal. Chem Pharm Bull. 1989;37(10):2687–2700. doi: 10.1248/cpb.37.2687. [DOI] [PubMed] [Google Scholar]
- 29.Sunazuka T, et al. Motilides, macrolides with gastrointestinal motor stimulating activity. II. Quaternary N-substituted derivatives of 8,9-anhydroerythromycin A 6,9-hemiacetal and 9,9-dihydroerythromycin A 6,9-epoxide. Chem Pharm Bull. 1989;37(10):2701–2709. doi: 10.1248/cpb.37.2701. [DOI] [PubMed] [Google Scholar]
- 30.Brown KA, Khanafer N, Daneman N, Fisman DN. Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection. Antimicrob Agents Chemother. 2013;57(5):2326–2332. doi: 10.1128/AAC.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uzun S, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(5):361–368. doi: 10.1016/S2213-2600(14)70019-0. [DOI] [PubMed] [Google Scholar]
- 32.Clement A, Tamalet A, Leroux E, Ravilly S, Fauroux B, Jais JP. Long term effects of azithromycin in patients with cystic fibrosis: A double blind, placebo controlled trial. Thorax. 2006;61(10):895–902. doi: 10.1136/thx.2005.057950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fan LC, Lu HW, Wei P, Ji XB, Liang S, Xu JF. Effects of long-term use of macrolides in patients with non-cystic fibrosis bronchiectasis: a meta-analysis of randomized controlled trials. BMC Infect Dis. 2015;15:160. doi: 10.1186/s12879-015-0872-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corey S, Clapham DE. Identification of native atrial G-protein-regulated inwardly rectifying K+ (GIRK4) channel homomultimers. J Biol Chem. 1998;273(42):27499–27504. doi: 10.1074/jbc.273.42.27499. [DOI] [PubMed] [Google Scholar]
- 35.Wang T, Rowland JG, Parmar J, Nesterova M, Seki T, Rainey WE. Comparison of aldosterone production among human adrenocortical cell lines. Horm Metab Res. 2012;44(3):245–250. doi: 10.1055/s-0031-1298019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology. 2012;153(4):1774–1782. doi: 10.1210/en.2011-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monticone S, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab. 2012;97(8):E1567–E1572. doi: 10.1210/jc.2011-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulte HM, Mönig H, Benker G, Pagel H, Reinwein D, Ohnhaus EE. Pharmacokinetics of aldosterone in patients with Addison’s disease: effect of rifampicin treatment on glucocorticoid and mineralocorticoid metabolism. Clin Endocrinol (Oxf) 1987;27(6):655–662. doi: 10.1111/j.1365-2265.1987.tb02948.x. [DOI] [PubMed] [Google Scholar]
- 39.Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 1997;82(11):3570–3573. doi: 10.1210/jcem.82.11.4381. [DOI] [PubMed] [Google Scholar]
- 40.Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 K(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab. 2015;100(8):E1089–E1095. doi: 10.1210/jc.2015-2149. [DOI] [PubMed] [Google Scholar]
- 41.Scholl UI, et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) 2015;83(6):779–789. doi: 10.1111/cen.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seiple IB, et al. A platform for the discovery of new macrolide antibiotics. Nature. 2016;533(7603):338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colao A, Savastano S. Medical treatment of prolactinomas. Nat Rev Endocrinol. 2011;7(5):267–278. doi: 10.1038/nrendo.2011.37. [DOI] [PubMed] [Google Scholar]
- 44.Chen AX, Nishimoto K, Nanba K, Rainey WE. Potassium channels related to primary aldosteronism: Expression similarities and differences between human and rat adrenals. Mol Cell Endocrinol. 2015;417:141–148. doi: 10.1016/j.mce.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lichtenauer U, et al. Establishment of an in vivo model for KCNJ5 dependent hyperaldosteronism. Exp Clin Endocrinol Diabetes. 2015;123(03):P09_25 [Google Scholar]
- 46.Xie CB, et al. Regulation of aldosterone secretion by Cav1.3. Sci Rep. 2016;6:24697. doi: 10.1038/srep24697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puri SK, Lassman HB. Roxithromycin: a pharmacokinetic review of a macrolide. J Antimicrob Chemother. 1987;20(Suppl B):89–100. doi: 10.1093/jac/20.suppl_b.89. [DOI] [PubMed] [Google Scholar]
- 48.Oki K, Plonczynski MW, Lam ML, Gomez-Sanchez EP, Gomez-Sanchez CE. The potassium channel, Kir3.4 participates in angiotensin II-stimulated aldosterone production by a human adrenocortical cell line. Endocrinology. 2012;153(9):4328–4335. doi: 10.1210/en.2012-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, Melton DW, Zhang Y, Hornsby PJ. Improved coinfection with amphotropic pseudotyped retroviral vectors. J Biomed Biotechnol. 2009;2009:901079. doi: 10.1155/2009/901079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaw SJ, et al. Structure-activity relationships of 9-substituted-9-dihydroerythromycin-based motilin agonists: optimizing for potency and safety. J Med Chem. 2009;52(21):6851–6859. doi: 10.1021/jm901107f. [DOI] [PubMed] [Google Scholar]
- 51.Flynn EH, Murphy HW, McMahon RE. Erythromycin. II. Des-N-methylerythromycin and N-methyl-C14-erythromycin. J Am Chem Soc. 1955;77(11):3104–3106. doi: 10.1021/ja01616a051. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.