

Abstract
Cell-surface-receptor pathways amplify weak, rare and local stimuli to induce cellular responses. This task is accomplished despite signaling components that segregate into nanometer-scale membrane domains. Here we describe a ‘catch-and-release’ mechanism that amplified and dispersed stimuli by releasing activated kinases from receptors lacking intrinsic catalytic activity. Specifically, we discovered a cycle of recruitment, activation and release for Zap70 kinases at phosphorylated T cell antigen receptors (TCRs). This turned the TCR into a ‘catalytic unit’ that amplified antigenic stimuli. Zap70 released from the TCR remained at the membrane, translocated, and phosphorylated spatially distinct substrates. The mechanisms described here are based on widely used protein domains and post-translational modifications; therefore, many membrane-associated pathways might employ similar mechanisms for signal amplification and dispersion.
Adaptive immune responses are based on the ability of T cells to discriminate between structurally similar stimulatory (agonist) and non-stimulatory (self) peptide–major histocompatibility complex (pMHC) molecules presented by antigen-presenting cells1. Full T cell responses are triggered by fewer than ten agonist pMHC molecules2–4. Because the affinities of T cell antigen receptors (TCRs) for agonist pMHC molecules and self pMHC molecules differ only slightly, T cell activation thresholds cannot be based solely on the number of ligand-engaged TCRs5. This suggests that stimuli from a few agonist pMHC molecules must be amplified above T cell– activation thresholds, while the overwhelming stimuli from self pMHC molecules are ignored. Hence, the amplification of TCR signaling has been attributed to the activation of multiple TCRs by a single agonist ligand (serial triggering)6 and prolonged binding of agonist ligands to TCRs (kinetic proofreading)7. Additional models for the co-activation of TCRs by self pMHC (pseudo-dimers)2,8,9 and pMHC-independent transactivation of TCRs10 have been proposed. However, imaging studies have shown that T cell signaling originates exclusively from TCRs bound to agonist pMHC molecules, which suggests that signal amplification is downstream of the TCR11.
The recognition of agonist pMHC by TCRs activates a downstream signaling cascade5,12. In brief, a pMHC-engaged TCR scans CD4 or CD8 co-receptors to find one paired with an activated Lck tyrosine kinase13,14. Lck phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) of invariant CD3 chains in complex with the TCR (TCR-CD3)15. Zap70 kinase is recruited from the cytosol to the TCR via interactions of its Src-homology 2 (SH2) domains with the doubly phosphorylated ITAMs16. Lck and trans-autophosphorylation activate TCR-bound Zap70 (refs. 17–19). Activated Zap70 phosphorylates its downstream substrates, including the adaptor LAT20. The phosphorylation of TCR and activation of Zap70 are controlled by kinetic proofreading mechanisms, which ensures that T cells remain quiescent in the absence of stimuli and become activated specifically by agonist pMHC molecules14,21. However, the mechanisms that amplify stimuli downstream of the TCR are poorly understood.
T cell activation is accompanied by a redistribution of T cell signaling molecules in the plasma membrane22,23. In quiescent T cells, the TCR signaling cascade and other membrane-associated pathways are segregated into membrane domains with widths of 50–200 nm (refs. 24–26). These domains are known as ‘protein islands’ or ‘nano-clusters’. Molecules that are part of the same signaling cascade (specifically, the TCR and LAT) can be separated into distinct protein islands (nanoclusters)25,26. When T cells are activated, microclusters form around ligand-engaged TCRs in an actin-dependent manner27–30. Microclusters contain signaling molecules involved in the early activation of T cells and are signaling hot spots; they are formed by the concatenation of protein islands (nanoclusters), which remain distinct and, specifically in the case of the TCR and LAT, do not intermingle their contents25. Microclusters move along microtubules toward the center of the contact site between the T cell and the antigen-presenting cell to form an immunological synapse31–33. Not all signaling molecules that form microclusters translocate to the synapse center29, which suggests that concatenated protein islands (nanoclusters) at least partly dissociate. However, the mechanisms by which the signaling sequence of the TCR pathway is maintained despite the segregation of its components are unknown.
Here we found that Zap70 was recruited to phosphorylated TCR-CD3 complexes via its SH2 domains, was activated by Lck and trans-autophosphorylation and was released from the TCR into the plane of the plasma membrane. Vacated TCR-binding sites became available for the activation of additional Zap70 molecules. This created a cycle that turned the TCR into a ‘catalytic unit’ and produced large amounts of active Zap70 to amplify antigenic stimuli. Analysis of Zap70 mutants revealed that phosphorylation and ATP binding promoted the release of Zap70 from the phosphorylated ITAMs of the TCR-CD3 complex. The released Zap70 remained associated with the plasma membrane and translocated to adjacent protein islands (nanoclusters), where it activated spatially distinct signaling molecules (i.e., LAT). The release and translocation of Zap70 was essential for conservation of the signaling chain of the TCR pathway. Our findings show that T cells are able to respond to very small numbers of agonist pMHC complexes because this ‘catch-and-release’ mechanism amplifies the signal and disperses it over large plasma membrane areas.
Results
Release of Zap70 from the TCR into the plasma membrane
TCR-associated Zap70 is activated by Lck; activated Zap70 then phosphorylates its substrates in membrane domains distinct from TCR domains25. We postulated that activated Zap70 must be released from the TCR into the plane of the membrane to disperse the TCR signal. A direct consequence of this release would be the recruitment and activation of additional Zap70 molecules via the vacated TCR-binding site. We analyzed the movement of Zap70 fused to a photoactivatable version of the red fluorescent protein TagRFP at the plasma membrane with nanometer precision by single-particle tracking (SPT) and total internal reflection microscopy. The TIRF microscopy illuminated fluorophores within 100–150 nm of glass surfaces (Supplementary Fig. 1a). Primary CD4+ T cells obtained from 5C.C7 mice (with transgenic expression of a TCR that recognizes agonist peptide derived from moth cytochrome c (MCC) presented by MHC class II molecules (specifically I-Ek)) and cultured short term were used for these analyses. 5C.C7 T cells were activated on glass surfaces coated with the extracellular domains of I-Ek–MCC complexes and the co-stimulatory molecule CD80. Such surfaces immobilize TCRs, with the concomitant result that TCR-bound molecules are co-immobilized21. Under these activation conditions, wild-type Zap70 showed movement within the membrane (Fig. 1a and Supplementary Video 1). These data demonstrated that Zap70 was released from the TCR and remained at the membrane. Next we tracked the tandem-SH2-domain module of Zap70 (tSH2), which consists of amino acids 1–256 of Zap70 and thus lacks interdomain B (which contains multiple regulatory sites) and the kinase domain34,35 (Fig. 1b). tSH2 binds phosphorylated ITAMs about tenfold more strongly than does wild-type Zap70 (ref. 21). In 5C.C7 T cells, tSH2 was immobilized after being recruited to the membrane (Fig. 1a and Supplementary Video 2), which suggested it stably associated with pMHC-bound TCR. Analysis of the mean square displacement (MSD) verified free diffusion for wild-type Zap70 and immobilization of tSH2 (Fig. 1c). We detected greater diffusion rates and spans for wild-type Zap70 than for tSH2 (Supplementary Fig. 1b–e and Supplementary Table 1). These data showed that the SH2 domains of Zap70 mediated its recruitment to the TCR, while features of the full-length protein controlled its release from the TCR.
Figure 1.
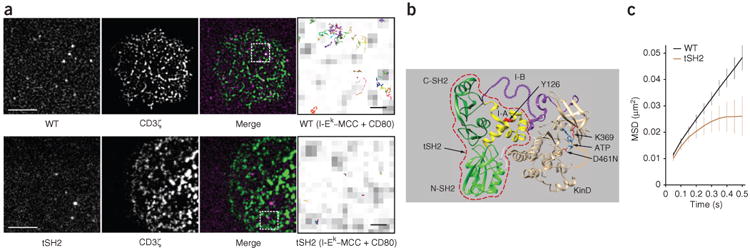
Zap70 is released from the TCR and remains at the plasma membrane. (a) SPT analysis of TagRFP-fused wild-type Zap70 (WT) (top) and tSH2 (bottom) in primary 5C.C7 T cells on stimulatory surfaces coated with I-Ek-MCC and CD80, presented as single molecules (left), TCR microclusters (specifically CD3ζ-GFP; middle left) and their overlay (middle right); far right, enlargement of outlined areas at left, showing single-molecule tracks (colored lines) overlaid on TCR microclusters (gray). Scale bars, 5 μm (far left) or 500 nm (far right); pixels, 160 nm × 160 nm. (b) The auto-inhibited structure of kinase-inactive Zap70 D461N, showing the SH2 domains at the amino terminus (N-SH2) and carboxyl terminus (C-SH2) and the tandem SH2 domain module (amino acids 1–256) (red dashed outline), interdomain B (I-B) and interdomain A (I-A) and the kinase domain (KinD): colors indicate protein domains; arrows indicate tSH2, ATP and amino acids (one-letter codes). (c) MSD plot for the tracks of wild-type Zap70 and tSH2 (key) in primary T cells. Data are from three or more independent experiments (average ± s.e.m. of at least 18 different cells in c).
Phosphorylation and ATP binding mediate Zap70 release
To determine whether tyrosine phosphorylation and/or conformational changes control the release of Zap70 from the TCR, we analyzed Zap70 mutants stably expressed in Zap70-deficient (P116) human Jurkat T cells. In T cells bound to surfaces coated with non-stimulatory poly-l-lysine (PLL), TCRs (specifically CD3ε) were mobile (Fig. 2, Supplementary Fig. 1f–i, Supplementary Video 3 and Supplementary Table 2). T cells were stimulated on glass surfaces coated with the OKT3 monoclonal antibody to human CD3ε. On these surfaces, the TCRs were immobilized (Fig. 2, Supplementary Fig. 1f–i; Supplementary Video 4 and Supplementary Table 2), which allowed us to study the effects of mutations on the movement of Zap70. The tracks of wild-type Zap70 on activating surfaces showed TCR-independent movement at the membrane, whereas tSH2 was immobilized at the TCR (Fig. 2, Supplementary Fig. 1f–i; Supplementary Videos 5 and 6 and Supplementary Table 2), consistent with our findings obtained with primary T cells. Wild-type Zap70 in paraformaldehyde-fixed T cells showed less average diffusion and shorter trajectory spans than those of the TCR or tSH2 in live T cells (Supplementary Fig. 1j and Supplementary Video 7). This was due to a small number of non-immobilized TCRs on OKT3-coated surfaces. However, the recruitment of Zap70 is restricted to ligand-engaged TCRs11 and was not detected at the membrane on PLL-coated surfaces11 (data not shown). tSH2 showed detectable association with the membrane on PLL-coated surfaces (Supplementary Fig. 1k and Supplementary Video 8), which suggested that nonspecific binding caused increased tSH2 diffusion and span trajectories36.
Figure 2.
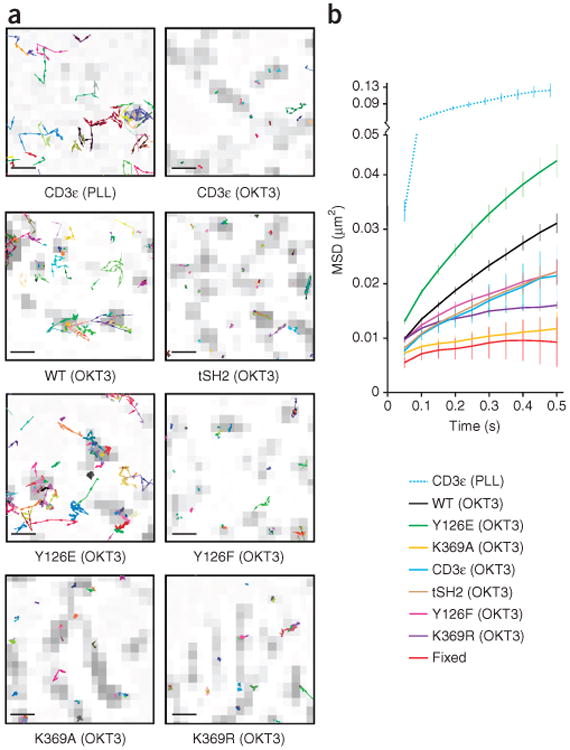
Tyr126 phosphorylation and kinase activity induce the release of Zap70 from the TCR. (a) Single-molecule tracks overlaid with TCR microclusters (presented as in Fig 1a, far right) in P116 Jurkat T cells stably expressing Zap70-TagRFP fusion proteins (CD3ε, wild-type Zap70, Zap70 Y126E, Zap70 Y126F, Zap70 K369A and Zap70 K369R; below plots), bound to PLL-coated (non-activating) surfaces (top left) or OKT3-coated (stimulatory) surfaces (other plots). Scale bars, 500 nm; pixels, 160 nm × 160 nm. (b) MSD plots of single-molecule tracks in P116 Jurkat T cells as in a (key) or paraformaldehyde fixed T cells (Fixed). Data are from three or more independent experiments (average ± s.e.m. of at least 18 different cells in b).
Phosphorylation of Tyr130 (located in interdomain A) in Syk kinase (which is Zap70's closest relative) causes diminished binding to phosphorylated ITAMs in the B cell receptor37,38. The equivalent residue in Zap70 (Tyr126) (Fig. 1b) can be phosphorylated in vitro17, and replacement of the tyrosine at position 126 with phenylalanine affects T cell activation39. Mass spectrometry and immunoblot analysis showed that phosphorylation of Zap70 at Tyr126 was induced by ligation of the TCR (Supplementary Fig. 2). We analyzed the mobility of Zap70 in which the tyrosine at position 126 was replaced with glutamic acid (Y126E) or phenylalanine (Y126F) to mimic phosphorylation or prevent it, respectively. Zap70 Y126E was more mobile than wild-type Zap70, whereas Zap70 Y126F showed less mobility than did wild-type Zap70, similar to tSH2 and CD3ε (Fig. 2, Supplementary Fig. 1f–i, Supplementary Videos 9 and 10 and Supplementary Table 2). We therefore concluded that phosphorylation of Tyr126 regulated the release of Zap70 from the TCR into the plasma membrane.
Only the release of active Zap70 from the TCR into the membrane contributes to signal dispersion. Binding of ATP and phosphate transfer depend on the active conformation of Zap70 (ref. 40). We postulated that the release of Zap70 from the TCR is controlled by ATP binding and catalytic activity. The Zap70 structure suggested that replacement of the lysine at position 369 (Fig. 1b) with alanine (K369A) would prevent the binding of ATP, whereas its replacement with arginine (K369R) would allow ATP binding but not phosphate transfer34,35. SPT demonstrated that Zap70 K369A and Zap70 K369R were immobilized after binding to the TCR (Fig. 2, Supplementary Fig. 1f–i, Supplementary Videos 11 and 12 and Supplementary Table 2). The differences in the diffusion rates of Zap70 K369A, Zap70 K369R and paraformaldehyde-fixed wild-type Zap70 were not significant (Supplementary Fig. 1h), in support of the published finding that only ligand-engaged TCRs recruit Zap70 (ref. 11). We concluded that kinase activity, not simply ATP binding, was required for the release of Zap70 from the TCR into the plasma membrane.
Zap70 release affects association with TCR microclusters
Overall exchange rates between TCR-bound Zap70 and cytosolic or mobile, membrane-localized Zap70 were analyzed by fluorescence recovery after photobleaching (FRAP). Microcluster fluorescence was monitored after photobleaching in P116 Jurkat T cells expressing Zap70 variants or TCR (specifically CD3ζ) fused to green fluorescent protein (GFP) (Supplementary Fig. 3a,b). Zap70 exchange rates are high during early T cell activation and lower at later stages21,27. Here, we quantified early exchange rates by determining the half-time of recovery (t1/2) and mobile fractions. Notably, binding to all ten phosphorylated ITAMs of the TCR-CD3-complex contributes to Zap70 clustering. The fluorescence recovery of CD3ζ was minimal due to the immobilization of TCRs by OKT3 on the surfaces (Fig. 3a,b and Supplementary Table 3). tSH2 showed a smaller mobile fraction and a greater recovery time than those of wild-type Zap70 but higher exchange rates than those of CD3ζ (Fig. 3a,b and Supplementary Table 3). Because SPT showed no differences between tSH2 and TCR in their membrane movement (Fig. 2 and Supplementary Fig. 1f–i), tSH2 exchanged exclusively with its cytosolic pool. The mobile fraction of Zap70 Y126E was greater, with a slightly lower recovery time, relative to those of wild-type Zap70 (Fig. 3a,b and Supplementary Table 3). In contrast, Zap70 Y126F showed a smaller mobile fraction and a greater recovery time than those of wild-type Zap70, similar to tSH2 (Fig. 3a,b and Supplementary Table 3), which suggested that phosphorylation of Tyr126 promoted the release of Zap70 from the TCR. Zap70 K369A and, to a lesser degree, Zap70 K369R showed smaller mobile fractions and greater recovery times than those of wild-type Zap70 (Fig. 3a,b and Supplementary Table 3), in support of our finding (above) that kinase activity was required for the release of Zap70 into the membrane. The intermediate exchange rate for Zap70 K369R indicated that binding of ATP promoted the release of Zap70, but only into the cytoplasm, since SPT did not show lateral movement of Zap70 K369R (Fig. 2 and Supplementary Fig. 1f–i). tSH2 Y126E showed a greater mobile fraction and a shorter recovery time than those of tSH2, while tSH2 Y126F displayed the opposite change in mobility (Fig. 3c,d; and Supplementary Table 4), which confirmed the concept that phosphorylation of Tyr126 promoted the release of Zap70. The difference between tSH2 and tSH2 Y126F in Zap70-deficient T cells suggested that Lck contributed to the phosphorylation of Tyr126.
Figure 3.

Tyr126 phosphorylation and ATP binding promote the release of Zap70 from the TCR. (a) FRAP analysis of GFP-fused wild-type or mutant Zap70 or the TCR (CD3ζ) (key) in P116 Jurkat T cells within 10 min of the binding of T cells to an OKT3-coated surface (data points and curves fit to an exponential function). (b) Mobile fraction (top) and recovery rate (t1/2; bottom) for the cells in a. (c) FRAP analysis of tSH2 and its variants (key) in P116 Jurkat T cells (assessed and presented as in a). (d) Mobile fraction and recovery rate for the cells in c (presented as in b). Data are from three or more independent experiments with at least 15 different cells (average ± s.e.m. in a,c; average + 95% confidence interval in b,d).
Trans-autophosphorylation controls Zap70 release
To determine if trans-autophosphorylation regulates the release of Zap70, we compared membrane movement (by SPT) and exchange rates (by FRAP) of the kinase-inactive Zap70 molecules (K369A and K369R) in Zap70-deficient P116 T cells with those in the parental E6.1 cells and in P116 cells stably re-expressing wild-type Zap70. In the presence of catalytically active Zap70, the membrane movement of Zap70 K369A and Zap70 K369R returned to that of wild-type Zap70 (Fig. 4a and Supplementary Table 5). FRAP recovery curves for Zap70 K369A and Zap70 K369R were closer to that of wild-type Zap70 in the presence of catalytically active Zap70 (Fig. 4a and Supplementary Table 3). However, results for mobile fractions and half-times of recovery were inconclusive because FRAP is vulnerable to the varying expression of mutant and wild-type Zap70 (Supplementary Fig. 3c). Still, the increased MSD and diffusion rates for Zap70 K369A and Zap70 K369R at the single-molecule level suggested that trans-autophosphorylation, probably at Tyr126, promoted the release of Zap70 from the TCR. Zap70 activity also facilitated membrane binding, since the released Zap70 K369A and Zap70 K369R moved within the membrane. Multiple Zap70 tyrosine residues are trans-autophosphorylated5,12 and could support membrane association through protein-protein interactions or conformational changes. The presence of catalytically active Zap70 had only minor effects or no effect on the membrane movement and exchange rates of tSH2, Zap70 Y126E or Zap70 Y126F (Supplementary Fig. 3 and Supplementary Tables 3 and 5).
Figure 4.
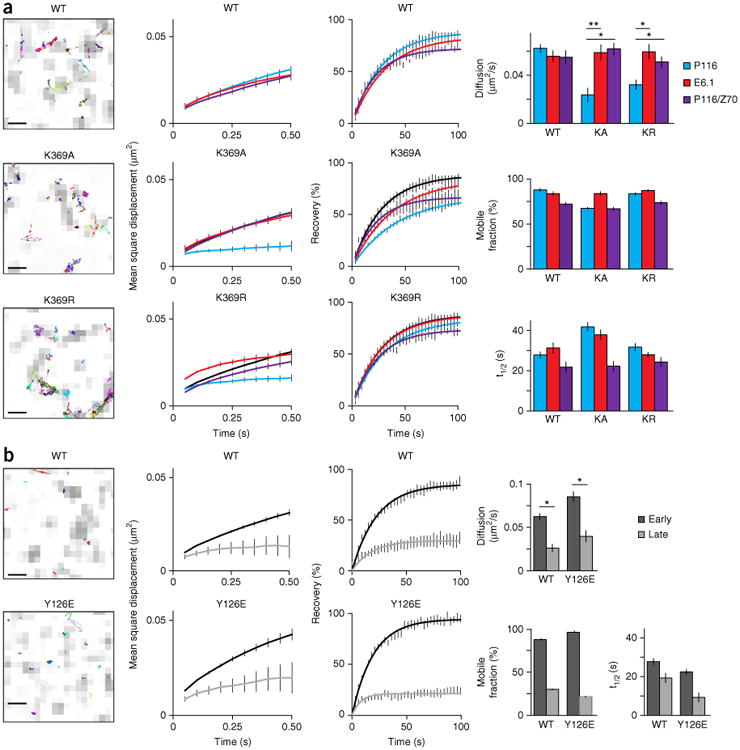
Trans-autophosphorylation mediates the release of Zap70 during early T cell activation. (a) Single-molecule trajectories of wild-type or mutant Zap70 (above plots) in E6.1 cells, presented as an overlay on TCR microclusters (CD3ζ-GFP) (far left; presented as in Fig 1a, far right); MSD plots for wild-type or mutant Zap70 (above plots) in P116 cells (cyan lines), E6.1 cells (red line) or P116 cells re-expressing wild-type Zap70 (purple lines), with wild-type Zap70 in P116 cells (black lines) included in each plot for comparison (middle left); FRAP curves for each cell line at left (middle right); and diffusion rates from tracking (top), plus the mobile fraction (middle) and recovery rate (t1/2; bottom) for the analyses at left (far right). (b) Single-molecule trajectories of wild-type Zap70 (top) and Zap70 Y126E (bottom) in P116 cells, assessed at a late time point (far left); MSD plots for wild-type or mutant Zap70 (above plots) in P116 cells at an early time point (<10 min; black line) or late time point (>30 min; gray line) (middle left); FRAP curves for wild-type Zap70 and Zap70 Y126E at early and late time points (middle right; as at left); and diffusion rates from tracking experiments, plus the mobile fraction and recovery rate (t1/2) (far right). Scale bars (far left), 500 nm. *P < 0.01 and **P < 0.001 (two-tailed unpaired t-test). Data are from three independent experiments (left and middle; average ± s.e.m. of at least ten cells (middle)) or are from three or more independent experiments with at least ten different cells (far right; average + s.e.m. (FRAP) or average ± 95% confidence interval (mobile fraction and recovery rate)).
If the role of Zap70's release from the TCR is to overcome signaling thresholds and induce signaling in spatially distinct membrane domains, it might occur only during initial T cell activation. We compared the exchange rates and membrane movement of wild-type Zap70 and Zap70 Y126E during early (<10 min) and late (>30 min) T cell activation. The exchange rates and membrane movements of wild-type Zap70 and Zap70 Y126E during late T cell activation mirrored TCR mobility (Fig. 4b and Supplementary Table 6), which suggested that the release of Zap70 was an early signaling event. The mechanisms that stabilize Zap70-TCR interactions during late signaling dominated over the Zap70 Y126E substitution and most probably the phosphorylation of Tyr126 (Fig. 4b and Supplementary Table 6). At later stages, the exchange rates of Zap70 K369R were diminished to those of Zap70 K369A in P116 Jurkat T cells, and both mutants dropped to the rate of wild-type Zap70 in E6.1 Jurkat T cells (Supplementary Fig. 4 and Supplementary Table 7). This suggested that Lck and trans-autophosphorylation were able to stabilize the TCR-Zap70 binding. Phosphorylation of Tyr315 and Tyr319 increases the affinity of Zap70 for the TCR21. Thus, the sequential phosphorylation of tyrosine residues or the dominance of one phosphorylation site over another might control and time the release of Zap70 during T cell activation.
Phosphorylation and ATP binding reduce Zap70 affinity
We assessed the capacity of various Zap70 forms to bind phosphorylated ITAMs in ELISA-based competition assays21. Specifically, Zap70 variants compete with tagged (Myc or hemagglutinin), kinase-inactive Zap70 (D461N-tagged) for binding to the phosphorylated ITAM of the cytosolic domain of CD3γ (refs. 21,41,42). The binding of Zap70 D461N-tagged to phosphorylated CD3γ was halved when it was in competition with equimolar amounts of wild-type Zap70 or Zap70 Y126F (Fig. 5a and Supplementary Table 8), which suggested that their in vitro binding capacities were comparable. Zap70 Y126E had to be at a concentration approximately ninefold higher than that of wild-type Zap70 or Zap70 Y126F to halve the binding of Zap70 D461N-tagged to phosphorylated CD3γ (Fig. 5a and Supplementary Table 8), which suggested that phosphorylation of Tyr126 regulated the release of Zap70 from the TCR. Since tSH2 Y126E had to be approximately ninefold higher concentrated than tSH2 or tSH2 Y126F to compete similarly with Zap70 D461N-tagged for binding to phosphorylated CD3γ (Fig. 5a and Supplementary Table 8), phosphorylation of Tyr126 directly weakened Zap70-TCR binding. In the absence of ATP, wild-type Zap70 and the kinase-inactive mutants Zap70 K369A, Zap70 K369R and Zap70 D461N competed comparably with Zap70 D461N-tagged for binding to phosphorylated CD3γ (Fig. 5b and Supplementary Table 8). In the presence of ATP, approximately ninefold lower concentrated Zap70 K369A than wild-type Zap70, Zap70 K369R or Zap70 D461N competed similarly with Zap70 D461N-tagged for binding to phosphorylated CD3γ (Fig. 5b and Supplementary Table 8. From these data, we concluded that binding of ATP diminished the affinity of Zap70 for phosphorylated ITAMs and thereby promoted the release of Zap70 from the TCR.
Figure 5.

Tyr126 phosphorylation and ATP binding decrease the efficiency of Zap70's binding to phosphorylated ITAMs. (a) Binding of Zap70 D461N-tagged to phosphorylated CD3γ in the presence of competitor full-length wild-type Zap70, Zap70 Y126E or Zap70 Y126F (left), or tSH2, tSH2 Y126E or tSH2 Y126F (right), assessed at three different ratios of Zap70 D461N-tagged to competitor (key). (b) Binding of Zap70 D461N-tagged to phosphorylated CD3γ in the presence of competitor Zap70 D461N, Zap70 K369A or Zap70 K369R in the presence (+ ATP) or absence (–ATP) of ATP (below plots). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01 and ***P < 0.001 (two-tailed unpaired t-test). Data are from three independent experiments (average ± s.e.m.).
We used biolayer interferometry (BLI) to determine the dissociation constant (KD), on rate (kon) and off rate (koff) for the binding of recombinant Zap70 forms to phosphorylated CD3γ. For the tSH2 and tSH2 Y126F, the data fit one-component binding models and yielded single sets of constants for each protein (Fig. 6a). The data for tSH2 Y126E fit a two-component binding model (Fig. 6a), which suggested it was in equilibrium between conformations21. We calculated effective binding constants for tSH2 Y126E by averaging weak constants and strong constants weighted by their contributions. tSH2 Y126E showed a ∼3.5-fold higher KD, a ∼2-fold lower kon and a >3-fold higher koff than those of tSH2 and tSH2 Y126F (Fig. 6b and Supplementary Table 9). These data supported the proposal that phosphorylation at Tyr126 accelerated the release of Zap70 from phosphorylated ITAMs by increasing its koff. BLI data for wild-type Zap70 and catalytically inactive Zap70 K389A, Zap70 K369R and Zap70 D461N fit two-component models and yielded sets of high- and low-affinity constants (Fig. 6c,d and Supplementary Table 10). This suggested that the Zap70-TCR interactions involved equilibria between conformations21. The presence of ATP accelerated the release of wild-type Zap70, Zap70 K369R and Zap70 D461N from phosphorylated CD3γ and increased the contribution of their low-affinity constants but did not affect Zap70 K369A (Fig. 6c,d and Supplementary Table 10). The effective binding constants showed over twofold higher effective KD values, over twofold higher effective kon values, and over threefold higher effective koff values for wild-type Zap70, Zap70 D461N and Zap70 K369R after the addition of ATP, whereas the constants for Zap70 K369A remained relatively unchanged (Fig. 6d and Supplementary Table 11). We concluded from these data that binding of ATP promoted the release of active Zap70.
Figure 6.

Tyr126 phosphorylation and ATP increase Zap70's koff for binding to phosphorylated ITAM. (a) BLI analysis of the binding of tSH2, tSH2 Y126E or tSH2 Y126F (above plots) to phosphorylated CD3γ, presented as raw data and fitted curves (key) for six different concentrations (lines: 100 nM, 50 nM, 25 nM, 12.5 nM, 6.25 nM, 3.125 nM and 1.5625 nM): tSH2 and tSH2-Y126F were fit with a one-component model; tSH2-Y126E) fit best with a two-component model (tSH2-Y126EΔ) component model (key). (b) KD, kon and koff for tSH2, tSH2 Y126E and tSH2 Y126F (horizontal axes), fit as noted in a. (c) BLI analysis of the binding of wild-type Zap70, Zap70 K369A, Zap70 K369R or Zap70 D461N (above plots) to phosphorylated CD3γ in the presence or absence of ATP (left margin), presented as in a. (d) Fraction of wild-type Zap70, Zap70 K369A, Zap70 K369R and Zap70 D461N in the high-affinity state (far left), plus the effective KD (KDeff), kon (koneff) and koff (koffeff) values (all fit with a two-component model). *P < 0.05 and **P < 0.001 (two-tailed unpaired t-test). Data are from five (a,b) or four (c,d) independent experiments (error bars, s.e.m.).
Zap70 release determines signal thresholds and amplitudes
We analyzed the phosphorylation of Zap70 and LAT in P116 Jurkat T cells matched for expression of GFP-fused wild-type Zap70, Zap70 Y126E or Zap70 Y126F (Supplementary Fig. 5a). Phosphorylation of Zap70 at Tyr493 is required for its full catalytic activity. After activation with various concentrations of OKT3, phosphorylation at Tyr493 was slightly lower for Zap70 Y126F and up to 60% lower for Zap70 Y126E than for wild-type Zap70 (Fig. 7a). This suggested that Zap70 Y126E mimicked the constitutive phosphorylation at Tyr126, which caused its rapid and premature release from the TCR. The diminished dwell times of Zap70 Y126E at the TCR would make encounters with upstream kinases (for example, Lck) less likely and consequently diminish phosphorylation at Tyr493. While wild-type Zap70 and Zap70 Y126F showed similar phosphorylation at Tyr493, phosphorylation of LAT was up to 50% lower for cells expressing Zap70 Y126F than for those expressing wild-type Zap70 (Fig. 7a). Despite their having up to 60% less phosphorylated Tyr493, cells expressing Zap70 Y126E showed LAT phosphorylation similar to that of cells expressing Zap70 Y126F (Fig. 7a). The molar activity (ratio of phosphorylated LAT to Zap70 phosphorylated at Tyr493) of Zap70 Y126E was 40% greater with weaker stimuli than that of wild-type Zap70, whereas Zap70 Y126F showed a general ∼20% lower molar activity than that of wild-type Zap70 (Fig. 7a). From these data, we concluded that the release of Zap70 allowed more frequent interactions with spatially distinct LAT. Since we observed no accumulation of phosphorylated Tyr493 for Zap70 Y126E (Fig. 7a), it is likely that released Zap70 was dephosphorylated. As expected, the ATP-binding mutants were not able to phosphorylate LAT (Supplementary Fig. 5b,c).
Figure 7.
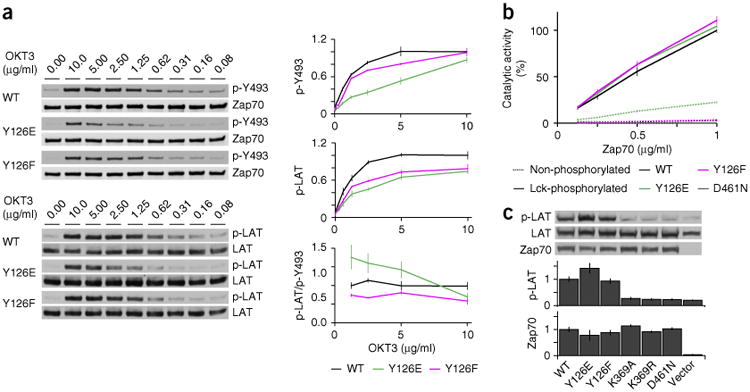
Release of Zap70 mutants affects TCR signaling. (a) Immunoblot analysis (left) of Zap70 phosphorylated at Tyr493 (p-Y493) and total Zap70 (top group) or LAT phosphorylated at Tyr171 and Tyr191 (p-LAT) and total LAT (bottom group) in P116 Jurkat T cell lines with matched expression of wild-type Zap70, Zap70 Y126E or Zap70 Y126F (left margins), stimulated with various concentrations (above lanes) of OKT3; right, densitometry analysis of phosphorylation, relative to total protein (top and middle), and molar activity, presented as the ratio of phosphorylated LAT to Zap70 phosphorylated at Tyr493 (bottom; molar activities < 1 μg/ml are not shown here because of the high s.e.m.), all normalized to results obtained for the stimulation of wild-type Zap70 with the highest concentration of OKT3. (b) In vitro kinase activity of recombinant Zap70 (key) not phosphorylated (dashed lines) or phosphorylated by Lck (solid lines), assessed at various concentrations of Zap70 (horizontal axis); results are normalized to those obtained with highest concentration of wild-type Zap70. (c) Immunoblot analysis (top) of phosphorylated and total LAT (as in a) in HEK293T cells co-expressing LAT plus empty vector (Vector; far right) or vector encoding wild-type or mutant Zap70 (below plots; match lanes above); below, phosphorylation of LAT relative to total LAT (top plot) and Zap70 expression (bottom plot). Data are from three independent experiments (a, right; error bars, s.e.m.), four independent experiments with duplicates (b; error bars, s.e.m.) or two independent experiments (c; error bars, s.e.m.).
We compared the in vitro phosphorylation of the cytosolic domain of LAT (expressed in bacteria and purified) by wild-type Zap70, Zap70 Y126E, Zap70 Y126F and Zap70 D461N (all expressed in baculovirus and purified) to exclude the possibility that such substitution of Tyr126 directly affected the catalytic activity of Zap70. Non-phosphorylated wild-type Zap70, Zap70 Y126F and Zap70 D461N showed no catalytic activity, whereas Zap70 Y126E showed some catalytic activity, with small amounts of phosphorylated LAT (Fig. 7b). After being phosphorylated by recombinant Lck, wild-type Zap70, Zap70 Y126E and Zap70 Y126F showed similar catalytic activity, whereas Zap70 D461N remained inactive (Fig. 7b). In addition, transient co-expression of LAT with various Zap70 molecules in HEK293T human embryonic kidney cells resulted in the phosphorylation of LAT in cells expressing wild-type Zap70, Zap70 Y126E or Zap70 Y126F but not in those expressing the ATP-binding mutants (Fig. 7c). Zap70 Y126E showed some basal activity in vitro (Fig. 7b), and cells expressing Zap70 Y126E showed more phosphorylation of LAT than that of cells expressing wild-type Zap70 (Fig. 7c). This was due to either background phosphorylation of Zap70 Y126E during expression or low constitutive activity of Zap70 Y126E (Fig. 7c). These data showed that after being phosphorylated by Lck, wild-type Zap70, Zap70 Y126E and Zap70 Y126F had similar catalytic activity. Consequently, the diminished cellular activity of Zap70 Y126E and Zap70 Y126F was not due to changes in their catalytic potential. We propose that optimized release of Zap70 amplifies signaling by activating multiple Zap70 molecules per phosphorylated ITAM of the TCR-CD3 complex and enables activated Zap70 to translocate to LAT-containing membrane domains.
We analyzed phosphorylation of the kinases Erk1 and Erk2 (collectively called ‘Erk1/2’ here) in T cells stimulated with either OKT3 or the superantigen SEE presented by the Raji human Burkitt's lymphoma cell line. With either stimulus, the phosphorylation of Erk1/2 was less in T cells expressing Zap70 Y126F than in those expressing wild-type Zap70 or Zap70 Y126E (Fig. 8a), which suggested that the release of Zap70 enhanced the activation of Erk1/2. In our experimental system, cells expressing wild-type Zap70, Zap70 Y126E or Zap70 Y126F showed no significant difference in their secretion of the cytokine IL-2 after being activated by SEE (Supplementary Fig. 6a,b).
Figure 8.
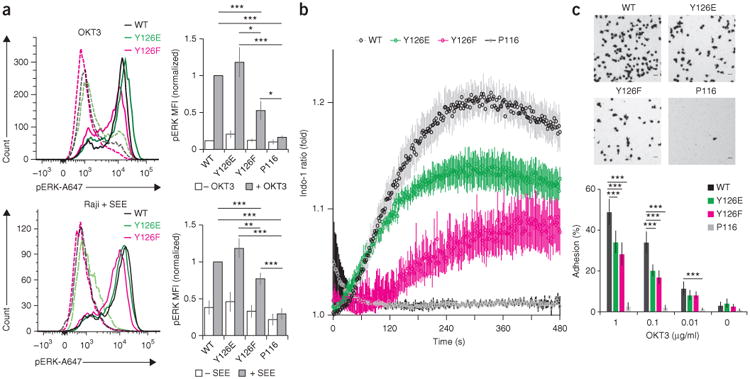
Zap70 release mutants affect T cell functions. (a) Phospho-flow analysis (left) of phosphorylated Erk1/2 (p-ERK) in P116 Jurkat T cell lines matched expression of wild-type Zap70, Zap70 Y126E or Zap70 Y126F (key), left unstimulated (dashed lines) or stimulated (solid lines) with OKT3 (top) or SEE-loaded Raji cells (bottom); right, median fluorescent intensity (MFI) of phosphorylated Erk1/2 in cells as at left. (b) Ca2+-flux analysis of P116 Jurkat T cell lines left untransduced (P116) or transduced to express wild-type Zap70, Zap70 Y126E or Zap70 Y126F (key), at various times (horizontal axis) after stimulation with OKT3 (10 ng/ml), presented as the ratio of the fluorescent Ca2+ indicator dye Indo-1 (violet fluorescence/blue fluorescence), relative to that in unstimulated cells. (c) Microscopy of P116 Jurkat T cell lines transduced as in b (above images), on ICAM-1-coated surfaces (top), and quantification of the adhesion of those T cells to surfaces coated with various concentrations (horizontal axis) of OKT3 (below). *P < 0.05, **P < 0.01 and ***P < 0.001 (two-tailed unpaired t-test). Data are from at least three independent experiments (a,c; average ± s.e.m.) or are from four independent experiments (b; average ± s.e.m.).
We also measured Ca2+ flux in response to stimulation with OKT3 in P116 Jurkat T cells expressing wild-type or mutant Zap70. After treatment with weak stimuli, cells expressing Zap70 Y126E or wild-type Zap70 had Ca2+ flux with similar kinetics, but the Ca2+ flux of cells expressing Zap70 Y126E had a lower maximum (Fig. 8b). Cells expressing Zap70 Y126F showed delayed and up to twofold lower responses than those of cells expressing wild-type Zap70 (Fig. 8b). These data suggested that release of Zap70 was required for the initiation of Ca2+ flux and that the amplitude of Ca2+ flux depended on the amount of active Zap70. Because effects were greatest at low stimuli, we propose that optimized release of Zap70 increased T cell sensitivity (Fig. 8b and Supplementary Fig. 6c). Complementary single-cell analyses of Ca2+ flux confirmed those findings (Supplementary Fig. 6d). Quantifying the adhesion of T cells to surfaces coated with the adhesion molecule ICAM-1 can be used to assess the effects of the Zap70 mutants on activation of the integrin LFA-1 (ref. 43). After being stimulated with various concentrations of OKT3, T cells expressing Zap70 Y126E or Zap70 Y126F showed 40–50% less adherence than those expressing wild-type Zap70, while T cells expressing kinase-inactive mutants did not adhere (Fig. 8c and Supplementary Fig. 7). These data showed that adhesion relied on signal amplification and dispersion through optimal release of Zap70. Collectively, our findings suggest a ‘catch-and-release’ model for signal amplification, dispersion and timing (Supplementary Fig. 8).
Discussion
We have shown that activated Zap70 was released from the phosphorylated ITAMs of the TCR-CD3 complex into the plane of the plasma membrane. The released Zap70 moved for distances within the membrane that allowed it to interact with spatially distinct signaling molecules (i.e., LAT). The release of Zap70 was controlled by at least two synergistic mechanisms. Phosphorylation of Tyr126 by Lck and/or trans-autophosphorylation diminished the binding affinity of Zap70 for phosphorylated ITAMs. ATP binding, a hallmark of active kinases40, also lowered the binding affinity of Zap70 for phosphorylated ITAMs and guaranteed that only activated kinases were released, which thereby maximized signal amplification and dispersion. The change in binding affinity was probably due to conformational changes transferred from the ATP-binding site to the SH2 domains by intramolecular interactions, which has been described as the ‘linker-kinase sandwich’34. Lateral movement was not due to release from and rebinding to the TCR, as Zap70 affinities and mobility did not correlate, and proteins with tandem SH2 domains have longer ‘survival times’ than the SPT track durations observed here44. Substitutions that modulated Zap70-release kinetics showed the greatest effects with weak stimuli, which suggested that T cell sensitivity originates in the optimized release of Zap70 from the TCR. Our data suggest that serial activation of Zap70 molecules at a single phosphorylated ITAM maximizes signal amplitude, while the translocation to spatially distinct signaling molecules ensures rapid response kinetics. The release of Zap70 is restricted to early T cell activation, when signal amplification and crosstalk with other pathways are essential to ensure binary T cell responses.
Imaging analyses have shown that the recruitment of Zap70 to the plasma membrane occurs exclusively via the phosphorylated ITAMs of agonist-pMHC-engaged TCRs and that signal amplification must occur downstream of the TCR11. This is exactly where release of Zap70 from the TCR facilitates the activation of multiple kinases at the same phosphorylated ITAM and thus signal amplification. This ‘catch-and-release’ mechanism turns the TCR into a ‘catalytic unit’ that activates sufficient numbers of Zap70 kinases to overcome activation thresholds. FRAP studies of T cells bound to pMHC surfaces have shown that Zap70's exchange rates with microclusters are several-fold higher than those with the TCRs21. Interactions of agonist peptide– MHC class II complexes with TCRs have half-lives of 0.1–0.2 s in situ45, and positive selecting MHC class I ligands and MHC class II ligands have minimum dwell times of 0.9 s and 0.2 s, respectively14. Such data, together with the koff values for phosphorylated Zap70 measured here, would suggest that large numbers of Zap70 molecules could be activated at and released from each TCR. However, the signal-amplification efficiency must be quantified in future studies. This mechanism is most probably the first amplification step of the TCR signaling cascade and would explain how fewer than ten agonist peptides can induce full T cell responses2,3.
After a T cell is activated, protein islands (nanoclusters) concatenate into microclusters but remain spatially distinct25. This raises the question of how the TCR signaling sequence is maintained when its components are not co-localized. Specifically, Zap70 is recruited to TCR protein islands (nanoclusters) that are adjacent to those containing its substrate LAT. Released and active Zap70 remains associated with the membrane and can transfer the signal to the spatially separated LAT. Our data suggest that Zap70 transfers signals between concatenated protein islands (nanoclusters), but Zap70 could reach as far as neighboring microclusters. The translocation of Zap70 molecules from the TCR to LAT membrane domains needs to be confirmed by imaging studies. TCR-independent binding of Zap70 to the plasma membrane could take place through interactions with other membrane-associated proteins via its SH2 domains or phosphorylated tyrosine residues46,47. Alternatively, the carboxy-terminal SH2 domain of Zap70 contains a lipid-binding site for phosphatidylinositol trisphosphate and phosphatidylinositol-(4,5)-bisphosphate that is required for T cell activation and could facilitate direct membrane binding upon Zap70's release36.
The release of Zap70 from the TCR occurs during early T cell activation. At later signaling stages, Zap70 functions exclusively in a TCR-bound form21,27. That conclusion is supported by the increasing number of Zap70 molecules associated with a specific TCR over the course of T cell activation11. The reduced release of Zap70 from the TCR at later stages of T cell activation depends, but most probably not exclusively, on trans-autophosphorylation of Zap70. Specifically, phosphorylation of Tyr315 and Tyr319 (ref. 11) enhances the affinity of Zap70 for phosphorylated ITAMs21. Future studies should investigate further if sequential phosphorylation, dominance of specific phosphorylation sites or the generation of distinctly phosphorylated Zap70 populations that accumulate at different rates triggers the switch or transition from release to stable TCR binding.
Our proposed ‘catch-and-release’ model could have implications for most cellular pathways involving membrane receptors that lack intrinsic catalytic activity or signaling components that are divided into distinct membrane domains25,26,48,49. SH2-domain-containing proteins are used for signal transduction, cytoskeletal regulation, transcription and protein degradation46,50. Moreover, paired SH2 domains, as studied here, can be found in many enzyme types, such as kinases, phosphatases, hydrolases and transcription factors. More generally, any protein-interaction domain with specificity for protein motifs, post-translational modifications, nucleic acids or lipids has the potential to be part of a similar mechanism47. Thus, the activation-dependent release of enzymes from receptors into restricted environments, such as the plasma membrane, is most probably a common mechanism for signal amplification and dispersion.
Online Methods
Cell culture and generation of Jurkat T cell lines
Primary T cells were isolated from 5C.C7 mice (homogenized lymph nodes and spleen). After homogenization and red blood cell lysis, T cells were stimulated with MCC peptide (amino acids 88-103: ANERADLIAYLKQATK) and IL-2 (added after 24 h in culture) in RPMI medium (Gibco) supplemented with 10% FCS, glutamine (Sigma-Aldrich), sodium pyruvate (Sigma-Aldrich) and non-essential amino acids (Sigma-Aldrich). The MCC peptide was synthesized and purified by the peptide synthesis core of the Salk Institute. In parallel, HEK293T cells were transfected with Fugene HD (Promega) to express retrovirus constructs encoding mouse Zap70-PATagRFP, Zap70-tSH2-PATagRFP, and CD3ζ-eGFP with a pCL-Eco packaging vector at a 1:1 ratio to DNA. T cells were spinoculated (2,500 r.p.m., 90 min) with 293T viral supernatant preincubated with 10 μg/ml Polybrene (Millipore) for 20 min 2 d after isolation. T cells were selected with antibiotics 36 h after infections and live cells were isolated after gradient centrifugation with Histopaque-1077 (Sigma-Aldrich) at least 4 h before imaging 6 or 7 d after initial T cell harvest.
Stable Jurkat T cell lines (P116 and E6.1) expressing fluorescent proteins were obtained by lentiviral infection. Virus was prepared from HEK 293T cells transfected with 7 μg vector of interest (in pRLL lentivirus vectors), 2.5 μg pVSVG, 2 μg pRSV/REV and 5 μg pMDL.G (RRE) packaging vectors in 1.5 ml Opti-MEM (Life Technologies). Lipofectamine 2000 (5 μl/μg of DNA; Life Technologies) was added to 1.5 ml Opti-MEM, incubated for 5 min at RT, and then combined with the DNA mixture. After 20 min of incubation, the mixture was added dropwise to HEK293T cells. After 2 d, the viral supernatant was extracted, mixed with 10 μg/ml polybrene for 20 min, and added to Jurkat T cell lines (∼1 × 106 cells/ml of viral mixture). Cells were spun for 90 min at 2,500 r.p.m., and media was replaced with regular growth media (RPMI plus 5% FCS). After 2 d of growth, cells were incubated with antibiotics [puromycin (bioWORLD) or blasticidin (Life Technologies)] to select for cells expressing constructs of interest. Dead cells were removed with Histopaque-1077 (Sigma-Aldrich). Relative expression of Zap70 molecules in all Jurkat cell lines were quantified by immunoblot analysis with LI-COR infrared antibodies. P116 cells expressing wild-type Zap70, Zap70 Y126E and Zap70 Y126F fused to GFP were sorted (BD Biosciences Influx) for equivalent expression for functional assays.
Jurkat P116 and E6.1 cells were transiently transfected by electroporation (Neon transfection system, Invitrogen). 350,000 cells grown in 5% FCS/RPMI media were washed with PBS, resuspended in 10 μl of resuspension buffer (Invitrogen) and mixed with 2 μg vector of interest (pIRES vectors). Cells were electroporated with a 10 μl Neon tip using an optimized setting for P116 cells (950 V, 30 ms, two pulses) and E6.1 cells (1,600 V, 10 ms, three pulses). After electroporation, cells were resuspended in 500 μl of 5% FCS and RPMI and incubated at 37 °C for 24 h.
SPT of Zap70 release from the TCR
For primary T cells, eight-well glass chambers (Lab-Tek II chambered coverglass #1.5 borosilicate, Nalgen Nunc International) were cleaned with plasma (Harric Plasma) and sequentially coated with 0.1 mg/ml biotinylated poly-l-lysine, 25 μg/ml streptavidin, and 100 nM biotinylated I-EK-MCC plus 300 nM biotinylated B7.1 (CD80; extracellular domains). For Jurkat T cells, eight-well glass chambers were cleaned with plasma and coated with 0.5 mg/ml poly-l-lysine, dried overnight, and then incubated with 10 μg/ml antibody to (anti-) CD3ε (OKT3, eBioscience) in HEPES-buffered saline (HBS: 20 mM HEPES pH7.4 plus 150 mM NaCl) for 2 h at 37 °C.
Prior to imaging cells were spun at 1,500 r.p.m., exchanged into imaging buffer (Hank's buffered saline solution (HBSS) with 0.5 mM CaCl2, 1 mM MgCl2, 1% FCS), and added to coated glass chambers containing imaging buffer equilibrated to 37 °C. Cells were maintained at 37 °C during image acquisition with a Tokai Hit Stage Top Incubator. Imaging was performed on a Nikon Ti-E inverted microscope platform with a 100× objective (CFI Apo TIRF oil immersion NA 1.49, Nikon) and Andor iXon 860E EMCCD camera. A 405-nm 100-mW diode laser was used for photoactivation of PATagRFP (CUBE, Coherent) and kept below 1% of power to achieve low molecular densities. Laser beams with excitation wavelengths of 488 nm and 568 nm (Genesis MX, Coherent) were controlled by acoustic optical modulators (Isomet) for stroboscopic illumination (2- and 5-ms pulses, respectively). Tracking experiments were performed at 20 Hz in streaming acquisition mode driven with MetaMorph imaging software (Molecular Devices). Data was acquired within 10 min (for early signaling) or after 20 min (for late signaling) of adding T cells to an imaging well. SPT in fixed Jurkat T cells (4% PFA (Electron Microscopy Sciences) for 10 min) established an experimental minimum for single-molecule movement in our system.
Single-molecule identification and tracking was performed with DiaTrack 3.03 software. Image series were imported and intensity spots were identified with a 2D Gaussian fit function. Images were background-subtracted and processed with a Gaussian filter of a half-width-half-max value of 1.2 pixels. Particle-identification thresholds were user determined for each data series, which included an upper and lower threshold to discard false-positive particles and particles too bright to be considered single molecules. 50–100 frames were previewed to minimize false particle identification. Particles were tracked with a maximum displacement of three pixels (480 nm) per 50 ms frame. Trajectories with lifetimes of less than three frames were eliminated from further analysis. The distribution of spans (distance between two furthest points in a track) were exported for analysis in Excel (Microsoft). The coordinate time series data was exported for further analysis in IgorPro 6.2 (WaveMetrics, Inc.). The coordinate time series for each data set was used to compute average mean square displacement curves. Non-confined diffusion represents a linear increase in MSD over time. Confined or immobile molecules cause the MSD to plateau over time relative to the localization uncertainty. The average diffusion was computed at a 500-ms time delay. A localization precision for each tracked molecule was computed from a five-by-five pixel summation of signal at each coordinate position, subtracting a standard computed background signal, and converting the intensity to photons from the conversion gain of the Andor iXon 860E EMCCD.
FRAP analysis of Zap70 exchange at TCR microclusters
FRAP experiments were performed in various Jurkat T cells lines (E6.1, P116 and P116 cells stably expressing Zap70-mCherry) stably expressing different Zap70 variants fused to GFP. FRAP was measured on a Zeiss LSM 780 confocal microscope with a 100× objective and Tokai Hit Stage Top Incubator to 37 °C. FRAP was performed on the same OKT3-coated surfaces used for SPT. Prior to imaging, cells were spun and resuspended in 100 μl of imaging media at ∼3.5 × 106 cells/ml and 10 μl were added to each well for immediate imaging. Once a cell landed on the OKT3-coated surface, a cluster was identified and was selected for bleaching along with a control and background area of equal size for measurement. Bleaching was performed until 80% of initial intensity was reached, after which the area was monitored for recovery every 3 s for 2 min. Background intensity was subtracted from the intensity of the bleach and reference area and the ratio of bleach to reference cluster was normalized over the entirety of the imaging period. Data were plotted as recovery over time. Curves were fit with a single exponential function [F* = A(1 – e–τt)] as previously described21. Means and errors for the mobile fraction (f) and recovery rate (t1/2) represent a 95% confidence interval derived from the fitting parameters.
Expression and purification of Zap70, CD3γ and Lck derived proteins. Baculovirus expression. With the exception of the Zap70 Y126E mutant, full-length Zap70 proteins were produced in insect cells with the Bac-to-Bac Baculovirus Expression System (Invitrogen). Full-length wild-type human Zap70 and mutant forms Zap70 Y126F, Zap70 K369A, Zap70 K369R and Zap70 D461N were cloned into the pFastBac Vector (Invitrogen). All constructs have a C-terminal linker (GAS) and a human rhinovirus (HRV) 3C protease cleavage site (LEVLFQGP) followed by a glycine serine linker (GGGS) and a 12-histidine (His) tag (HHHHHHHHHHHH). Mutations in the gene encoding wild-type Zap70 were introduced by site-directed mutagenesis and confirmed by DNA sequencing. To prevent autophosphorylation, we generated clones to produce wild-type Zap70 and Zap70 Y126F with a C-terminally fused human SHP-1 phosphatase domain (amino acids 265-522) between the glycine serine linker and the 12-His tag. In addition, we expressed Zap70 D461N with C-terminal tags for detection by placing a Myc tag (EQKLISEFELN) or a hemagglutinin (HA) tag (YPYDVPDYA) between Zap70 and the HRV 3C protease-cleavage site (D461N-tagged). Baculovirus stocks were made according to the Bac-to-Bac manufacturer's protocol. Recombinant ‘bacmid’ DNA (baculovirus shuttle vector) was isolated from transfected DH10bac cells by maxi-prep DNA purification (Invitrogen), and bacmids were screened by blue-white selection and subsequently by PCR. Sf9 insect cells were transfected with Cellfectin II reagent according to the manufacturer's manual for obtaining viral stock. We amplified baculovirus by infecting Sf9 cells with 1:2,000 viral stock at 27 °C in Sf900 media (Invitrogen) supplemented with 10% FCS (HiClone), 1× pen-strep, 2× glutamine, Fungizone 1:400 by maintaining the cell number at 2 × 106/ml and collecting virus at 50% cell death. The viral titer of P1 stocks was individually optimized by small-scale Zap70-expression analysis (48 h after infection) with Hi5 insect cells in protein-expression media (Sf900/Insect Express (Lonza) at 1:3 ratio) supplemented with 1× pen-strep, 2× glutamine, Fungizone 1:400). All insect cell cultures were grown at 27 °C.
Zap70 molecules’ expression in and purification from insect cells
For preparative-scale expression, 2 × 106 Hi5 cells/ml were infected with P1 viral stocks at dilutions ranging from 1:100 to 1:500 and grown at 27 °C for 64 h with agitation (125 r.p.m). Cells were collected by centrifugation (1,800g, 15 min). Cells were lysed at 4 °C for 30 min in 100 ml Lysis Buffer (50 mM HEPES, pH 7.4, 0.5 M NaCl, 10% glycerol, 10 mM MgCl2, 1.3% NP-40, 1 mM DTT, 20 mM imidazole, 2 μg/ml leupeptin, 1 μg/ml pepstatin, 0.1 mM PMSF, 0.1 μM staurosporine and 20 μM genistein) per liter of culture volume and stirring at 4 °C. Lysates were clarified by centrifugation at 20,000g for 1 h at 4 °C. Cleared lysate was treated with 10 μg/ml DNaseI (Roche) for 25 min at room temperature (with continuous stirring). Zap70 was purified by binding to nickel–nitrilotriacetic acid (Ni-NTA) agarose beads (Thermo Scientific) overnight at 4 °C. Ni-NTA beads were washed on a gravity flow column with at least 50 column volumes of Wash Buffer (50 mM HEPES, pH 7.4, 0.5 M NaCl, 10% glycerol, 0.1% octyl-β-D-glucopyranoside, 1 mM DTT, 50 mM imidazole and 0.05 mM PMSF). Zap70 was subsequently cleaved off the beads in wash buffer (two times the bead volume) with HRV 3C protease (75 μg per ml of Ni-NTA beads) at 4 °C overnight. Zap70-containing elution fractions were concentrated and purified by S200 size-exclusion chromatography (SEC) into SEC Buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT, 0.1% octyl-β-D-glucopyranoside, 10% glycerol and 1 mM Na3VO4). Pure Zap70 was dialyzed against SEC buffer supplemented with 50% glycerol and stored at −20 °C. Final protein preparations were verified as lacking tyrosine phosphorylation by both ELISA and immunoblot analysis with antibodies to specific phosphorylated tyrosine residues (i.e., Tyr292, Tyr319, Tyr492 and Tyr493 (Supplementary Table 12)) as well as antibody 4G10, to all phosphorylated tyrosine residues.
Zap70 Y126E expression in and purification from HEK293T cells
Ful-length Zap70 Y126E was not amenable to production in insect cells and was instead produced in HEK293T cells. Sequence encoding Zap70 Y126E constructs (catalytically active and Zap70 D461N) with carboxy-terminal linker, HRV 3C protease sites and 12-His tags (as described above for pFastBac constructs) were inserted into pcDNA3.1 (Invitrogen). HEK293T cells were plated onto poly-l-lysine-coated 150-mm plates the day before transfection to achieve 75% density at the time of transfection. After 24 h, cells were transfected with a mixture of 4 μg polyethylenimine (PEI; Polysciences, Inc.) per μg of DNA in 150mM NaCl to achieve 25 μg of DNA per plate in 2 ml. Fresh medium containing 5% FBS was added to the cells (12 ml/plate), and the DNA:PEI mixture was added dropwise to the dishes. After 24 h, an additional 13 ml of fresh medium containing 10% FBS and 30 mM HEPES, pH7.4, was added to the cultures. 72 h after transfection, cells were treated with kinase inhibitors (0.5 μM staurosporine and 100 μM genistein) for 45 min at 37 °C before harvesting of cell lysates directly from the plates. Media was harvested and 4 ml/plate Lysis Buffer was applied before scraping the plates and recovering additional cells by rinsing with another 2 ml Lysis Buffer. Lysates were incubated for 1 h at 4 °C with agitation and then insoluble material was clarified as described above for insect cell lysates, and additional NaCl was added to a final concentration of 500 mM. Ni-NTA resin equilibrated with lysis buffer was then added to the lysate and further purification was carried out as described for insect cell production. We treated the Ni-NTA-immobilized catalytically active Zap70 Y126E protein with Lambda Phosphatase (Lambda PP; New England BioLabs) to remove any phosphorylation. After extensive washing of the resin with Wash Buffer, the resin was equilibrated to Lambda PP buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 2 mM DTT, 0.01% Brij-35) supplemented with 1 mM MnCl2. Lambda PP was added approximately at an 1:20 molar ratio with the Zap70 Y126E (estimated from assessing the resin-bound Zap70 against a known titration of purified Zap70 on SDS-PAGE) for 1 h at 30 °C. Lambda PP was removed by further washing and re-equilibration of the resin into Wash Buffer for subsequent HRV 3C protease cleavage and further purification as described above for insect-cell-produced proteins. Final protein preparations were analyzed for tyrosine phosphorylation by both ELISA and immunoblot analysis with antibodies to specific phosphorylated tyrosine residues (i.e., Y292, Y319, Y492, Tyr493) as well as antibody 4G10 to all phosphorylated tyrosine residues (Supplementary Table 12). We observed a small amount of residual phosphorylation for catalytically active Zap70 Y126E.
ΔN-Lck expression and purification from insect cells
His-tagged ΔN-Lck (which lacks the first 60 amino acids) was purified from baculovirus-transfected insect cells. 2 × 106 Hi5 insect cells per milliliter were transfected with 1:200 baculovirus P1 and grown for 64 h in a shaker (27 °C, 125 rpm). Before being collected, Hi5 cells were treated with 0.4 mM pervanadate for 30 min at 27 °C with agitation (125 rpm) to phosphorylate and activate Lck. We made fresh pervanadate by oxidizing 20 mM Na3VO4 with 1.5% H2O2 at room temperature for 10 min and subsequently removing access peroxide by catalase treatment. After collecting cells by centrifugation (1,800 g, 15 min), we lysed them in Lysis Buffer (50 mM HEPES, pH 7.4, 200 mM NaCl, 10% glycerol, 10 mM MgCl2, 1.3% NP-40, 1 mM DTT, 20 mM imidazole, 2 μg/ml leupeptin, 1 μg/ml pepstatin, 0.1 mM PMSF and 0.5 mM Na3VO4) by suspending them in 1/10 the original culture volume and stirring for 30 min at 4 °C. Lysate was cleared by centrifugation at 20,000g for 1 h at 4 °C. Cleared lysate was treated with 10 μg/ml DNAseI (Roche) for 25 min at room temperature with stirring. ΔN-Lck was purified by binding to Ni-NTA (Thermo Scientific) overnight at 4 °C. Ni-NTA beads were washed on a gravity flow column with at least 50 column volumes of wash buffer (50 mM HEPES, pH 7.4, 500 mM NaCl, 10% glycerol, 0.1% octyl-β-D-glucopyranoside, 1 mM DTT, 40 mM imidazole, 0.5 mM Na3VO4 and 0.05 mM PMSF), and ΔN-Lck was eluted with wash buffer supplemented with 300 mM imidazole. Fractions containing ΔN-Lck were concentrated and purified by SEC in Lck SEC buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT, 0.1% octyl-β-D-glucopyranoside, 10% glycerol and 1 mM Na3VO4). Pure ΔN-Lck was dialyzed against SEC buffer supplemented with 50% glycerol and stored at −20 °C.
tSH2 and CD3γ expression in and purification from Escherichia coli
All constructs were cloned into a modified pET30 vector resulting in an N-terminal 12-His tag followed by GST, a linker (AAGAAG), an HRV 3C protease cleavage site (LEVLFQGP) and another linker (GT) N-terminal to the target proteins. Human Zap70's tSH2 module (amino acids 1–256; wild-type Zap70 or with replacement of the tyrosine at position 126 with glutamic acid or phenylalanine); human CD3γ amino acids 137–182 (preceded by Gly-Gly; CD3γCD); and CD3γ amino acids 137–182 containing a cysteine inserted immediately after the HRV 3C cleavage site, yielding an essentially N-terminal cysteine upon cleavage (cysCD3γCD) were transformed into BL21-DE3 bacteria and grown in TB media with 10 μg/ml kanamycin after 1:20 inoculation from an overnight culture in a shaker (37 °C, 220 r.p.m.). After the culture reached an absorbance of 0.7 at 600 nm, we induced expression with 0.5 mM IPTG and allowed cells to grow for 4 h (30 °C, 220 r.p.m.). Cells were collected by centrifugation (4,500g, 15 min, 4 °C), suspended in Lysis Buffer (25 mM HEPES, pH 7.4, 300 mM NaCl, 5% glycerol, 20 mM imidazole, 5 mM β-mercaptoethanol and 0.1 mM PMSF) and lysed by three passes through a microfluidizer cell disruptor (Microfluidics). Lysates were cleared by centrifugation (30,000g, 30 min, 4 °C) and immobilized on Ni-NTA affinity resin. After overnight incubation with Ni-NTA at 4 °C, beads were washed with 50 column volumes of Wash Buffer (25 mM HEPES, pH 7.4, 300 mM NaCl, 40 mM imidazole, 5% glycerol and 5 mM β-ME) on a gravity flow column. GST-CD3γCD was subsequently eluted with wash buffer supplemented with 375 mM imidazole, concentrated and purified by S200 SEC (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT, 0.1% octyl-β-D-glucopyranoside, 10% glycerol and 1 mM Na3VO4). For tSH2 constructs, CD3γCD and cysCD3γCD, proteins were subsequently cleaved off the beads in wash buffer (two times the bead volume) with HRV 3C protease overnight at 4 °C (75 μg per ml of beads). Elution fractions containing tSH2 (wild-type Zap70, Zap70 Y126E and Zap70 Y126F) were concentrated and purified by SEC. CD3γCD and cysCD3γCD fractions were either acetone-precipitated to concentrate them or loaded directly onto a Superdex Peptide column without concentration. Pure tSH2 proteins were dialyzed against SEC buffer supplemented with 50% glycerol and stored at −20 °C.
Pure GST-CD3γCD, CD3γCD and cysCD3γCD were phosphorylated with recombinant Lck as follows: SEC-purified proteins (at 1–2 mg/ml) were incubated with a 1:4-molar-ratio-purified ΔN-Lck for 60 min at 30 °C in SEC buffer supplemented with 5 mM ATP, 25 mM MgCl2, 1 mM Na3VO4. After phosphorylation, GST-phosphorylated CD3γCD was further purified at 4 °C overnight on glutathione-agarose affinity resin (Bioworld) to remove Lck. Glutathione beads were washed with 50 column volumes of wash buffer on a gravity column, and bound proteins were eluted with wash buffer supplemented with 10 mM reduced glutathione. GST-phosphorylated CD3γCD-containing fractions were concentrated, purified by S200 SEC and stored at −20 °C after dialysis into SEC buffer supplemented with 50% glycerol. phosphorylated CD3γCD was cleared of Lck by acetone precipitation and then subjected to SEC on a Superdex Peptide column. phosphorylated CD3γCD was acetone precipitated and stored at −20 °C. cys-phosphorylated CD3γCD was purified by Superdex Peptide SEC into PBS buffer containing 10 mM EDTA. DMSO (20% final) was added to cys-phosphorylated CD3γCD (1 mg/ml), which were then biotinylated by overnight incubation at RT with 2 mg/ml EZ Link Maleimide-PEG2-Biotin (Thermo Scientific). After biotinylation, bio-phosphorylated CD3γCD was cleared of free biotin by purification on Superdex Peptide SEC with subsequent acetone precipitation and stored at −20 °C.
Determining Zap70 concentrations for affinity measurement
All purified proteins were analyzed by SDS-PAGE with Coomassie Brilliant Blue staining and by immunoblot analysis for tyrosine phosphorylation. We estimated protein stock concentrations by measuring absorbance at 280 nm. We achieved rough verification of these values by running 1 μg of stock (predicted from the A280) on a Coomassie-stained SDS-PAGE gel and comparing it to a known standard, and correcting the concentrations. On the basis of these assessments, we prepared twofold dilutions ranging from 1 μg/ml to 7.5 ng/ml in 50 mM Na2CO3, pH 9.2, and used 50 μl of each to coat duplicate wells of Immulon 4HBX plates. We allowed proteins to adsorb at room temperature for 1 h, at which time we removed unbound protein by washing wells with TBS plus 0.05% Tween-20 (TBST). Next, the antigen-coated wells were blocked for 1 h at room temperature with 1% BSA in TBST. Adsorbed proteins were subsequently detected with 1 μg/ml anti-Zap70 clone 29 (BD Biosciences) to detect the carboxyl terminus or clone 2F3.2 (Upstate) to detect the amino terminus of Zap70. Detection antibodies were incubated for 1 h at room temperature in 0.5% BSA in TBST with agitation. Wells were subsequently washed with TBST and then detected with 0.8 μg/ml horseradish peroxidase (HRP)-labeled goat anti-mouse (ThermoFisher #32430) for 30 min. Wells were washed again with TBST, and then bound detection antibody was visualized colorimetrically with Fast-OPD (Sigma); color development was stopped with 3 N HCl according to the manufacturer's instructions. Absorbance at 490 nm was determined, and concentrations were corrected on the basis of comparison of multiple points in the linear portion of the curves. New protein lots were corrected using the Myc- or HA-tagged preparations as a standard.
ELISA-based Zap70 competition assay
Immulon 4HBX microtiter plates were coated overnight with 50 ng/ml GST-phosphorylated CD3γCD plus 400 ng/ml GST in 50 mM Na2CO3, pH 9.2. Wells were washed with TBST to remove unadsorbed protein and then blocked for 1 h at room temperature with TBST plus 1% BSA. Competitor proteins were mixed, at the ratios indicated in Figure 5, with 5 μg/ml Zap70 D461N-tagged in exchange buffer (50 mM HEPES, pH 7.4, 140 mM KCl, 10 mM NaCl, 5 mM MgCl2, 1% glycerol, 0.2% NP-40, 1 mM DTT and 0.5% BSA with or without 1 mM ATP). Protein mixtures were then added to the blocked wells and allowed to bind for 1 h at room temperature. Unbound protein was removed by washing with exchange buffer. Bound Zap70 D461N-tagged was detected with 1 μg/ml antibody to the Myc tag (clone 9E10, Fisher Scientific) or antibody to the HA tag (clone 12CA5, Becton Dickinson). Primary antibodies (Supplementary Table 7) were incubated for 1 h at room temperature and then washed with exchange buffer to remove unbound antibody. Primary antibodies were detected by incubation for 30 min at room temperature with 4 μg/ml secondary HRP-labeled anti-mouse (ThermoFisher #32430) in exchange buffer. Wells were washed, and then bound detection antibody was visualized colorimetrically with Fast-OPD (Sigma); color development was stopped with 3 N HCl according to the manufacturer's instructions. Absorbance at 490 nm was determined on a VersaMax microplate reader (Molecular Devices).
BLI analysis
Zap70 stock solutions were adjusted for exact concentration by ELISA as described above. Stock Zap70 samples were diluted in BLI buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 0.25 mM TCEP, 5 mM MgCl2, 5% glycerol, 0.05 mM Na3VO4, 0.5% BSA and 0.1% Tween-20, with or without 1 mM ATP) in twofold dilutions between 100 nM and 1.5625 nM. For each experiment, eight streptavidin Dip and Ready Biosensors (FortéBio) were coated with 2 μg/ml bio-phosphorylated CD3γCD for 1 min in BLI buffer. Each Zap70 protein was measured with a fresh set of biosensors. Association of Zap70 with biophosphorylated CD3γCD-covered biosensors was measured for 5 min with 100 nM, 50 nM, 25 nM, 12.5 nM, 6.25 nM, 3.125 nM, 1.5625 nM and no Zap70 (reference well). Dissociation in BLI buffer was measured for 10 min. We processed binding data with Savitzky-Golay filtering after subtraction of the reference well data, y-axis alignment to baseline and interstep alignment correction to dissociation. Graphs were fit globally for association and dissociation over the full range of the experiment (Rmax UnLinked by sensor) using a 1:1 or 2:1 (heterogeneous ligand) binding model as determined by curve fit using χ2. For the 2 component binding model, the fractions of both binding constants (a × KD1, b × KD2) were calculated as a = Req1 / Req1+2 and b = Req2 / Req1+2. Effective binding constants were determined from the relative contribution of each component. Goodness of fit was determined with χ2 and R2 tests (Octet software v.7).
ELISA-based kinase activity assay
The potential of wild type and mutant Zap70 to activate LAT was determined in a two-step in vitro kinase assay. Zap70 concentrations were determined by Qubit protein assay (Life Technologies), ELISA and quantitative immunoblot analysis to ensure that all forms of Zap70 were the same concentration. Each recombinant Zap70 was phosphorylated and/or activated by incubation with recombinant Lck bound to Ni-NTA magnetic beads (G Bioscience). Specifically, 2.5 μg of recombinant and active Lck were bound 7.5 μl of washed magnetic beads in 40 μl binding buffer (50 mM HEPES pH7.4, 140 mM KCl, 10 mM NaCl, 10 mM MgCl2, 0.5 mM Na3VO4, 1 mM dithiothreitol, 0.2% NP-40, 5% glycerol, 10 mM imidazole and 1 mg/ml bovine serum albumin (BSA)) for 2 h at 8 °C in an Thermomixer C (Eppendorf) at 1,250 r.p.m. The magnetic bead were washed in binding buffer supplemented with 40 mM imidazole and resuspended in 100 μl kinase buffer without ATP (50 mM HEPES pH8, 140 mM KCl, 10 mM NaCl, 10 mM MgCl2, 0.5 mM Na3VO4, 1 mM dithiothreitol, 0.2% NP-40, 1% glycerol and 1 mg/ml bovine serum albumin (BSA)). Zap70 was activated at 5 μg/ml with 20 μl Lck-bound magnetic beads in 60 μl kinase buffer supplemented with 2 mM ATP for 30 min at 32 °C and 1,250 r.p.m. Lck was removed using a magnet, and the supernatants containing phosphorylated Zap70 molecules were collected. The catalytic activity of non-phosphorylated and phosphorylated Zap70 molecules were measured at various concentrations (1 μg/ml, 0.5 μg/ml, 0.25 μg/ml and 0.125 μg/ml) and 12.5 μg/ml recombinant Myc-tagged LAT (amino acids 34-262, Myc-LAT) as substrate in 100 μl kinase buffer supplemented with 2 mM ATP for 30 min at 32 °C and 1,250 r.p.m. The kinase reaction was stopped with 50 μl of 0.5 M EDTA. The reaction was diluted to 2.5 μg/ml LAT followed by twofold dilution steps in Tris-buffered saline solution, pH 7.4, supplemented with 0.01% Tween-20 (TBST) plus Na3VO4 plus 0.5 mM 0.5% BSA to yield six different concentrations of Myc-LAT. Each Myc-LAT concentration was added in duplicate onto ELISA plates (Immulon4 HBX from Thermo Scientific) that had been coated overnight at 4 °C with 5 μg/ml antibody to Myc-tag (9E10; Fisher Scientific) in 50 mM Na2CO3, pH 9.2, and subsequently blocked with TBST plus 1% BSA for 2 h at RT. Myc-LAT was captured for 2 h and washed three times with 300 μl TBST plus 0.1 mM Na3VO4. The plate was incubated for 2 h with 0.5 μg/ml HRP-conjugated antibody 4G10 to phosphorylated tyrosine (identified above) in TBST buffer plus 0.5 mM Na3VO4 plus 0.5% BSA. The plate was washed three times with 300 μl TBST plus 0.1 mM Na3VO4 buffer and developed with 150 μl o-phenylenediamine dihydrochloride substrate (SigmaFAST OPD). The reaction was stopped after 10 min with 50 μl 3N HCl. The absorbance at 492 nm was immediately measured. Kinase activity was determined by plotting of the kinase concentration against normalized ELISA absorbance and fitting the data points with a nonlinear fitting model using Excel (Microsoft Office). The activity for different non- and phosphorylated Zap70 were compared by normalizing 100% as the highest activity of phosphorylated wild-type Zap70.
In-cell kinase assay
HEK293T cells were plated at 5 × 105/well of a six-well dish 24 h before transfection with a combination of LAT and the indicated Zap70 constructs, or LAT and empty vector as a control. A stock solution of LAT-GFP/pIRESpuro3 (10.5 ng/μl) was divided into seven tubes (200 μl each) and then 0.9 μg Zap70-eGFP/pIRESpuro3 (wild-type Zap70, Zap70 Y126E, Zap70 Y126F, Zap70 K369A, Zap70 K369R or Zap70 D461N) or pIRESpuro3 (empty vector) was added to each tube. PEI (9 μg/tube) was added and the tubes were mixed vigorously and then allowed to incubate at room temperature for 15 min to allow DNA:PEI complex formation. The entire reaction volume was then added dropwise with agitation to 2 ml freshly replaced growth medium/well, and the cells were cultured for 48 h before removal of culture supernatant and lysis directly on the plate as described for protein production in HEK293T cells. Recovered lysates were clarified by centrifugation and the protein concentration of each lysate determined by BCA Assay (ThermoFisher). Equal amounts of cell lysate were separated by SDS-PAGE and immunoblot analysis as described below.
Immunoblot analysis of activated Zap70 cell lines
For Jurkat activation, 2× concentrations of activating antibody solutions were prepared as follows: OKT3 (eBioscience) was diluted to 20 μg/ml in HBSS+ (Hank's Balanced Salt Solution containing 0.5 mM CaCl2 and 2 mM MgCl2), and then serial twofold dilutions were prepared in HBSS+. Stable P116 cell lines were pelleted and washed once with HBSS+ before being resuspended in HBSS+ at 1 × 108 cells/ml. Cells (100 μl per condition) were added to an equal volume of activating antibody solution, and tubes were incubated at 37 °C for 2 min before addition of an equal volume (200 μl) of 2× Lysis Buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 2% NP-40, 20% glycerol, 2 mM DTT, 2 mM EDTA, 2 mM Na3VO4, 20mM NaF, 2 μg/ml pepstatin and 4 μg/ml leupeptin). For Raji B cell conjugation experiments, 0 or 2 μg/ml superantigen (SEE from Toxin Technology) was used to prime Raji cells for two hours before Jurkat T cell activation. Raji and Jurkat T cells were mixed at a 1:1 ratio and spun for 30 s to enhance cell-cell contacts and lysed as described above after 2 min of activation. Cell lysates were incubated on ice for 30 min, and then insoluble material was clarified by centrifugation. For the detection of LAT, lysates were directly processed for immunoblot analysis; for the detection of ectopically expressed Zap70, lysates were immunoprecipitated overnight with 1 μg biotinylated 9F9 monoclonal antibody to GFP (Rockland) per sample. Antibody-antigen complexes were precipitated for 2 h with 10 μl Streptavidin-Agarose (Novagen) before extensive washing with Lysis Buffer. In all cases, reducing Laemmli sample buffer was added to a final concentration of 1× and samples were boiled and analyzed as described below.
Cell lysates or recombinant proteins were diluted in 1× reducing Laemmli sample buffer and heated for 7 min at 95 °C before being loaded onto 4–12% BisTris gels (Novex). Gels were run in 1× MES-buffer and then blotted onto PVDF membrane. Membranes were blocked with 0.1% casein in 0.2× PBS for 2 h to overnight. Membranes were then incubated with primary antibodies (Supplementary Table 12) in blocking buffer containing 0.1% Tween-20 for 1 h to overnight before being washed with TBST, and primary antibodies were detected with secondary HRP-conjugated anti-mouse (ThermoFisher #32430) or anti-rabbit (ThermoFisher #32460), where appropriate. HRP-labeled secondary antibodies were visualized with Pierce ECL Western Blotting Substrate (ThermoFisher) and exposure to HyBlot CL autoradiography film (Denville Scientific).
Immunoblots were quantified by scanning (Epson Perfection V750 Pro) in transluminescence and linear mode. Multiple exposure times (three to six) were used for quantification. Exposure times causing saturation of the signal were eliminated from the analyses. Scans were quantified using ImageJ software and the gel-analysis tool. A minimum of three exposure times from two independent experiments were averaged and used to determine the s.e.m.
Mass spectrometry analyses of Tyr126 phosphorylation
P116 cells stably expressing Zap70-GFP (3.5 × 108 cells/sample) were activated with crosslinked OKT3-OKT4 antibodies (eBiosciences; 10 μg/ml each with 20 μg/ml goat anti-mouse IgG (ThermoFisher #32430), Fc fragment (Pierce)) for 2 min. Zap70 was immunoprecipitated as described above. Proteins were reduced with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich) and alkylated. Proteins were digested for 18 h at 37 °C in 2 M urea and 100 mM TEAB, pH 8.5, with Lys-C (Wako). Analysis was performed using an EASY-nLC 1000 (Thermo) and a Thermo Fusion Orbitrap Tribrid mass spectrometer with a 30-cm, 75-μm ID column packed with BEH 1.7um C18 resin (Waters) and using an in-house built electrospray stage51. Protein and peptide identification and protein quantitation were done with Skyline and Integrated Proteomics Pipeline - IP2 (Integrated Proteomics Applications, Inc., San Diego, CA http:// www.integratedproteomics.com/). Tandem mass spectra were extracted from raw files using RawConverter52 and were searched against UniProt protein database with reversed sequences using ProLuCID53,54. The search space included all peptide candidates for the digest with static modification of 57.02146 on cysteine (carbamidomethylation) and differential modification of 79.9663 on STY (phosphorylation). Peptide candidates were filtered using DTASelect, with the following parameters: -p 2 -y 1–trypstat–pfp 0.1 –modstat– extra–pI -DM 5–DB–dm -in–brief–quiet52,55.
Analysis Of Phosphorylated Erk1/2 In Activated Zap70-Expressing T Cell Lines
Phospho-flow cytometry was used to analyze phosphorylated Erk1/2 in P116 cells expressing wild-type Zap70 and Zap70 Tyr126 mutants. For OKT3 activation, cells were spun at 1,500 r.p.m. and resuspended at 1 × 107 cells/ml. An equal volume of 20 μg/ml OKT3 (eBiosciences) was added in the same media (HBSS, 0.5mM CaCl2, 1 mM MgCl2 and 1% FCS) for 5 min of activation. Cells were washed once with ice-cold PBS and fixed for 10 min with 1.5% PFA. Cells were transferred into ice-cold methanol and stored at −20 °C until staining was performed. Cells were blocked in 1% BSA in PBS for 1 h before staining with primary rabbit antibody to phosphorylated Erk1/2 (Cell Signaling, #4370) for 1 h at room temperature. Cells were stained with an Alexa Fluor 647 goat secondary antibody to rabbit (Jackson ImmunoResearch; #111-606-144) and analyzed on an LSR II flow cytometer (BD Bioscience). Median fluorescent intensity of the same gated GFP fluorescence were used across experimental conditions, representing an equal number of cells (15–25% of total). For Raji B cell conjugation experiments, 2 μg/ml superantigen (SEE) was used to prime Raji cells for 2 h before Jurkat T cell activation. Raji and Jurkat T cells were mixed at a 1:1 ratio and spun at 1,500 r.p.m. for 30 s to enhance cell-cell contacts. Cells were fixed after 2 min and processed for analysis of phosphorylated Erk as described above.
IL-2 quantification from T cell media supernatant
IL-2 was assessed with a human IL-2 ELISA based kit (eBioscience) from the supernatant of Jurkat T cells stimulated for various times. T cells were stimulated either by 10 μg/ml OKT3 (eBiosciences) crosslinking or Raji B cells pre-incubated with SEE for 2 h. Concentrations of IL-2 were quantified per the ELISA kit's protocol relative to recombinant human IL-2.
Ca2+-flux analysis
Flow cytometry was used to analyze Ca2+ flux of the P116 Jurkat T cell lines described above. Indo-1 (ThermoFisher) ratiometric dye was loaded into cells at 5 × 106 cells/ml in media (HBSS, 0.5 mM CaCl2, 1 mM MgCl2 and 0.5% BSA) for 30 min at 37 °C. Cells were washed and left to rest for at least 15 min in media at room temperature before analysis on the LSR II flow cytometer (BD Bioscience). Cells were analyzed in non-activating media for the first 30 s and then switched to the stimulating media condition with variable concentrations of OKT3 in the same media. Ca2+ flux was measured for 10 min. The Indo-1 ratio (violet/blue, 355-nm excitation, 405-nm/485-nm emission ratio) was analyzed with FlowJo software (FlowJo) and values were exported to Excel (Microsoft).
Single-cell depletion of ER calcium was measured on activating on OKT3 surfaces. P116 Jurkat T cell lines matched for Zap70 expression were loaded with 4 μM Fura-2 (Biotium) for 30–45 min and then washed into imaging media without calcium (HBSS, 1% BSA, 2 mM MgCl2, and 0.2 mM EGTA) and allowed to recover for 15–30 min. T cells were sequential illumination at 380 nm and 340 nm using a Lambda LS lamp (Sutter Instruments) and MetaMorph's multidimensional acquisition mode at 1 Hz for five minutes. Ratiometric calcium flux curves were analyzed with the ImageJ cookbook ratio profiler plugin.
Adhesion analysis of activated Zap70 T cell lines to ICAM-1-coated surfaces
24-well plates were cleaned with plasma and coated with 0.1 mg/ml biotinylated poly-l-lysine for 30 min at room temperature, washed with water, and dried overnight. Blocking with 5% BSA in HEPES-buffered saline (HBS) for 2 h. Plates were washed with HBS and coated with 25 μg/ml streptavidin in HBS with 2.5% BSA for 2 h. After washing with HBS, plates were coated with 2.5 μg/ml biotinylated ICAM-1 (extracellular domain) in HBS plus 2.5% BSA overnight at 4 °C. Wells were washed with HBS and loaded with 0.5 ml of 2× stimulating solutions. Specifically, either 10 mM MgCl2 and 2 mM EGTA solution was used to activate LFA-1 nonspecifically (control and standard for 100% binding of T cell lines) or 4 mM MgCl2 and 1 mM CaCl2 with various concentrations of OKT3 was used to activate TCRs. Jurkat cells were re-suspended in HBSS with 1% BSA, counted to ensure precisely 0.5 ml of 2 × 106 cells/ml were added to each well, and mixed thoroughly to ensure an even distribution of cells. Plates were incubated at 37 °C for 15 min to allow for cell activation and adhesion to ICAM-1-coated surfaces. Each plate was washed by submerging three times in HBS with 2 mM MgCl2 and 0.5 mM CaCl2 and then fixed in the same solution with 1 ml of 2% paraformaldehyde per well and incubated for 30 min at room temperature. Plates were washed by submersion in PBS and incubated with 1 ml of 0.1% Crystal Violet (Sigma C0775) over night at RT to stain adhered T cells. Plates were washed multiple times in water and imaged on an Axio Vert.A1 microscope (Zeiss). Three representative images per well were taken and areas covered by cells in percent were analyzed in each field with ImageJ software. Liquid from each well was aspirated and crystal violet dye was extracted with 0.25 ml 10% acetic acid for 10 min. Absorbance was measured at 590 nm on a VERSAmax microplate reader (Molecular Devices).
Supplementary Material
Acknowledgments
We thank J. Williamson (the Scripps Research Institute) for use of the BLI instrument; the Salk Biophotonics Core for assistance with FRAP acquisition; the Salk Flow Cytometry Core Facility for assistance with flow-cytometry-based analysis of calcium flux and cell sorting; Jolene Diedrich and James Moresco for mass spectrometry analyses; M. Hetzer, T. Hunter, P. Hogan, G. Lemke, and J. Karlseder for critical reading of the manuscript; A. Weiss (University of California San Francisco, San Francisco, California, USA) for parental Jurkat cell lines; M.M. Davis (Stanford University, Stanford, California) for HEK293T cells; and the Nomis Foundation, the Waitt Foundation and the James B. Pendleton Charitable Trust for support. Supported by the US National Institutes of Health (1DP2GM105455-01 to B.F.L. and T32 CA009370 to Z.B.K.), the Salk Institute Cancer Center core facilities (funded by CA014195 from the National Cancer Institute), the Mass Spectrometry Core of the Salk Institute (supported by P30 014195 from the National Cancer Institute Cancer Center Support Grants of the US National Institutes of Health) and the Helmsley Center for Genomic Medicine.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Author Contributions: Z.B.K. and B.F.L. designed the study and wrote the manuscript; Z.B.K. performed SPT, Ca2+-flux measurements, phospho-flow cytometry and statistical analyses; L.N. performed FRAP experiments; L.N. and A.B. established expression systems for recombinant proteins; and B.F.L. performed kinase and adhesion assays.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 2.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 3.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 4.Huse M, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 5.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 6.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 7.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wülfng C, et al. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat Immunol. 2002;3:42–47. doi: 10.1038/ni741. [DOI] [PubMed] [Google Scholar]
- 9.Krogsgaard M, et al. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 10.Cooper JA, Qian H. A mechanism for SRC kinase-dependent signaling by noncatalytic receptors. Biochemistry. 2008;47:5681–5688. doi: 10.1021/bi8003044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT. Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. eLife. 2013;2:e00778. doi: 10.7554/eLife.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, et al. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veillette A, Caron L, Fournel M, Pawson T. Regulation of the enzymatic function of the lymphocyte-specific tyrosine protein kinase p56lck by the non-catalytic SH2 and SH3 domains. Oncogene. 1992;7:971–980. [PubMed] [Google Scholar]
- 14.Stepanek O, et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell. 2014;159:333–345. doi: 10.1016/j.cell.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baniyash M, Garcia-Morales P, Luong E, Samelson LE, Klausner RD. The T cell antigen receptor zeta chain is tyrosine phosphorylated upon activation. J Biol Chem. 1988;263:18225–18230. [PubMed] [Google Scholar]
- 16.Hatada MH, et al. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature. 1995;377:32–38. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]
- 17.Watts JD, et al. Identification by electrospray ionization mass spectrometry of the sites of tyrosine phosphorylation induced in activated Jurkat T cells on the protein tyrosine kinase ZAP-70. J Biol Chem. 1994;269:29520–29529. [PubMed] [Google Scholar]
- 18.Chan AC, et al. Activation of ZAP-70 kinase activity by phosphorylation of tyrosine 493 is required for lymphocyte antigen receptor function. EMBO J. 1995;14:2499–2508. doi: 10.1002/j.1460-2075.1995.tb07247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brdicka T, Kadlecek TA, Roose JP, Pastuszak AW, Weiss A. Intramolecular regulatory switch in ZAP-70: analogy with receptor tyrosine kinases. Mol Cell Biol. 2005;25:4924–4933. doi: 10.1128/MCB.25.12.4924-4933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horejsí V, Zhang W, Schraven B. Transmembrane adaptor proteins: organizers of immunoreceptor signalling. Nat Rev Immunol. 2004;4:603–616. doi: 10.1038/nri1414. [DOI] [PubMed] [Google Scholar]
- 21.Klammt C, et al. T cell receptor dwell times control the kinase activity of Zap70. Nat Immunol. 2015;16:961–969. doi: 10.1038/ni.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. doi: 10.1146/annurev-biophys-042910-155238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klammt C, Lillemeier BF. How membrane structures control T cell signaling. Front Immunol. 2012;3:291. doi: 10.3389/fimmu.2012.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc Natl Acad Sci USA. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lillemeier BF, et al. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman E, et al. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35:705–720. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunnell SC, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokosuka T, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 29.Cambi A, et al. Organization of the integrin LFA-1 in nanoclusters regulates its activity. Mol Biol Cell. 2006;17:4270–4281. doi: 10.1091/mbc.E05-12-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 32.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto-Tane A, et al. Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation. Immunity. 2011;34:919–931. doi: 10.1016/j.immuni.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Deindl S, et al. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 2007;129:735–746. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 35.Yan Q, et al. Structural basis for activation of ZAP-70 by phosphorylation of the SH2-kinase linker. Mol Cell Biol. 2013;33:2188–2201. doi: 10.1128/MCB.01637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park MJ, et al. SH2 domains serve as lipid-binding modules for pTyr-signaling proteins. Mol Cell. 2016;62:7–20. doi: 10.1016/j.molcel.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keshvara LM, Isaacson C, Harrison ML, Geahlen RL. Syk activation and dissociation from the B-cell antigen receptor is mediated by phosphorylation of tyrosine 130. J Biol Chem. 1997;272:10377–10381. doi: 10.1074/jbc.272.16.10377. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, et al. Tyr130 phosphorylation triggers Syk release from antigen receptor by long-distance conformational uncoupling. Proc Natl Acad Sci USA. 2008;105:11760–11765. doi: 10.1073/pnas.0708583105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szabo M, et al. Fine-tuning of proximal TCR signaling by ZAP-70 tyrosine residues in Jurkat cells. Int Immunol. 2012;24:79–87. doi: 10.1093/intimm/dxr105. [DOI] [PubMed] [Google Scholar]
- 40.Lovering F, et al. Identification of type-II inhibitors using kinase structures. Chem Biol Drug Des. 2012;80:657–664. doi: 10.1111/j.1747-0285.2012.01443.x. [DOI] [PubMed] [Google Scholar]
- 41.Isakov N, et al. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: the tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J Exp Med. 1995;181:375–380. doi: 10.1084/jem.181.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bu JY, Shaw AS, Chan AC. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc Natl Acad Sci USA. 1995;92:5106–5110. doi: 10.1073/pnas.92.11.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans R, et al. Integrins in immunity. J Cell Sci. 2009;122:215–225. doi: 10.1242/jcs.019117. [DOI] [PubMed] [Google Scholar]
- 44.Oh D, et al. Fast rebinding increases dwell time of Src homology 2 (SH2)-containing proteins near the plasma membrane. Proc Natl Acad Sci USA. 2012;109:14024–14029. doi: 10.1073/pnas.1203397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawson T, Gish GD, Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001;11:504–511. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 47.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 48.Wilson BS, Pfeiffer JR, Oliver JM. Observing FcεRI signaling from the inside of the mast cell membrane. J Cell Biol. 2000;149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattila PK, et al. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 2013;38:461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 50.O'Brien R, et al. Alternative modes of binding of proteins with tandem SH2 domains. Protein Sci. 2000;9:570–579. doi: 10.1110/ps.9.3.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolters DA, Washburn MP, Yates JR., III An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 52.He L, Diedrich J, Chu YY, Yates JR., III Extracting accurate precursor information for tandem mass spectra by RawConverter. Anal Chem. 2015;87:11361–11367. doi: 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 54.Xu T, et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics. 2015;129:16–24. doi: 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.