

Cell plate formation in dividing plant cells follows a highly consistent pattern involving precisely choreographed vesicle motion and functionally distinct stages.
Abstract
Cell division in plant cells requires the deposition of a new cell wall between the two daughter cells. The assembly of this plate requires the coordinated movement of cargo vesicles whose size is below the diffraction-limited resolution of the optical microscope. We combined high spatial and temporal resolution confocal laser scanning microscopy with advanced image-processing tools and fluorescence fluctuation methods and distinguished three distinct phases during cell plate expansion in tobacco (Nicotiana tabacum) ‘Bright Yellow-2’ cells: massive delivery of preexisting vesicles to a disk-shaped region at the equatorial plane precedes a primary rapid expansion phase followed by a secondary, slow expansion phase during which the extremity of the circular plate seeks contact with the mother wall and brings about the separation of the two portions of cytoplasm. Different effects of pharmacological inhibition emphasize the distinct nature of the assembly and expansion mechanisms characterizing these phases.
Cytokinesis is a highly regulated process that separates the cytoplasm of the two daughter cells following the formation of two nuclei during mitosis. In both animal and plant cells, this process entails precisely coordinated events that are mediated by cytoskeletal dynamics. However, the mechanism differs significantly between the wall-less cells of animals and plant cells, which are enveloped in a polysaccharidic wall. While, in animal cells, the separation of the cytoplasm is accomplished by a constriction of the plasma membrane, in plant cells, a plate is constructed starting in the center of the equatorial region. This newly assembled wall, the cell plate, expands radially and eventually connects to the existing wall to completely separate the two daughter cell protoplasts. The process begins after the mitotic division of the nucleus and the migration of the two sets of chromosomes to the opposite poles of the cell pulled by the spindle microtubules. The cell plate is formed from cargo vesicles that fuse in the equatorial region to first form a tubulovesicular network that expands in diameter through the further addition of vesicles. While expanding at the periphery, the more mature, central region of the disk starts assembling a very dense fibrous coat composed of matrix polysaccharides such as pectin and hemicellulose (synthesized in the Golgi) as well as cellulose and extensins onto the membrane (Samuels et al., 1995; Cannon et al., 2008; Miart et al., 2014) and subsequently deposits callose for mechanical stabilization (Samuels et al., 1995; Staehelin and Hepler, 1996). Gradually, the mesh-like tubulovesicular network is filled in, giving rise first to a tubular network and subsequently to a planar fenestrated sheet (Seguí-Simarro et al., 2008). Eventually, all gaps in the fenestrated sheet are closed and the mature cell plate consists of two parallel plasma membranes sandwiching a perfectly planar polysaccharidic cell wall that fuses with the parental cell wall.
The initiation of the cell plate and its formation necessitate the accurate targeting and regulation of vesicle transport to the equatorial target site to ensure the correct positioning and mechanical functionality of both the new membrane and cell wall material. Both cargo transport toward the newly forming cell plate and the positioning of this structure in the three-dimensional (3D) space of the cell are controlled by a cytoskeletal array known as the phragmoplast. The phragmoplast is composed of microtubules, actin microfilaments, and the cell plate assembly matrix (CPAM). The microtubules enable cell plate formation by creating a scaffold that directs cell plate-forming vesicles. Their (+)-ends face the cell plate and contain protein complexes that control microtubule dynamics as well as their interactions with the CPAM, organelles, and proteins. Phragmoplast actin filaments form two opposing sets that are oriented parallel to the phragmoplast microtubules (Higaki et al., 2008), with their barbed end toward the cell plate (Zhang et al., 2009). Actin barbed ends point toward each other but do not interdigitate at the equatorial plate, as observed in somatic cells of Arabidopsis (Arabidopsis thaliana; Austin et al., 2005). They are proposed to restrain the accumulation and fusion of vesicles to the midzone or initiation site (Esseling-Ozdoba et al., 2008; Higaki et al., 2008) and to guide the phragmoplast microtubule-binding myosin VIII, thus guiding the margin of the expanding cell plate toward the cortical division site (Hepler et al., 2002; Molchan et al., 2002; Van Damme et al., 2004; Panteris, 2008; Wu and Bezanilla, 2014). The precise role of actin in cytokinesis is poorly understood and controversial, since latrunculin B (LatB), an inhibitor of actin polymerization, was observed to inhibit cell plate expansion or impede its straightening by some (Nebenführ et al., 2000; Higaki et al., 2008; Zhang et al., 2009; Wu and Bezanilla, 2014) but not by others (Van Damme et al., 2011). Additional evidence for the role of actin stems from the administration of the myosin ATPase inhibitor 2,3-butanedion monoxime (BDM; Nebenführ et al., 2000; Higaki et al., 2008; Zhang et al., 2009), which inhibits cell plate expansion, suggesting an important role for an actomyosin-based mechanism in the centrifugal expansion and the alignment of the cell plate.
The trans-Golgi network (TGN) is considered the cornerstone compartment through which either newly synthesized Golgi-derived proteins and polysaccharides, or endocytosed material (proteins, polysaccharides, and lipids) from the plasma membrane and cell wall, and potentially from late endosomes, build the new cell plate (Chow et al., 2008; Thellmann et al., 2010; Richter et al., 2014; Rybak et al., 2014). 3D electron tomography has yielded exquisite ultrastructural details about the anatomy of the growing cell plate and the fusion of vesicles between each other and to the plate (Samuels et al., 1995; Austin et al., 2005). However, electron micrographs are static snapshots in time and cannot reveal dynamics, which necessitates live cell imaging. An optical microscopy approach, on the other hand, is severely challenged by the small size of the vesicles, which is approximately 50 to 80 nm (Samuels et al., 1995; Austin et al., 2005). While their size does not, per se, prevent their positional tracking via optical microscopy, the combination of their small dimensions, rapid movement, and dense crowding around the cell plate renders tracking impossible with conventional microscopy methods. Approximately 95,000 vesicles fuse to initiate the formation of the tubulovesicular network in the cell plate (Seguí-Simarro and Staehelin, 2006); hence, novel strategies need to be employed to assess their movements. We circumvent these challenges by combining high spatial and temporal resolution confocal laser scanning microscopy with advanced fluorescence fluctuation and image analysis methods. Using a specially developed, automated image analysis algorithm, we monitored the diameter of the cell plate over time and measured the intracellular transport of vesicles using spatio-temporal image correlation spectroscopy (STICS; Hebert et al., 2005). We correlate these vesicle transport data with those of the structural elements forming the phragmoplast, actin filaments and microtubules, to reveal mechanistic details about the assembly process as it proceeds from the time of nuclear separation to the full expansion of the cell plate separating the two daughter cells.
RESULTS
Cell Plate Formation in Tobacco ‘Bright Yellow-2’ Cells Occurs in Distinct Phases
In order to characterize the dynamics of cell plate formation in dividing plant cells, we stably transformed tobacco (Nicotiana tabacum) ‘Bright Yellow-2’ (BY-2) cells with pKN::GFP-KN. Knolle is a syntaxin protein or Qa-SNARE (target-SNARE) that is specifically required for the fusion of cell plate vesicles (Lauber et al., 1997; Waizenegger et al., 2000). The fusion protein specifically marks the vesicles that are delivered to form the cell plate and was imaged in three-dimensions over time using both point and line laser scanning confocal microscopy. As is the case for all GFP-based localization studies, some of the signal may derive from unbound protein.
To accurately measure the diameter of the expanding cell plate using an extended time-lapse image series, we developed FluMOS (Fluorescence Morphological Operators Software), a fully automated image-processing pipeline capable of identifying cell plates within fluorescence images and computing their diameter (Fig. 1A; Supplemental Movie S1). Plotting cell plate diameter over time revealed that the expansion of this disk-shaped structure occurs in three phases (Fig. 1B). First, vesicles accumulated and aggregated in a centrally positioned region oriented perpendicular to the length axis of the cell. The diameter of this disk-shaped region was typically around 5.5 µm, with only very small variations between individual cells (5.6 ± 0.84 µm; n = 6). We refer to this phase as IPA. Once this initial disk is assembled, it rapidly expands radially (Fig. 1C) during a phase we call PCG. This rapid phase (1.44 ± 0.44 µm min−1) proceeded until the plate reached a diameter of approximately 15.5 µm (15.6 ± 1.9 µm; n = 12) within the first 10 min after the beginning of the PCG. At this point, the plate margins had not reached any of the parental cell wall boundaries. Subsequently, radial cell plate expansion proceeded at a reduced rate until the circular margin started fusing with the parental plasma membrane. We termed this phase SCG. The average expansion rate of the plate diameter during SCG was less than one-third of the rate observed during PCG (Fig. 1C; 0.35 ± 0.13 µm min−1), with significant variability between individual cells. It is remarkable that, although the absolute sd for the expansion rate over the sampled cell population (n = 14) was smaller for the SCG phase, its relative value was much higher (47% of the average expansion rate) than that of the PCG phase (34%; Fig. 1D). The final diameter of the cell plate was between 20 and 35 µm, depending on the size of the mother cell. The biphasic pattern of centrifugal cell plate expansion was distinctly different from the dynamics of expansion that would be expected if the volume of delivered material was constant over time. To model this, we simplified the geometrical growth pattern by assuming a constant rate of material supply to a smoothly expanding disk that is solid and has constant thickness. The increase in diameter for the cell plate shown in Figure 1B can be calculated with
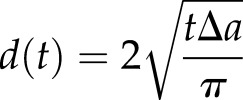 |
where t is time in minutes and ∆a is the increase in the surface area of the plate per minute. Given d (1 min) = 5 µm at the beginning of PCG and d (22 min) = 22 µm at the end of SCG, this yields ∆a = 19 µm2. Comparison between the calculated and the observed dynamics shows that the expansion rate during PCG is faster than the theoretical rate, whereas, during SCG, the plate expands more slowly than calculated. This suggests that the rate of material delivery changes during the developmental steps of cell plate formation, with a higher than predicted rate during PCG and a lower rate during SCG. To assess whether the observed phenomena are representative for plant cells in general, and whether they also apply to cells in a tissue context, we analyzed time-lapse series of Arabidopsis root cells published by others (Supplemental Table S1; Supplemental Fig. S1). The cells of the Arabidopsis root system differ from BY-2 cells by the facts that they are embedded into a well-organized tissue and that they have only few or small vacuoles. Our analyses of the published data revealed that the initiation of the cell plate in these cells occurs on a similarly sized disk with an average diameter of 5.7 ± 0.75 µm (n = 9). The growth of the cell plate displays clear biphasic behavior, with an average PCG rate of 0.87 ± 0.26 µm min−1 and an average SCG rate of 0.25 ± 0.09 µm min−1 (paired Student’s t test, two tailed, P = 0.0001). The switch point between the two growth phases in the Arabidopsis system consistently occurs within the first 10 min after the beginning of the PCG, when the cell plate is approximately 15 µm in diameter. This confirms that the biphasic behavior is a global phenomenon and not specific to cells with large vacuoles or cells growing in the absence of a surrounding tissue.
Figure 1.

Cell plate diameter tracked using FluMOS and 3D representation. A, Typical confocal laser scanning micrographs of median optical sections of a GFP-KN-labeled cell plate (top row) detected by FluMOS software (overlay of the red detection line; bottom row). B, Typical cell plate growth dynamics based on FluMOS analysis (dashed line; IPA, initial plate assembly; PCG, primary centrifugal growth; SCG, secondary centrifugal growth) and theoretical expansion assuming constant vesicle supply (solid black line). C, Radial growth rate during PCG and SCG (average ± sd; n = 14). D, Variability in growth rates between cells, shown as the ratio between sd and average values. E, Surface rendering of z-stacks of a growing cell plate labeled with GFP-KN. Bars = 10 µm.
3D surface rendering of confocal image stacks confirms the presence of distinct phases (Fig. 1E; Supplemental Movies S2 and S3). During IPA, vesicle accumulation onto the initial 5-µm-diameter disk is dense, and the supply remained high and evenly distributed during PCG. Once the cell plate assembly entered SCG, the cell plate displayed an inner circle that was relatively smooth, whereas the outer margin showed the characteristic rough surface that represented arriving and fusing vesicles. In the example shown, another region of 11.5 μm diameter appeared in the center while SCG proceeded, separated from the margin of the rough plate by a 5-μm-wide smoother region. This architecture is consistent with the centrifugal propagation of developmental processes that ensure the formation of the cell plate from the center outward.
Vesicle Dynamics during Cell Plate Assembly
Given the apparent changing rates of material delivery during cell plate assembly, we wanted to investigate in more detail the dynamics of vesicle motion in and around the expanding plate. To this end, we performed experiments based on fluorescence recovery after photobleaching (FRAP). To generate a baseline for the transport dynamics of different markers, we first assessed the behavior of FM4-64, a lipophilic styryl dye that associates with the plasma membrane and is readily endocytosed by BY-2 cells. After its incorporation, FM4-64 labels most endomembrane systems and eventually becomes incorporated into the growing cell plate.
To measure the delivery rate of FM4-64, we photobleached a rectangular region of interest (ROI) on the plasma membrane of BY-2 cells (Supplemental Fig. S2A, inset). Prior to photobleaching, the cells were washed to remove any unincorporated dye. Thus, fluorescence recovery in the plasma membrane had to derive from lateral diffusion within the membrane or from the delivery of new material from the inside of the cell. Photobleaching was adjusted to reduce fluorescence intensity in the ROI to 20% (Supplemental Fig. S2, A and B). FM4-64 label in the plasma membrane showed a high degree of recovery, with a mobile fraction (Mf) of 82% (Supplemental Fig. S2B; Supplemental Movie S4), which is consistent with previously published values (Dhonukshe et al., 2006) and a half-time of equilibration (T1/2) of 45 ± 9.5 s (se; Supplemental Fig. S2A). By contrast, the same analysis made on GFP-KN revealed a much lower recovery (Mf of 45%; Supplemental Fig. S2B; Supplemental Movie S5) but with statistically indistinguishable T1/2 (46.4 ± 8.4 s; Supplemental Fig. S2A) compared with FM4-64. This means that the recovery rate of FM4-64 on the parental plasma membrane is twice as fast as that of GFP-KN. The observed difference in mobility suggests that GFP-KN fluorescence recovery on the plasma membrane is likely to result from lateral diffusion within the plasma membrane rather than from exocytosis. In contrast, FM4-64 is a small lipidic probe that diffuses faster than the large GFP-KN protein within the bilipid layer, and several processes are likely to contribute to its recovery in the plasma membrane after photobleaching, including exocytosis and membrane diffusion.
Next, we wanted to investigate the dynamics of vesicles that are specifically targeted to the cell plate. To do so, we performed FRAP on BY-2 cells expressing GFP-KN at different stages of cell plate formation (Fig. 2; Supplemental Movie S6). Small rectangular ROIs directly on the cell plate were photobleached for this purpose. The T1/2 measured during PCG was clearly shorter than during SCG, and the Mf was higher during the early expansion. During SCG, we distinguished the center and the margin of the cell plate and found that the Mf was higher in the growing margins (51%) than in the center (36%). These differences were significant (unpaired Student’s t test, two tailed, P < 0.001) and are indicative of spatial and temporal differences in the rates of vesicle delivery during the formation of the cell plate.
Figure 2.

FRAP analysis during cell plate growth. A, Typical FRAP curves at the extremities and in the center of a GFP-KN-labeled cell plate during SCG. Fluorescence intensity is normalized to background and prebleach values. Bar = 10 µm. B, Mobile fraction after FRAP on a GFP-KN-labeled cell plate during PCG and SCG at the extremities and in the center of the plate (average ± se; n = 6).
STICS Maps Characteristic Patterns of Vesicle Motion
The FRAP results suggest that the transport of vesicles toward the expanding margin of the cell plate is more dynamic than in the center of the plate. Therefore, it would be interesting to map the spatial and temporal differences in vesicle motion during cell plate expansion. However, with a diameter of 50 to 80 nm, these vesicles are below the diffraction limit resolution of the optical microscope. Moreover, they cannot be tracked individually due to their high density near the cell plate. To circumvent this problem, we used STICS, an imaging domain extension of fluorescence correlation spectroscopy (Hebert et al., 2005). To determine the reliability of this method in a cellular system, we conducted a series of validation tests. We compared velocity values of larger and fast-moving cellular features (e.g. Golgi body dynamics during pollen tube growth) with measurements done by conventional particle-tracking methods, and we compared the speed of movement of cytoskeletal elements in BY-2 cells with previously published values (Supplemental Figs. S3 and S4A). For full details about the parameters used for STICS measurements of the velocities and output vector maps, see “Materials and Methods” and Supplemental Figures S7 to S9.
Thus, convinced of the reliability of STICS in the cellular context, we analyzed FM4-64- or GFP-KN-labeled vesicle dynamics during cytokinesis (Fig. 3). FM4-64 is a lipophilic dye and does not associate with the cytosol. The similarity in labeling pattern between the two markers, therefore, suggests that GFP-KN is unlikely to be expressed ectopically in the cells (FM4-64-stained vesicles; Supplemental Movie S7). The analyses showed that, during anaphase, GFP-KN vesicles within the phragmoplast display low-velocity magnitudes around 1 to 1.5 μm min−1 (Fig. 3B). Moreover, vesicle flow coming from the cytoplasm in contact with the plasma membrane was detected (Fig. 3B, inset). As soon as cell plate formation was initiated during IPA, vesicle movement occurred, with a speed of up to 6 μm min−1 (Fig. 3C; Supplemental Movies S8 and S9). During PCG, the velocity magnitude of KN-marked organelles remained high, between 3 and 4 μm min−1 in the vicinity of the cell plate (Fig. 3D). During SCG, vesicles displayed slower motion in the vicinity of the central region of the cell plate, with velocity magnitudes of approximately 2 μm min−1 (Fig. 3, E and F; Supplemental Movies S8 and S10). The highest speeds measured during this phase were observed at the margin of the cell plate, where they reached 3 to 4 μm min−1 (Fig. 3E). As a negative control, we included cells treated with BDM, a noncompetitive myosin ATPase inhibitor that interferes with actin-based organelle transport (Samaj et al., 2000; Tominaga et al., 2000; Higaki et al., 2008). In our hands, the drug (20 mm) completely blocked cell plate growth. STICS analysis revealed that all vesicle movement in the vicinity of the cell plate was arrested in the presence of BDM (Fig. 3A). In agreement with this, FRAP analysis revealed that the mobile fraction of KN-labeled structures dropped from 55% to 37.4% upon BDM treatment. The remaining recovery is likely to result from the lateral diffusion of GFP-KN from the portions of the cell plate that are outside of the bleached ROI. Such horizontal dynamics within the cell plate can actually be observed on the STICS vector maps (Fig. 3, C–F) and is nonnegligible, with speeds of 1 to 1.5 µm min−1. BDM also affected cytoskeletal dynamics, with differential effects on microtubules and actin filaments whose activities play important and different roles in cell plate formation (Fig. 4; Supplemental Fig. S4).
Figure 3.

STICS analysis of the vesicle supply to allow cell plate growth. Velocity maps of GFP-KN-labeled vesicles were analyzed over a 75-s period (100 frames). Numbers on the color bar at top right in A are movement rates in µm min−1. STICS analysis was not performed on complete images but on ROIs identified by the presence of vectors or blue dots. The color bar in A applies to all images (low magnification and insets) except for the dashed inset in B. ROI (yellow square) corresponds to 16 × 16 pixels (2.7 × 2.7 µm). The pixel shift was 2, except for B and E, where the pixel shift was 4. A, Negative control in the presence of BDM. B to F, Different stages of cell plate formation. Numbers at bottom right in each image indicate time in minutes.
Figure 4.

Effects of pharmacological interference with cytoskeletal dynamics on cell plate formation. A to J, Cells expressing GFP-MBD (A–C), GFP-KN (D–G), or Lifeact-GFP (H–J) were treated with 0.1% dimethyl sulfoxide (DMSO [control]; A, D, and H), 10 μm oryzalin (B and E), 2.5 µm LatB (F and I), or 20 mm BDM (C, G, and J). K to M, 3D surface rendering of a GFP-KN-labeled cell plate in a sample treated with oryzalin. N and O, 3D surface rendering of a GFP-KN-labeled cell plate in a sample treated with LatB. Bars = 10 µm. P, Representative examples of cell plate growth detected with FluMOS on GFP-KN-labeled cells treated with cytoskeleton inhibitors. Q, Mobile fraction during FRAP of GFP-KN on the cell plate of treated cells with 0.1% DMSO, 10 µm oryzalin, 1.25 µm LatB, or 20 mm BDM. se significance is as follows: *, highly significant; **, according to Student’s t test.
IPA Relies on Preexisting Vesicles Whose Delivery Is Spatially Confined by Actin
During normal IPA, the cell plate is consistently restricted to a disk with a diameter of 5.5 µm. This spatial restriction was lifted when cells were treated with inhibitors of actin polymerization or of the endocytic pathway. Treatment with 1.25 µm LatB for 45 min caused a complete depolymerization of the actin cytoskeleton, as visualized in BY-2 cells expressing Lifeact-GFP (Fig. 4I). During the IPA stage of the LatB-treated cells, an excessively large disk was initiated, as shown in cells expressing GFP-KN (14 µm in contrast to 5.5 µm). The successful initiation of IPA under these conditions shows that actin is not required to accomplish vesicle delivery, but it seems to be involved in confining the accumulation of vesicles to a smaller area. In addition, we observed that the diameter of the microtubule array initiating the cell plate was, in fact, narrower than the mitotic spindle (Supplemental Fig. S5). Clearly, there was a reorganization occurring, but the cell plate started to be built after the chromosomes moved to opposite ends of the cell (Supplemental Movie S8; Supplemental Fig. S5).
To assess whether de novo protein synthesis is required for cell plate initiation, we applied cycloheximide (CHX), a specific inhibitor of this process. Actively dividing cells treated with CHX for 30 min did not show any impairment of IPA formation, and the size of the initial disk was identical to the control, with a diameter of 5.5 µm (data not shown). Taken together, proper IPA formation on a confined site with a diameter of 5.5 µm does not require de novo protein synthesis but seems to involve the actin cytoskeleton and preexisting vesicles.
Completion of the Cell Plate Requires Actin
The cytoskeletal elements are essential for vesicle delivery during cell plate expansion (Samuels et al., 1995; Kutsuna and Hasezawa, 2002; Austin et al., 2005), and both FluMOS and FRAP analyses of oryzalin-treated BY-2 cells are consistent with the important role of microtubules (Fig. 4; Supplemental Fig. S4). The role of actin is less well understood. The depolymerization of actin resulting from LatB treatment did not affect the rapid cell plate expansion during PCG or the transition to SCG upon reaching a diameter of 15.5 µm. However, LatB treatment lowered the average expansion rate during SCG from 0.35 to 0.22 μm min−1. In addition, vesicle supply was only marginally affected, since the Mf observed during FRAP of GFP-KN labeled structures in LatB-treated samples was reduced only slightly (Fig. 4Q). However, as suggested previously (Hepler et al., 2002), LatB prevented the expanding cell plate from contacting the parental plasma membrane either by arresting its expansion prior to it reaching the final size or because the plate bent (Fig. 4, N and O). Typically, the shape of cell plates formed in the presence of LatB displayed aberrant morphology, with irregular protrusions normal to the plane of the plate (Fig. 4, I, N, and O).
Endocytic Pathways Are Involved in the Expansion of the Cell Plate
The origin of the material that contributes to cell plate assembly has been the subject of intense discussion (Dhonukshe et al., 2006; Reichardt et al., 2007; Chow et al., 2008; Lam et al., 2008; McMichael and Bednarek, 2013; Richter et al., 2014). It was shown that Golgi-derived vesicles are the principal but not the only source contributing to the expanding cell plate, as other compartments composing the endocytic pathway seem to be involved as well (Reichardt et al., 2011; Richter et al., 2014). Since GFP-KN-labeled vesicles derive from the Golgi but not from late endosome or plasma membrane endocytosis, we used this marker here. To probe vesicle supply routes during PCG and SCG, we pharmacologically targeted the different trafficking pathways during these cell plate expansion phases (Fig. 5). In tobacco cells used at low concentration (50 μm), the ARF-GEF inhibitor brefeldin A (BFA) blocks secretory vesicle trafficking (Grebe et al., 2003; Dhonukshe et al., 2006; Tse et al., 2006; Reichardt et al., 2007; Lam et al., 2009) and induces the formation of a hybrid endoplasmic reticulum (ER)-Golgi compartment as well as a BFA compartment consisting of a mixture of the TGN vesicles and endosomal vesicles. However, it does not affect the late endosomal compartment labeled with YFP-ARA7 (Ueda et al., 2004; Dhonukshe et al., 2006; Fig. 5C). BFA treatment slowed down the PCG to an expansion rate of 0.13 μm min−1 (compared with 1.8 μm min−1 in 0.1% (v/v) DMSO control cells; Fig. 5G), and there was no distinct transition from PCG to SCG, as the rate was reduced only very gradually when passing the transition point at 15.5 µm. The expansion rate during SCG also was reduced compared with the control, to 0.026 μm min−1 (versus 0.453 μm min−1 in control cells; Fig. 5G). During this phase, the GFP-KN mobile fraction corresponded to 37.8% ± 0.02% (Fig. 5H), compared with 55% ± 0.03% for the control. Even after 1 h of treatment with BFA (50 μm), cell plate expansion proceeded, albeit slower, and eventually the two cytoplasms were completely separated.
Figure 5.

Effects of vesicle-trafficking inhibitors on cell plate growth. A to C, ARA-7 localization in cells treated with 0.1% DMSO (A), 33 µm wortmannin (Wort; B), or 50 µm BFA (C). D to F, GFP-KN-expressing cells treated in the same conditions. G, Cell plate growth detected with FluMOS on treated GFP-KN cells: 0.1% DMSO (control), 50 µm BFA, 33 µm Wort, or 50 µm CHX. H, Average mobile fraction after FRAP on GFP-KN-labeled cell plates after drug treatment. se is highly significant (**) according to Student’s t test. Scale bar = 10 μm.
In order to inhibit the endocytosis pathway, we used Wort, a phosphatidylinositol-3-kinase inhibitor, which is known to inhibit trafficking at the level of late endosomes/MVB and partially affect the endocytotic recycling from the plasma membrane (Reichardt et al., 2007). Treatment with 11.7 μm Wort slowed down cell plate expansion after 2 h. Treatment for 30 min with 33 μm induced the swelling of MVB (diameter of 1.3 µm versus 0.7 µm in control cells) labeled with YFP-Ara7 (Fig. 5, A and B). FRAP experiments (Fig. 5H) during the SCG showed a limited impact of Wort on fluorescence recovery, with an Mf of 39.5% ± 0.02%, in contrast to FRAP performed during PCG, where the Mf was approximately 60% (data not shown), similar to the control. This is in agreement with the FluMOS results (Fig. 6G), showing a PCG expansion rate of 0.86 μm min−1 in the presence of Wort. However, the expansion rate during SCG slowed down to 0.12 μm min−1, much slower than in the control. This suggests that the membrane recycling pathway is involved in the SCG phase of cell plate formation.
Figure 6.

Overview of the temporal time course of cell plate formation. Vesicle dynamics characterizing the three phases of cell plate assembly and drug sensitivities studied here are temporally correlated with cytoskeletal dynamics and ultrastructural stages of cell plate architecture identified previously.
Secondary Centrifugal Growth and Fusion Require de Novo Protein Synthesis
Dividing cells treated with CHX for 30 min entered PCG, and cell plate expansion proceeded until the diameter reached 15.5 μm. During this relatively short period of CHX treatment, the mobile fraction of GFP-KN was not affected (60.7% ± 0.05%; data not shown), and the cell plate was eventually completed and fused with the plasma membrane. However, during SCG, expansion was significantly slower than in the control, at 0.05 μm min−1 (Fig. 5G). Simultaneously, the mobile fraction of GFP-KN-labeled structures dropped to 36.5% ± 0.05% (Fig. 5H). In samples that were exposed to CHX for 1 h, however, the expansion of existing cell plates was arrested and new plates were not initiated. This means that de novo protein synthesis is required to deliver new material to the expanding cell plate. If protein synthesis is blocked for a sufficiently long period prior to the onset of mitosis, this will also inhibit the formation of the preexisting vesicles that are used to initiate the assembly of the cell plate.
DISCUSSION
Since plant cells generally do not move relative to each other, the dividing wall built during cytokinesis typically remains a shared wall between the two daughter cells. Therefore, the initiation, generation, and position of this dividing wall are important for all subsequent morphogenetic processes in terms of both tissue growth and organ differentiation (Müller, 2012; Rasmussen et al., 2013).
In this study, we developed and used a suite of image-processing tools that allowed us to analyze the formation of the cell plate in real time, revealing three distinct phases: IPA, PCG, and SCG (Fig. 6). We defined these phases in BY-2 cells based on the lateral expansion rate of the growing cell plate as determined by FluMOS, but our quantitative analyses of published data for Arabidopsis root cells revealed that they are consistent with the kinetics of cells in a tissue context. It is safe to hypothesize that the phases we identified coincide with those based on other cellular parameters, such as actin dynamics, sensitivity to caffeine (Valster and Hepler, 1997), and microtubule dynamics (Seguí-Simarro et al., 2008), although an exact correlation warrants experimental proof.
While the chromosomes migrate to the poles, the mitotic spindle reorganizes into a phragmoplast array. During this initial phragmoplast stage, a CPAM region starts to form at the location of the future cell plate, allowing the fusion of vesicles into dumbbell structures identified by transmission electron microscopy (Seguí-Simarro et al., 2004, 2008) and presumed to be apparent in fluorescence microscopy. The solid phragmoplast stage is then associated with the assembly of the dumbbell structures and vesicles to initiate the formation of the cell plate. Our data show that, in both BY-2 cells and Arabidopsis root cells, the initiation of the formation of the plate during IPA consistently occurs in a disk-shaped region with an average diameter of 5.5 μm (BY-2) and 5.7 µm (Arabidopsis). STICS demonstrated that, during this phase, vesicle trafficking to the disk region is rapid, with a speed of up to 6 μm min−1. Given that this is only a local average value that integrates traffic both toward and away from the cell plate, the velocity of individual vesicles might be much higher. Vesicle movement from the cytoplasm in the vicinity of the plasma membrane detected by STICS and pharmacological evidence suggest that, during this initial stage, primarily preexisting rather than newly formed vesicles are recruited mainly from Golgi/TGN compartments. These vesicles are delivered by the phragmoplast, which is in its initial phase (Fig. 6). Actin filaments seem to play an important role in focusing the localization of the primary aggregation and vesicle fusion (Esseling-Ozdoba et al., 2008; Higaki et al., 2008; Panteris, 2008). We observed that the diameter of the microtubule array forming the IPA stage cell plate was somewhat narrower than the mitotic spindle, which might result from forces exerted by the actin cytoskeleton, as suggested previously by Higaki et al. (2008).
Evidence for a Golgi-based origin of vesicles stems from the presence of methylesterified pectins (as deduced from JIM7 label; Rybak et al., 2014) in the early stage of the cell plate. Furthermore, during this early stage, cell plate formation is prevented when ER-to-Golgi transport is inhibited by low BFA concentration (Ritzenthaler et al., 2002; Lam et al., 2009; Langhans et al., 2011) or when CHX is administered prior to DNA condensation to interfere with de novo protein synthesis. When CHX was applied after DNA segregation, the cell plate was initiated unhindered. This means that the initiation of the cell plate relies on preexisting vesicles.
The initiation of the formation of the cell plate corresponds to the fusion of vesicles in the CPAM enriched in exocyst-tethering complexes (including EXO70A1 and EXO84b; Fendrych et al., 2010; Rybak et al., 2014). It is intriguing that the exocyst complexes move back and forth between the plasma membrane and the cytoplasm (Fendrych et al., 2010, 2013). In addition, FM4-64 is observed at the cell plate as early as the IPA stage, and phosphatidylinositol-4-phosphate (originating from both the Golgi and the plasma membrane) is integrated into the growing cell plate (Vermeer et al., 2009), with the accumulation of YFP-2x-FYVE-positive endosomes at the periphery of the growing cell plate (Vermeer et al., 2006, 2009).
The GTPase Rab-A2/A3 proteins colocalize with KN and partially colocalize with VHA-a1, another early endosomal marker, on the pathway leading to MVBs (Chow et al., 2008). The vesicles that initiate the formation of the cell plate may originate from the RabA2/A3-positive early endosomal/TGN compartment, a key intersection between secretion and endocytosis pathways.
Based on recent studies (Chow et al., 2008; Richter et al., 2014), it is reasonable to assume that recycled components converge into the late-secretory pathway at the TGN. This means that, during IPA, the components forming the cell plate will have a mosaic origin and can derive from both the ER and the Golgi that localize in the vicinity of the future cell plate (Nebenführ et al., 2000; Rybak et al., 2014) as well as from the outer envelope of the cell (plasma membrane and parental cell wall), although the respective contribution of one source may dwarf the other.
In Arabidopsis, BFA inhibits the recycling pathway while the secretory pathway is insensitive to the drug. BIG3 is a BFA-resistant ARF-GEF protein involved in vesicle trafficking between the TGN and the cell plate in dividing cells (Richter et al., 2014). BFA treatment of wild-type plants does not affect the localization of KN at the cell plate but inhibits that of PEN1, a cycling plasma membrane syntaxin that normally accumulates at the cell plate in dividing cells. In big3 mutants, BFA treatment results in the accumulation of both KN and PEN1 in the BFA compartments, inhibiting the secretory pathway as well as the endocytic pathway. The convergence of the pathways of recycled and newly synthesized material at the TGN compartment forces us to draw conclusions from pharmacological inhibition cautiously. The reason is that any drug inhibiting secretion or the endocytosis pathway (like BFA and Wort) may indirectly affect the early endosomal/TGN/RABA2/3 compartments (akin to a congested roundabout), which would hamper the distinction between the pathways converging at the TGN and skew their respective roles.
Following initiation, the cell plate expands radially in two distinct stages defined by their different cell plate expansion rates, the speed and spatial distribution of vesicle delivery. While the resolution of optical micrographs does not allow identifying the anatomy of the cell plate at this stage, electron microscopy-based studies suggest that, during the PCG, corresponding to the solid phragmoplast stage, the dumbbell-shaped intermediates fuse to form the tubulovesicular network (Seguí-Simarro et al., 2004, 2008). Rybak et al. (2014) recently proposed that TRAPPII is the main tethering complex that drives the fusion of the TGN-derived vesicles to build the expanding cell plate. The biogenesis stage undoubtedly correlates with the intense vesicle traffic that we observe during IPA and PCG, as indicated by both the rapid and nearly complete recovery of cell plate fluorescence after photobleaching and the extremely rough cell plate surface observed in four-dimensional (4D) reconstruction series.
Pharmacological inhibition suggests that actin is not involved in this early rapid phase of expansion, whereas microtubule depolymerization leads to a dramatic drop of vesicle supply and arrest in cell plate expansion. However, the initiation of the cell plate has created a new membrane compartment from which endocytotic trafficking occurs to counterbalance the excess of internalized membrane. Inhibiting the BFA-dependent secretion pathway reduced the expansion rates during both PCG and SCG by a factor 13.5 and 17.4, respectively, but did not completely block cell plate expansion or its fusion with the parental plasma membrane. While we cannot exclude that BFA may not have completely inhibited the secretion pathway, these results suggest that endomembrane recycling pathways play a sustained role during cell plate expansion and, most importantly, for ensuring a rapid primary growth of the cell plate.
Contrary to other models, which posit a gradual, time-dependent slowing of the expansion rate (Higaki et al., 2008), we clearly observed a biphasic behavior with a marked phase transition between PCG and SCG. The surface rendering of the 4D image series suggests that this change in expansion rate coincides with the transitional phragmoplast phase, as defined based on microtubule configuration (Seguí-Simarro et al., 2008; Fig. 6; Supplemental Fig. S5). 3D electron tomography of apical meristem cells of Arabidopsis observed during similar stages of development show the cell plate to consist of a tubular network at this point (Samuels et al., 1995; Seguí-Simarro et al., 2004; Austin et al., 2005).
The question is what causes the sudden slowdown of the expansion rate during the transition from PCG to SCG. One hypothesis could be that the slowdown is caused by a squeezing of the vacuoles between the expanding cell plate and the parental wall. However, although the high abundance of vacuoles in BY-2 cells is consistent with this notion, the fact that Arabidopsis meristematic root cells show a similar biphasic behavior despite being much less vacuolated does not support a role for vacuoles in bringing about a transition between PCG and SCG. Rather than being caused by external constraints, our data suggest that the transition from PCG to SCG correlates with the cell plate reaching a dimension-based switch point that is defined by its diameter, which is consistently around 15 to 16 µm, in both BY-2 and Arabidopsis cells. When the cell plate reaches this critical size, a restructuring of the phragmoplast is likely to occur, combined with a reorganization of the vesicle supply pathways. To determine a causal relationship between cytoskeletal reorganization and the drop in cell plate expansion, it will be important to temporally correlate these events and test this correlation by the controlled administration of cytoskeletal drugs occurring at precisely timed moments during the process.
During SCG, vesicle delivery is targeted to the margin of the expanding cell plate, which is consistent with the phragmoplast reorganization from a solid into a ring-shaped array (Samuels et al., 1995; Seguí-Simarro et al., 2004; Austin et al., 2005; Fig. 6). Surface rendering revealed two concentric, ring-shaped rough regions on the surface of the cell plate toward the final stage of SCG, approximately 20 min after the beginning of cell plate formation. This seems to suggest the presence of two concentric regions of targeted vesicle delivery. Without simultaneous ultrastructural evidence, it is impossible to correlate these regions to a particular architecture of the cell plate, but given the general centrifugal expansion pattern, we posit that the inner ring corresponds to the front of the transition from the tubular network to the planar fenestrated sheet, whereas the outer ring might represent the transition from the tubulovesicular network to the tubular network (Seguí-Simarro et al., 2008). The rough margins seem to represent the locations of the elevated vesicle supply that fuels these formation steps (Austin et al., 2005).
Drugs targeting either de novo protein synthesis or endocytic pathways showed that both pathways are involved in SCG. A short CHX treatment (30 min) did not significantly affect the PCG growth rate but was sufficient to decrease the amount of GFP-KN-labeled vesicle supply and the rate of SCG cell plate expansion. After prolonged CHX treatment (more than 1 h), the inhibition of protein synthesis prevented Golgi-derived vesicle supply to the cell plate, stopping the expansion of existing cell plates or impeding new plates to initiate. Wort is an inhibitor of the endomembrane recycling pathway and leads to the swelling of MVBs. Upon Wort treatment, the PCG rate decreased by half without significantly reducing the GFP-KN-labeled vesicle supply. However, the SCG growth rate decreased by 21-fold accompanied by a significant decrease of vesicle supply, as shown by our FRAP experiments. Although we cannot rule out that Wort affected the recycling pathway at the cell plate level, this highlights the importance of membrane recycling in the second stage of its growth compared with the PCG stage. These mechanical/dynamic data are in agreement with the general idea that excess cell plate membrane is recycled from the center and redelivered to its leading edges.
Similar to observations made in the highly vacuolated shoot cells of Arabidopsis, some cell plates in BY-2 cells expanded asymmetrically, anchoring to one flank of the cell prior to continuing their expansion. This is postulated to provide stronger protection against cytoplasmic and vacuole disturbances (Cutler and Ehrhardt, 2002). Unlike the very uniform expansion rate during PCG, the growth rates observed during SCG vary significantly between individual cells. One of the putative reasons for this heterogeneity during SCG could be the fact that, in some cells, the cell plate expands perfectly symmetrically to attach simultaneously to the parental plasma membrane over its entire circumference, whereas in other cells, the contact with the parental plasma membrane is made early at one side followed by an asymmetric expansion toward the opposite side. However, no such correlation between SCG growth rate and symmetric or asymmetric expansion was observed.
Importantly, in contrast to PCG, actin filaments play a crucial role during SCG. Pharmacological interference with actin functioning during this phase reduced the growth rate of the cell plate by half and impeded its proper fusion with the mother plasma membrane. This is in agreement with the behavior of plant cells upon the administration of inhibitors of actin polymerization, such as bistheonellide A (Hoshino et al., 2003; Higaki et al., 2008) and ML-7 (Hepler et al., 2002). The myosins VIII, ATM1 (Van Damme et al., 2004), and Myo8A (Wu and Bezanilla, 2014) and the class II formin For2a (van Gisbergen et al., 2008) are localized in the forming cell plate of BY-2 cells; however, direct evidence of the involvement of the actomyosin system in vesicle transport has been elusive. Kinetic studies on plant-derived motor proteins have revealed that myosins are fast motors with a translocation velocity close to 60 µm s−1 (Henn and Sadot, 2014), while kinesins have a velocity around 0.2 µm s−1 (Zhu and Dixit, 2011; Kong et al., 2015). The velocity of the cell plate-forming vesicles measured by STICS, therefore, is more consistent with a kinesin-based mechanism rather than myosin.
Taken together, our results have provided a detailed time course of cell plate formation in conjunction with the associated spatial and temporal patterns of vesicle supply. Intriguingly, cell plate formation occurs in three distinct phases that are characterized by very different dynamics of both vesicle delivery and assembly (Fig. 6). Our data refine the existing model of cell plate expansion based on transmission electron microscopy (Seguí-Simarro et al., 2004) and molecular studies (Rybak et al., 2014). A clear distinction of different phases is likely to ensure a combination of optimal efficiency and precision of the process. While the phragmoplast enters the solid stage, it becomes narrow at the site where dumbbell-shaped intermediates and vesicles fuse. This vesicle accumulation, defined here as the IPA stage, occurs at high velocity without producing any cell plate expansion. A subsequent rapid initiation and early growth phase (PCG) ensures that the process advances quickly once it is initiated. During the transitional phragmoplast stage and until the end of the ring stage, a slower and actin-dependent second expansion phase (SCG) serves to adjust the cell plate diameter to the mother cell size and to precisely position the connection between the margins of the new wall and the parental cell wall. As this position is crucial for the morphogenesis of the entire tissue and organ, it is likely subject to numerous feedback mechanisms that steer the process. The differences in sensitivity toward the battery of drugs we used corroborates that fundamentally different processes operate during these distinct, successive phases of cell plate formation. Our spatiotemporal characterization of the process represents a benchmark that will serve to characterize the many pathways that, combined, provide a sturdy and fail-safe mechanism driving plant cell division.
MATERIALS AND METHODS
Transformation and Culture of BY-2 Cells
The tobacco (Nicotiana tabacum) BY-2 cell line was cultured in the dark (25°C, 150 rpm) as described (Nagata et al., 1992; Samuels et al., 1995; Thiele et al., 2009). Cells were subcultured weekly by transferring 1 mL of a 7-d-old culture into 50 mL of fresh BY-2 medium (Murashige and Skoog medium, pH 5, enriched with 0.2 g L–1 KH2PO4 and containing 30 g L−1 Suc, 100 mg L–1 myoinositol, 1 mg L–1 thiamine hydrochloride, and 0.22 mg L–1 2,4-dichlorophenoxyacetic acid as an auxin source).
Stable transformations of BY-2 cells were performed using Agrobacterium tumefaciens strain LBA4404 following the protocol by Higaki et al. (2008). Stably transformed BY-2 cell lines were maintained independently by culturing them every week (0.1:4) in BY-2 medium on a six-well plate with shaking (25°C, 150 rpm). All lines were selected and subcultured in BY-2 medium containing carbenicillin (150 mg L−1; to kill agrobacteria) and the appropriate antibiotic: kanamycin for pSYP111::GFP-SYP111 (Nagata et al., 1992), hygromycin for p35S::GFP-MBD, p35S::GFP-Lifeact (cloned for this study in pMDC32 [Curtis and Grossniklaus, 2003] using a Gateway strategy), and p35s::Ara7-YFP.
Inhibitor Treatments
FM4-64 (Life Technology, Molecular Probes), dissolved in water, was applied at a 2.5 µm final concentration for 30 min to BY-2 cells. Excess dye was washed away with medium just before observations. All drugs were used from DMSO-dissolved 1,000× stock solutions and were applied to cells at final concentrations of 50 µm BFA (Bioshop), 33 µm Wort (Bioshop), 50 µm CHX (Gold Biotechnology), 20 mm BDM (Sigma), 10 µm oryzalin (Sigma), and 1.2 µm LatB (Van Gestel et al., 2002; Enzo Life Science) for the indicated periods of time.
Live Imaging
Confocal laser scanning microscopy imaging was performed with a Zeiss LSM 510 META/LSM 5 LIVE/Axiovert 200 M system. The microscope was fitted with a 40×/1.3 oil EC Plan Neofluar and a 63×/1.4 oil DIC Plan Apochromat. To observe GFP-fused proteins, we used the 488-nm diode laser (100 mW) with an LP505 emission filter. For FRAP analyses of GFP-KN with the META mode, a specific region was bleached using two iterations at 70% of the diode, while image acquisition required 1.9% of the laser power, which results in a decrease of 80% of the fluorescence intensity. Live imaging was performed using 19% of the diode laser power. Images were 1,024 × 1,024 pixels, with a pixel size of 0.11 µm in the META mode and 0.17 µm in the live mode.
Image Analysis
Surface renderings and 3D reconstructions were created from z-stacks (full stacks were acquired at a z-distance of 0.3 μm and a rate of one per 25 s) with the software IMARIS 7.3 (Bitplane; http://www.bitplane.com). For FRAP analyses, quantification of fluorescence intensity in the bleached ROI was performed using AIM software (Zeiss) and data were analyzed in Excel (Microsoft Office). To correct for photobleaching while imaging the fluorescence recovery, a reference background ROI was monitored in parallel. To obtain the final recovery curve, we first normalized the ROI fluorescence curve against the background curve; then the fluorescence curve was normalized to the prebleach fluorescence.
The T1/2 of the recovery was calculated as follows:
where Thalf corresponds to the time of half recovery and Tpost to the time of maximum bleaching.
The Mf is defined by:
where Fpre corresponds to the prebleach fluorescence, Fpost to the fluorescence intensity after bleaching, and Fend to the recovered fluorescence intensity.
We used Excel (Microsoft Office) to calculate averages, se, and Student’s t tests (two tailed; two-sample unequal variance).
FluMOS
Time-lapse series of fluorescence micrographs were processed using our in-house FluMOS, which relies on the Pandore image-processing library (a library of image-processing operators [version 6.6]; GREYC Laboratory; https://clouard.users.greyc.fr/Pandore). FluMOS was designed as a fully automated pipeline of arithmetic, morphological, and thresholding operators and is capable of recognizing the cell plate and computing its length. FluMOS relies on advanced segmentation operators and a morphological analysis of the characteristics of the cell plates. The principal power of this algorithm is its ability to determine the diameter of the cell plate despite variations in shape and continuous bleaching of the structure.
More specifically, the pipeline consists of four main steps (Supplemental Fig. S6).
(1) The grayscale image is preprocessed to enhance the brightest regions using the power operator, which has the added benefit of reducing the noise in darker regions. The order of the power, as well as other parameters of the pipeline, was determined empirically. Typical values range between 2 and 3, with higher coefficients useful to preprocess images with low contrast. The image was then normalized so that all pixel intensities were in the range between 0 and 100.
(2) The normalized image is thresholded to delete darker areas of the cell and to preserve the brightest areas. The value of the threshold is automatically computed so that 5% to 10% of the original image is preserved, depending on the anticipated size of the cell plate. The resulting image is segmented into two classes by performing binary thresholding. The threshold value depends on the order of the power: the higher the power, the lower the threshold, with typical values ranging between 20 and 30. Morphological dilatation and erosion with a circular structuring element was performed to smooth the binary image.
(3) The binary image is segmented to build regions from sets of connected pixels using a neighborhood connexity (also known as connectivity) of 8. The shape and area of the different regions are analyzed to automatically filter out isolated blobs that do not present the typical morphological characteristics of cell plates. The remaining region corresponds to the cell plate and is further smoothed by calculating its convex hull.
(4) Finally, the skeleton of the convex hull is built, and short barbs are removed to reduce noise. The cell plate diameter was extracted by computing the length of the corresponding skeleton.
The output is saved in a CSV file for further analysis. The skeleton also is superimposed onto the original image to facilitate quality control. Once parameters are empirically determined, FluMOS is fully automated. We achieved a detection rate over 95%, thus requiring very little manual curation, effectively allowing us to process high-throughput microscopy imaging data and perform reliable statistical analysis of cell plate expansion over extended periods of time.
STICS
STICS is a member of a family of image correlation techniques that was initially developed to measure the directed transport or flow of proteins inside living cells. STICS involves calculating the complete space-time correlation function of the intensity fluctuations in images of a time series recorded using a fluorescence microscope. The changes in this function directly reflect the underlying transport dynamics of the fluorescently labeled macromolecules in the imaged cell. We can define a generalized spatio-temporal correlation function with the independent spatial lag variables ξ and η and the temporal lag variable τ for one image detection channel a as:
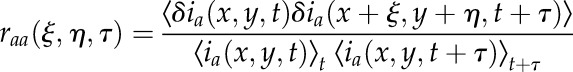 |
where 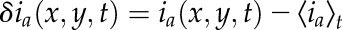 is the intensity fluctuation in channel a at image pixel position x,y and time t, the angular brackets in the denominator represent spatial averaging over images at time t and t + τ in the time series, while the numerator is an ensemble average spatial correlation over all pixel fluctuations in pairs of images separated by a time lag of τ.
is the intensity fluctuation in channel a at image pixel position x,y and time t, the angular brackets in the denominator represent spatial averaging over images at time t and t + τ in the time series, while the numerator is an ensemble average spatial correlation over all pixel fluctuations in pairs of images separated by a time lag of τ.
In practice, what we calculate is the discrete approximation to the generalized spatiotemporal correlation function:
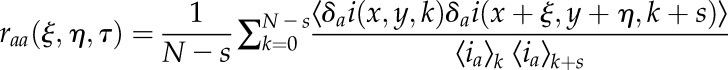 |
where N is the total number of images in the time series or time window of interest (TOI) analyzed, the spatial lag variables ξ and η represent pixel shifts in x and y, and s is the discrete integer frame lag between pairs of images in the time series. The real discrete time lag, Δt ≈ τ, is simply the product of the integer frame lag and the sampling time per image frame: Δt = s δt (where δt is the sampling time per frame). This discrete correlation function is calculated by averaging the spatial cross-correlation of intensity fluctuations between all image pairs separated by the time lag Δt in a fluorescence image time series. The function raa represents the average correlation function for channel a for all pairs of images separated within the image time series by a lag time of Δt.
If there is concerted translational motion of some of the fluorescent proteins during the imaging, it may be observed in the time evolution of the spatiotemporal correlation function. In the STICS calculation, the correlation function is first calculated for every frame in the series, with Δt = 0. For every image frame, this correlation function will appear as a two-dimensional Gaussian function with its peak centered at zero (spatial lag variables ξ = 0 and η = 0). Similarly, the correlation functions for all pairs of images separated by lag times ranging from 1 to N − 1 are calculated and averaged for each discrete time lag to yield a mean correlation function over the TOI. A longer TOI improves temporal sampling if the flow process remains constant as more fluctuations of flowing particles are sampled for a given Δt and averaged. The optimal TOI for this study was determined empirically by testing TOIs of 25, 50, 75, and 100 image frames. If the spatial distribution of labeled particles has changed between frames, due to the movement of particles, the Gaussian peak of the correlation function will change in location and/or width. For a flowing population of particles, the Gaussian function will stay constant in width but its peak position will translate in space. The displacement distance and direction of this correlation peak can be tracked giving the underlying flow vector (i.e. magnitude and direction) of the directed movement. In the case of random diffusion of the particles, no directional flow component peak is observed in the correlation function, and the correlation peak stays centered at the origin while it decays in amplitude and spreads out uniformly in space as Δt increases and the particles diffuse randomly in all directions. A beam radius threshold of 0.8 µm was used to set the maximum e−2 radius allowed for the correlation functions. Additionally, a correlation local maxima of 0.5 was applied. This is the ratio of the noise correlation peak amplitudes (off center) to the amplitude of the main correlation function peak from the fluorescence fluctuation signal, which ensures that the noise correlation peaks are not larger than the signal correlation peak; otherwise, fitting is terminated. These filters are used when analyzing the shape of the correlation functions for every lag time, such that tracking is terminated if the signal peak of the function cannot be reliably located or if the width of the peak reaches the radius threshold. As well, a maximum time lag parameter is set to restrict the correlation function peak fitting and tracking to lower time lag values. For the work presented here, the maximum time lag parameter was 10 for TOIs of 25 and 50 and 20 for TOIs of 75 and 100. Note that the fitting could be terminated before the maximum time lag value if the radius or the peak signal-to-noise criterion was not met.
The presence of an immobile population of particles in the region of analysis can introduce an unwanted static contribution to the correlation functions that can mask the dynamics of transport we wish to measure. To overcome this, we filter the image time series prior to calculating the correlation functions, such that the immobile components are removed from the raw images. This is done in time frequency space, where the lowest temporal frequency components of each pixel’s intensity time trace are removed before reverting back to the time domain, as has been described in detail (Hebert et al., 2005). In order to enable STICS analysis of vesicle trafficking, images were recorded with the live mode of the Zeiss LSM 510, every 1 s for microtubules and actin (except when mentioned in Supplemental Fig. S4) and every 0.735 s per frame for GFP-KN-labeled vesicles, with a pixel size of 0.17 μm. However, the vector maps were generated by performing STICS analysis in parallel on multiple smaller ROIs that were 16 × 16 pixels and 100 image frames in time. The vector maps were spatially oversampled by shifting adjacent ROI subregions by a pixel shift of 2 or 4 in the x and y directions.
Computer Simulations
Image time series of flowing particle populations were generated via MATLAB (The Mathworks) following similar procedures as described previously in detail (Hebert et al., 2005). We will outline the key details here for understanding the results presented. Image matrices of 256 × 256 pixels grid size and 120 total frames were populated with point particles at random grid positions with a user-defined density. The pixel size was set to 0.1715 μm per pixel, and sequential image frame iterations were 0.735 s per frame to match experimental microscopy collection parameters. Particle transport was preset by user input, which was a constant velocity translation at 6 μm min−1 in this case. Periodic boundary conditions were applied in order to ensure the constant number of particles in the simulation area. The particle position matrix was mathematically convolved with a two-dimensional Gaussian function of e−2 radius of 0.4 μm at each time step, and the output for each convolution step was an image frame for the time series. The convolution with the Gaussian function simulated diffraction-limited imaging and acted as the spatial correlator for the underlying particles. Simulated image series were analyzed by STICS as described above with variable TOIs (Supplemental Figs. S7 and S8).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Cell plate diameter of a dividing Arabidopsis root meristem cell.
Supplemental Figure S2. FRAP analysis at the plasma membrane.
Supplemental Figure S3. Golgi dynamics in a pollen tube processed with STICS.
Supplemental Figure S4. STICS analysis of the vesicle supply to allow cell plate growth.
Supplemental Figure S5. Microtubule array from metaphase to telophase.
Supplemental Figure S6. FluMOS pipeline.
Supplemental Figure S7. STICS vector maps obtained for TOIs of 25, 50, and 100 frames with time-shift oversampling.
Supplemental Figure S8. STICS analysis of computer-simulated image series with uniform particle flow with different TOI temporal sampling and different particle densities.
Supplemental Figure S9. Statistical testing of selected TOI sizes used in STICS measurements via dispersion analysis.
Supplemental Table S1. References used to quantify cell plate diameter in an Arabidopsis meristematic root cell.
Supplemental Movie S1. Detection of cell plate growth with FluMOS.
Supplemental Movie S2. GFP-KN-labeled cell plate expansion used for the surface rendering.
Supplemental Movie S3. 4D representation of a cell plate centrifugal growth.
Supplemental Movie S4. FRAP on a plasma membrane labeled with FM4-64.
Supplemental Movie S5. FRAP on a pKN::GFP-KN-labeled plasma membrane.
Supplemental Movie S6. FRAP on a pKN::GFP-KN-labeled cell plate.
Supplemental Movie S7. FM4-64-labeled cell plate-forming vesicles.
Supplemental Movie S8. GFP-KN-labeled cell plate expansion used for the STICS analysis.
Supplemental Movie S9. Tracking with STICS of the motion of vesicles forming the cell plate during IPA and PCG.
Supplemental Movie S10. Tracking with STICS of the motion of vesicles forming the cell plate during SCG.
Acknowledgments
We thank Dr. Hugo Zhang and Dr. Masa H. Sato for providing p35s::ARA7-YFP and pSYP111::GFP-SYP111 (Enami et al., 2009) GFP-KN plasmids, respectively, Dr. Andreas Nebenführ for the G-RK plasmid (Nelson et al., 2007), and Dr. Richard Cyr for GFP-MBD (from MAP4; Granger and Cyr, 2000).
Glossary
- 3D
three-dimensional
- CPAM
cell plate assembly matrix
- BDM
2,3-butanedion monoxime
- TGN
trans-Golgi network
- STICS
spatiotemporal image correlation spectroscopy
- BY-2
Bright Yellow-2
- FluMOS
Fluorescence Morphological Operators Software
- IPA
initial plate assembly
- PCG
primary centrifugal growth
- SCG
secondary centrifugal growth
- FRAP
fluorescence recovery after photobleaching
- ROI
region of interest
- Mf
mobile fraction
- LatB
latrunculin B
- CHX
cycloheximide
- BFA
brefeldin A
- ER
endoplasmic reticulum
- DMSO
dimethyl sulfoxide
- Wort
wortmannin
- 4D
four-dimensional
- TOI
time window of interest
Footnotes
This work was supported by the Fonds de Recherche du Québec, Nature et Technologies (to A.G. and P.W.W.), and by the Natural Sciences and Engineering Research Council of Canada (Discovery Grants to A.G. and P.W.W.).
Articles can be viewed without a subscription.
References
- Austin JR II, Seguí-Simarro JM, Staehelin LA (2005) Quantitative analysis of changes in spatial distribution and plus-end geometry of microtubules involved in plant-cell cytokinesis. J Cell Sci 118: 3895–3903 [DOI] [PubMed] [Google Scholar]
- Cannon MC, Terneus K, Hall Q, Tan L, Wang Y, Wegenhart BL, Chen L, Lamport DTA, Chen Y, Kieliszewski MJ (2008) Self-assembly of the plant cell wall requires an extensin scaffold. Proc Natl Acad Sci USA 105: 2226–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CM, Neto H, Foucart C, Moore I (2008) Rab-A2 and Rab-A3 GTPases define a trans-Golgi endosomal membrane domain in Arabidopsis that contributes substantially to the cell plate. Plant Cell 20: 101–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MD, Grossniklaus U (2003) A Gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133: 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler SR, Ehrhardt DW (2002) Polarized cytokinesis in vacuolate cells of Arabidopsis. Proc Natl Acad Sci USA 99: 2812–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhonukshe P, Baluska F, Schlicht M, Hlavacka A, Samaj J, Friml J, Gadella TWJ Jr (2006) Endocytosis of cell surface material mediates cell plate formation during plant cytokinesis. Dev Cell 10: 137–150 [DOI] [PubMed] [Google Scholar]
- Enami K, Ichikawa M, Uemura T, Kutsuna N, Hasezawa S, Nakagawa T, Nakano A, Sato MH (2009) Differential expression control and polarized distribution of plasma membrane-resident SYP1 SNAREs in Arabidopsis thaliana. Plant Cell Physiol 50: 280–289 [DOI] [PubMed] [Google Scholar]
- Esseling-Ozdoba A, Vos JW, van Lammeren AAM, Emons AMC (2008) Synthetic lipid (DOPG) vesicles accumulate in the cell plate region but do not fuse. Plant Physiol 147: 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrych M, Synek L, Pecenková T, Drdová EJ, Sekeres J, de Rycke R, Nowack MK, Zársky V (2013) Visualization of the exocyst complex dynamics at the plasma membrane of Arabidopsis thaliana. Mol Biol Cell 24: 510–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrych M, Synek L, Pecenková T, Toupalová H, Cole R, Drdová E, Nebesárová J, Sedinová M, Hála M, Fowler JE, et al. (2010) The Arabidopsis exocyst complex is involved in cytokinesis and cell plate maturation. Plant Cell 22: 3053–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger CL, Cyr RJ (2000) Microtubule reorganization in tobacco BY-2 cells stably expressing GFP-MBD. Planta 210: 502–509 [DOI] [PubMed] [Google Scholar]
- Grebe M, Xu J, Möbius W, Ueda T, Nakano A, Geuze HJ, Rook MB, Scheres B (2003) Arabidopsis sterol endocytosis involves actin-mediated trafficking via ARA6-positive early endosomes. Curr Biol 13: 1378–1387 [DOI] [PubMed] [Google Scholar]
- Hebert B, Costantino S, Wiseman PW (2005) Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys J 88: 3601–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A, Sadot E (2014) The unique enzymatic and mechanistic properties of plant myosins. Curr Opin Plant Biol 22: 65–70 [DOI] [PubMed] [Google Scholar]
- Hepler PK, Valster A, Molchan T, Vos JW (2002) Roles for kinesin and myosin during cytokinesis. Philos Trans R Soc Lond B Biol Sci 357: 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki T, Kutsuna N, Sano T, Hasezawa S (2008) Quantitative analysis of changes in actin microfilament contribution to cell plate development in plant cytokinesis. BMC Plant Biol 8: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino H, Yoneda A, Kumagai F, Hasezawa S (2003) Roles of actin-depleted zone and preprophase band in determining the division site of higher-plant cells, a tobacco BY-2 cell line expressing GFP-tubulin. Protoplasma 222: 157–165 [DOI] [PubMed] [Google Scholar]
- Kong Z, Ioki M, Braybrook S, Li S, Ye ZH, Lee YRJ, Hotta T, Chang A, Tian J, Wang G, et al. (2015) Kinesin-4 functions in vesicular transport on cortical microtubules and regulates cell wall mechanics during cell elongation in plants. Mol Plant 8: 1011–1023 [DOI] [PubMed] [Google Scholar]
- Kutsuna N, Hasezawa S (2002) Dynamic organization of vacuolar and microtubule structures during cell cycle progression in synchronized tobacco BY-2 cells. Plant Cell Physiol 43: 965–973 [DOI] [PubMed] [Google Scholar]
- Lam SK, Cai Y, Hillmer S, Robinson DG, Jiang L (2008) SCAMPs highlight the developing cell plate during cytokinesis in tobacco BY-2 cells. Plant Physiol 147: 1637–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SK, Cai Y, Tse YC, Wang J, Law AHY, Pimpl P, Chan HYE, Xia J, Jiang L (2009) BFA-induced compartments from the Golgi apparatus and trans-Golgi network/early endosome are distinct in plant cells. Plant J 60: 865–881 [DOI] [PubMed] [Google Scholar]
- Langhans M, Förster S, Helmchen G, Robinson DG (2011) Differential effects of the brefeldin A analogue (6R)-hydroxy-BFA in tobacco and Arabidopsis. J Exp Bot 62: 2949–2957 [DOI] [PubMed] [Google Scholar]
- Lauber MH, Waizenegger I, Steinmann T, Schwarz H, Mayer U, Hwang I, Lukowitz W, Jürgens G (1997) The Arabidopsis KNOLLE protein is a cytokinesis-specific syntaxin. J Cell Biol 139: 1485–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael CM, Bednarek SY (2013) Cytoskeletal and membrane dynamics during higher plant cytokinesis. New Phytol 197: 1039–1057 [DOI] [PubMed] [Google Scholar]
- Miart F, Desprez T, Biot E, Morin H, Belcram K, Höfte H, Gonneau M, Vernhettes S (2014) Spatio-temporal analysis of cellulose synthesis during cell plate formation in Arabidopsis. Plant J 77: 71–84 [DOI] [PubMed] [Google Scholar]
- Molchan TM, Valster AH, Hepler PK (2002) Actomyosin promotes cell plate alignment and late lateral expansion in Tradescantia stamen hair cells. Planta 214: 683–693 [DOI] [PubMed] [Google Scholar]
- Müller S. (2012) Universal rules for division plane selection in plants. Protoplasma 249: 239–253 [DOI] [PubMed] [Google Scholar]
- Nagata T, Nemoto Y, Hasezawas S (1992) Tobacco BY-2 cell line as the “HeLa” cell in the cell biology of higher plants. Int Rev Cytol 132: 1–30 [Google Scholar]
- Nebenführ A, Frohlick JA, Staehelin LA (2000) Redistribution of Golgi stacks and other organelles during mitosis and cytokinesis in plant cells. Plant Physiol 124: 135–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BK, Cai X, Nebenführ A (2007) A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J 51: 1126–1136 [DOI] [PubMed] [Google Scholar]
- Panteris E. (2008) Cortical actin filaments at the division site of mitotic plant cells: a reconsideration of the ‘actin-depleted zone.’ New Phytol 179: 334–341 [DOI] [PubMed] [Google Scholar]
- Rasmussen CG, Wright AJ, Müller S (2013) The role of the cytoskeleton and associated proteins in determination of the plant cell division plane. Plant J 75: 258–269 [DOI] [PubMed] [Google Scholar]
- Reichardt I, Slane D, El Kasmi F, Knöll C, Fuchs R, Mayer U, Lipka V, Jürgens G (2011) Mechanisms of functional specificity among plasma-membrane syntaxins in Arabidopsis. Traffic 12: 1269–1280 [DOI] [PubMed] [Google Scholar]
- Reichardt I, Stierhof YD, Mayer U, Richter S, Schwarz H, Schumacher K, Jürgens G (2007) Plant cytokinesis requires de novo secretory trafficking but not endocytosis. Curr Biol 17: 2047–2053 [DOI] [PubMed] [Google Scholar]
- Richter S, Kientz M, Brumm S, Nielsen ME, Park M, Gavidia R, Krause C, Voss U, Beckmann H, Mayer U, et al. (2014) Delivery of endocytosed proteins to the cell-division plane requires change of pathway from recycling to secretion. eLife 3: e02131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzenthaler C, Nebenführ A, Movafeghi A, Stussi-Garaud C, Behnia L, Pimpl P, Staehelin LA, Robinson DG (2002) Reevaluation of the effects of brefeldin A on plant cells using tobacco Bright Yellow 2 cells expressing Golgi-targeted green fluorescent protein and COPI antisera. Plant Cell 14: 237–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak K, Steiner A, Synek L, Klaeger S, Kulich I, Facher E, Wanner G, Kuster B, Zarsky V, Persson S, et al. (2014) Plant cytokinesis is orchestrated by the sequential action of the TRAPPII and exocyst tethering complexes. Dev Cell 29: 607–620 [DOI] [PubMed] [Google Scholar]
- Samaj J, Peters M, Volkmann D, Baluska F (2000) Effects of myosin ATPase inhibitor 2,3-butanedione 2-monoxime on distributions of myosins, F-actin, microtubules, and cortical endoplasmic reticulum in maize root apices. Plant Cell Physiol 41: 571–582 [DOI] [PubMed] [Google Scholar]
- Samuels AL, Giddings TH Jr, Staehelin LA (1995) Cytokinesis in tobacco BY-2 and root tip cells: a new model of cell plate formation in higher plants. J Cell Biol 130: 1345–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguí-Simarro JM, Austin JR II, White EA, Staehelin LA (2004) Electron tomographic analysis of somatic cell plate formation in meristematic cells of Arabidopsis preserved by high-pressure freezing. Plant Cell 16: 836–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguí-Simarro JM, Otegui MS, Austin JR (2008) Plant cytokinesis: insights gained from electron tomography studies. In Verma DPS, Hong Z, eds, Cell Division Control in Plants. Springer, Berlin, pp 251–287 [Google Scholar]
- Seguí-Simarro JM, Staehelin LA (2006) Cell cycle-dependent changes in Golgi stacks, vacuoles, clathrin-coated vesicles and multivesicular bodies in meristematic cells of Arabidopsis thaliana: a quantitative and spatial analysis. Planta 223: 223–236 [DOI] [PubMed] [Google Scholar]
- Staehelin LA, Hepler PK (1996) Cytokinesis in higher plants. Cell 84: 821–824 [DOI] [PubMed] [Google Scholar]
- Thellmann M, Rybak K, Thiele K, Wanner G, Assaad FF (2010) Tethering factors required for cytokinesis in Arabidopsis. Plant Physiol 154: 720–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele K, Wanner G, Kindzierski V, Jürgens G, Mayer U, Pachl F, Assaad FF (2009) The timely deposition of callose is essential for cytokinesis in Arabidopsis. Plant J 58: 13–26 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Yokota E, Sonobe S, Shimmen T (2000) Mechanism of inhibition of cytoplasmic streaming by a myosin inhibitor, 2,3-butanedione monoxime. Protoplasma 213: 46–54 [Google Scholar]
- Tse YC, Lo SW, Hillmer S, Dupree P, Jiang L (2006) Dynamic response of prevacuolar compartments to brefeldin A in plant cells. Plant Physiol 142: 1442–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Uemura T, Sato MH, Nakano A (2004) Functional differentiation of endosomes in Arabidopsis cells. Plant J 40: 783–789 [DOI] [PubMed] [Google Scholar]
- Valster AH, Hepler PK (1997) Caffeine inhibition of cytokinesis: effect on the phragmoplast cytoskeleton in living Tradescantia stamen hair cells. Protoplasma 196: 155–166 [Google Scholar]
- Van Damme D, Bouget FY, Van Poucke K, Inzé D, Geelen D (2004) Molecular dissection of plant cytokinesis and phragmoplast structure: a survey of GFP-tagged proteins. Plant J 40: 386–398 [DOI] [PubMed] [Google Scholar]
- Van Damme D, Gadeyne A, Vanstraelen M, Inzé D, Van Montagu MCE, De Jaeger G, Russinova E, Geelen D (2011) Adaptin-like protein TPLATE and clathrin recruitment during plant somatic cytokinesis occurs via two distinct pathways. Proc Natl Acad Sci USA 108: 615–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gestel K, Köhler RH, Verbelen JP (2002) Plant mitochondria move on F-actin, but their positioning in the cortical cytoplasm depends on both F-actin and microtubules. J Exp Bot 53: 659–667 [DOI] [PubMed] [Google Scholar]
- van Gisbergen PAC, Esseling-Ozdoba A, Vos JW (2008) Microinjecting FM4-64 validates it as a marker of the endocytic pathway in plants. J Microsc 231: 284–290 [DOI] [PubMed] [Google Scholar]
- Vermeer JEM, Thole JM, Goedhart J, Nielsen E, Munnik T, Gadella TWJ Jr (2009) Imaging phosphatidylinositol 4-phosphate dynamics in living plant cells. Plant J 57: 356–372 [DOI] [PubMed] [Google Scholar]
- Vermeer JEM, van Leeuwen W, Tobeña-Santamaria R, Laxalt AM, Jones DR, Divecha N, Gadella TWJ Jr, Munnik T (2006) Visualization of PtdIns3P dynamics in living plant cells. Plant J 47: 687–700 [DOI] [PubMed] [Google Scholar]
- Waizenegger I, Lukowitz W, Assaad F, Schwarz H, Jürgens G, Mayer U (2000) The Arabidopsis KNOLLE and KEULE genes interact to promote vesicle fusion during cytokinesis. Curr Biol 10: 1371–1374 [DOI] [PubMed] [Google Scholar]
- Wu SZ, Bezanilla M (2014) Myosin VIII associates with microtubule ends and together with actin plays a role in guiding plant cell division. eLife 3: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang W, Baluska F, Menzel D, Ren H (2009) Dynamics and roles of phragmoplast microfilaments in cell plate formation during cytokinesis of tobacco BY-2 cells. Chin Sci Bull 54: 2051–2061 [Google Scholar]
- Zhu C, Dixit R (2011) Single molecule analysis of the Arabidopsis FRA1 kinesin shows that it is a functional motor protein with unusually high processivity. Mol Plant 4: 879–885 [DOI] [PubMed] [Google Scholar]