

Abstract
Activation of the neuro-hormonal system is a pathophysiological consequence of heart failure. Neuro-hormonal activation promotes metabolic changes, such as insulin resistance, and determines an increased use of non-carbohydrate substrates for energy production. Fasting blood ketone bodies as well as fat oxidation are increased in patients with heart failure, yielding a state of metabolic inefficiency. The net result is additional depletion of myocardial adenosine triphosphate, phosphocreatine and creatine kinase levels with further decreased efficiency of mechanical work. In this context, manipulation of cardiac energy metabolism by modification of substrate use by the failing heart has produced positive clinical results. The results of current research support the concept that shifting the energy substrate preference away from fatty acid metabolism and towards glucose metabolism could be an effective adjunctive treatment in patients with heart failure. The additional use of drugs able to partially inhibit fatty acids oxidation in patients with heart failure may therefore yield a significant protective effect for clinical symptoms and cardiac function improvement, and simultaneously ameliorate left ventricular remodelling. Certainly, to clarify the exact therapeutic role of metabolic therapy in heart failure, a large multicentre, randomised controlled trial should be performed.
Keywords: Adrenergic system, beta-blockers, free fatty acids inhibitors, heart failure, left ventricular function, metabolic therapy, myocardial metabolism, perhexiline, renin-angiotensin-aldosterone system, trimetazidine
The development of heart failure is rarely dependent on primary alterations of cardiac metabolism. The majority of heart failure cases result from diseases of the cardiac muscle, most frequently ischaemic heart disease. However, whatever the cause of heart failure, the net result will be depletion of myocardial adenosine triphosphate (ATP), phosphocreatine and creatine kinase levels with decreased efficiency of mechanical work. Once heart failure has developed, the neurohormonal axis is activated with the aim to sustain haemodynamic failure. Activation of adrenergic and renin-angiotensin-aldosterone systems indirectly determine specific metabolic alterations in the cardiac and skeletal muscles. Over the last two decades, despite the adoption of drugs able to block neuro-hormonal activation in heart failure dramatically improving the overall prognosis of this deadly disease, mortality and morbidity remain a critical problem. In fact, apart from the well-known effects on chronotropism, inotropism, vascular tone and blood volume, the residual physiological effects of neuro-hormones indirectly determine a state of low metabolic efficiency in both the skeletal and cardiac muscles. The aim of this review is to analyse the metabolic derangement in the failing heart and, on this basis, speculate on possible new therapeutic targets.
Deranged Cellular Metabolism in Heart Failure
Under normal conditions, the healthy heart derives most of its energy from the free fatty acid (FFA) pathway that accounts for approximately two-thirds of energy production; the other source of energy being derived from glucose oxidation and lactate.[1,2] FFA and glucose metabolism inter-regulate each other, a process referred to as the Randle cycle.[3] Increasing FFA oxidation in the heart decreases glucose oxidation, while increasing glucose oxidation inhibits FFA oxidation. However, energy being derived from FFA oxidation is a less efficient source of energy than glucose oxidation (in terms of ATP produced per O2 molecules consumed) and determines a reduction of cardiac efficiency. In fact, the amount of ATP produced per O2 consumed is greater when glucose is oxidised compared with FFA and, therefore, FFA is a less efficient energy substrate than glucose. Elevated FFA oxidation can result in up to a 30 % decrease in cardiac efficiency.[2]
Progressive heart failure induces an imbalance between the requirement of cardiac tissue for oxygen and metabolic supplies and their availability, resulting in functional, metabolic and morphological alteration of the myocardium. At a cellular level, glucose uptake is decreased and conversion to lactate is increased; lactate uptake by the heart is switched to lactate production, and pyruvate is mostly transformed into lactate, thereby increasing cell acidosis. The FFA pathway is also slowed down, yet most of the produced energy comes from FFA oxidation, resulting in less ATP production. These metabolic changes lead to disruption of cell homeostasis, alterations in membrane structure and, ultimately, cell death. The following sections will attempt to clarify the mechanism at the base of these metabolic changes.
Effects of the Neuro-hormonal Activation on Metabolism of the Failing Heart
Neuro-hormonal activation significantly contributes to cardiac mechanical and metabolic inefficiency of the cardiac muscle and whole body of patients with heart failure. This vicious circle is likely mediated by increased use of non-carbohydrate substrates for energy production,[2] resulting from different mechanisms. Adrenergically mediated increased peripheral lipolysis (wasting of subcutaneous fat and skeletal muscle) results in grossly augmented FFA availability. In fact, fasting blood ketone bodies[4] as well as fat oxidation during exercise[5] have been shown to be increased in patients with heart failure. Adrenergic activation may also induce insulin resistance, which is also associated with heart failure[6] and may further contribute to increased circulating FFA levels by the development of ketosis and consequent impaired suppression of lipolysis. Indirect effects of augmented adrenergic tone in heart failure include increased heart rate, vasoconstriction and inotropism, which, in turn, may also indirectly contribute to a state of functional and metabolic inefficiency.
Angiotensin II is also an important regulator of cardiac energy metabolism and function.[7] There are several mechanisms through which angiotensin II contributes to heart failure occurrence and persistence. Angiotensin II damages mitochondria in the cardiomyocyte by increasing reactive oxygen species production[8] and affects mitochondrial oxidative phosphorylation, including FFA oxidation.[9,10] These data suggest that angiotensin II affects FFA oxidation. There is also evidence that angiotensin II regulates glucose oxidation[7,11] and its inhibition may exert beneficial effects. In addition, by decreasing oxidative metabolism, angiotensin II can compromise ATP production, thus reducing its availability.[12] In this context, angiotensin II antagonism represents an attractive therapeutic approach. Studies using the euglycaemic insulin clamp technique have indicated that the beneficial effect of angiotensin II is exerted on insulin sensitivity. In fact, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor antagonists have been shown to improve both left ventricular function and glucose homeostasis.[13,14] Increased blood flow in skeletal muscle, accumulation of bradykinin or more efficient insulin release may be suggested as potential modes of action.
Endothelial dysfunction, a critical component in the progression of heart failure, may result from increased oxidative stress, secondary to activation of the adrenergic and the renin-angiotensin systems and to the production of inflammatory cytokines.[15] In heart failure, the role of reduced bioavailability of nitric oxide (NO) is still under debate,[16,17] while increased endothelin-1 (ET-1) levels are a mainstay.[18] Growth factors, vasoactive substances and mechanical stress contribute to the increased ET-1 levels in patients with heart failure. Despite the known adaptative aspect of supporting contractility of the failing heart, persistent increases in cardiac ET-1 expression in the failing heart have a pathophysiological maladaptive aspect and are associated with the severity of myocardial dysfunction.[19] It has been observed that trimetazidine could reduce endothelin release in patients with cardiac disease.[20,21] Trimetazidine-induced reduction of intracellular acidosis in ischaemic myocardium might not only influence myocardial but also endothelial membranes.[22] By decreasing endothelial damage, trimetazidine could inhibit ET-1 release that, in turn, may decrease myocardial damage. A second hypothesis is that, by just decreasing the effects of chronic myocardial ischaemia, trimetazidine could inhibit ET-1 release. Therefore, the observed decrease in ET-1 release with trimetazidine, could likely be linked to trimetazidine-induced reduction of myocardial ischaemia. Finally, keeping in mind the close relation between endothelium and insulin sensitivity, the observed effects of trimetazidine on endothelial function could also explain the beneficial action of trimetazidine on glucose metabolism.
In the same context, the potential beneficial effect of 6 weeks of oral L-arginine supplementation on endurance exercise, an important determinant of daily-life activity in patients with chronic stable heart failure, has been assessed.[23] L-Arginine is the precursor of endogenous NO, which is a potent vasodilator acting via the intracellular second-messenger cyclic guanosine monophosphate. In healthy individuals, L-arginine induces peripheral vasodilation and inhibits platelet aggregation due to an increased NO production. The results of this study show that arginine enhanced endurance exercise tolerance, reducing both heart rate and circulating lactate levels, suggesting that chronic arginine administration might be useful as a therapeutic adjuvant to improve the patient’s physical fitness.
In summary, in the failing heart neuro-hormonal activation determines a combination of direct and indirect haemodynamic and metabolic actions, which, despite a potential teleological purpose, will eventually lead to further deterioration of cardiac function, mainly mediated by the resulting decreased metabolic efficiency of the cardiomyocytes. Specific therapies may attenuate these effects.
Abnormal Glucose Metabolism in Heart Failure
As glucose and lactate are more efficient fuels for aerobic respiration, increasing the use of these substrates can improve the oxygen consumption efficiency of the myocardium by 16–26 %.[24] In addition, skeletal muscle glucose uptake in the heart and arm is inversely related to serum FFA levels[25] and increased FFA flux from adipose tissue to non-adipose tissue amplifies metabolic derangements that are characteristic of the insulin resistance syndrome.[26] Further findings suggest that raised FFA levels not only impair glucose uptake in heart and skeletal muscle but also cause alterations in the metabolism of vascular endothelium, leading to premature cardiovascular disease.[27]
Global Energy Expenditure in Heart Failure
Energy consumption at rest appears higher in patients with heart failure than in healthy subjects.[28–30] It has been shown that increased rate of energy expenditure is related to increased serum FFA oxidation and that both energy expenditure and serum FFA oxidation are inversely correlated with left ventricular ejection fraction and positively correlated with growth hormone, epinephrine and norepinephrine concentrations.[31] Norepinephrine increases whole-body oxygen consumption, circulating FFA concentrations, and FFA oxidation.[32] These changes have been attributed to stimulation of hormone-sensitive lipase in adipose tissue, and to stimulation of oxygen consumption independent of lipolysis by norepinephrine.[33] These data, together with close correlations between plasma norepinephrine concentrations, energy expenditure at rest and FFA oxidation, make increased sympathetic activity the most likely explanation for alterations in fuel homeostasis in patients with heart failure.[33] Therefore, intervention strategies aimed at optimising global and cardiac metabolism could be useful for interrupting the vicious circle of reduced function at greater metabolic expenses in different cardiac conditions.
Pharmacological Implications of Impaired Myocardial Metabolism in Heart Failure
Given the above-described pathophysiological background and the difficulty of standard treatment to control the total symptomatic and prognostic burden in many patients with heart failure, it seems logical to consider pharmacological manipulation of cardiac energy metabolism as an adjunctive therapeutic option. Optimisation of cardiac energy metabolism is based on promoting cardiac glucose oxidation. Stimulation of myocardial glucose oxidation can be achieved either directly with stimulation of glucose metabolism, or indirectly through inhibition of fatty acid beta-oxidation, in order to shift energy substrate utilisation away from fatty acid metabolism and towards glucose metabolism which, as explained above, is more efficient in terms of ATP production per mole of oxygen used. Therefore, metabolic therapy could play a beneficial role in terms of glucose metabolism homeostasis.
The concept that drugs able to promote the use of glucose and non-fatty substrates by the mitochondria may increase metabolic efficiency and function of the failing heart has prompted several clinical studies. Experimental studies have first shown that stimulation of pyruvate dehydrogenase activity leads to enhanced glycolysis and use of lactate by the myocardium for aerobic respiration.[34] Myocardial consumption of FFA is simultaneously inhibited, with the overall effect of a change of substrate use from predominantly non-esterified FFA to glucose and lactate,[35] finally resulting in improved left ventricular mechanical efficiency.[36]
Trimetazidine (1-[2,3,4-trimethoxybenzyl]piperazine dihydrochloride) has been shown to directly inhibit FFA oxidation by blocking 3-ketoacyl-coenzyme A thiolase (3-KAT), the last enzyme involved in beta-oxidation,[37] although this issue remains controversial.[38,39] Trimetazidine affects myocardial substrate use by inhibiting oxidative phosphorylation and by shifting energy production from FFA to glucose oxidation (see Figure 1).[40] Several studies have outlined the potential benefits of this agent on regional and global myocardial dysfunction.[41–49] 3-KAT inhibitors could also play a beneficial role in terms of glucose metabolism homeostasis at both cardiac and skeletal muscle level. The beneficial effect of trimetazidine on left ventricular function, has been attributed to preservation of phosphocreatine (PCr) and ATP intracellular levels.[50] Clinical studies using phosphorus-31 magnetic resonance spectroscopy to measure PCr:ATP ratios in human myocardium have shown that this ratio is reduced in failing human myocardium.[51] The PCr:ATP ratio is a measure of myocardial energetics and its reduction may depend on imbalance of myocardial oxygen supply and demand,[52] and reduction of the total creatine pool, a phenomenon known to occur in heart failure.[53] In a study performed in patients with heart failure of different aetiologies receiving full standard medical therapy, it was observed that the trimetazidine-induced improvement of functional class and left ventricular function was associated with an improvement of PCr:ATP ratio, supporting the hypothesis that trimetazidine may preserve myocardial high-energy phosphate intracellular levels.[54] These results appear particularly interesting, especially in view of previous evidence indicating the PCr:ATP ratio as a significant predictor of mortality.[55] In fact, imetazidine has been shown to improve prognosis in patients with heart failure in a multicentre retrospective cohort study[56] and in two meta-analyses.[57,58] On this basis, its use in patients with heart failure has been advocated in a recently published position paper.[59]
Figure 1: Metabolic Vicious Circle in Heart Failure.
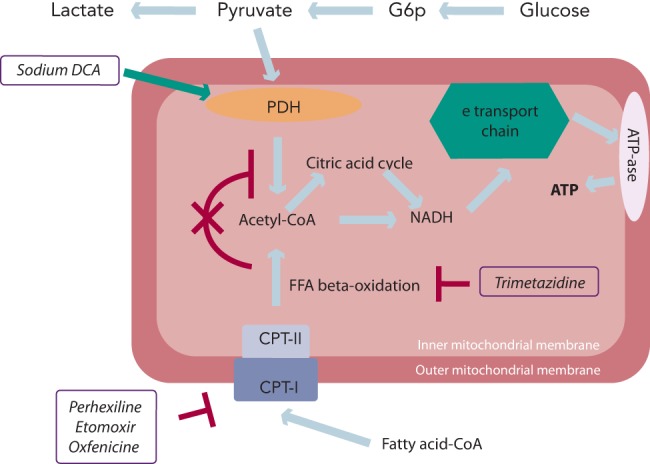
FFAs inhibit glycolysis and glucose uptake by the heart. Plasma FFA taken up by the heart is activated and transported by CPT-I into the mitochondria to uncouple respiration with oxygen wastage. In addition, hyperadrenergic state downregulates beta-adrenergic receptors. Acetyl CoA = acetyl coenzyme A; ATP = adenosine triphosphate; CPT = carnitine palmitoyltransferase; DCA = dichloroacetate; FFA = free fatty acid; PDH = pyruvate dehydrogenase.
Similarly to trimetazidine, ranolazine has also been shown to significantly improve left ventricular performance in experimental models of heart failure.[60–63] Sabbah et al. measured haemodynamics before and 40 minutes after intravenous ranolazine administration in a canine model of heart failure.[60] Results in 13 experimental dogs were compared with those obtained in eight normal healthy dogs. Ranolazine significantly decreased left ventricular end-diastolic pressure and increased left ventricular ejection fraction in the absence of any effects on heart rate or blood pressure. In subsequent experiments from the same laboratory, Chandler et al. reproduced these findings and determined that the improvement in left ventricular performance was not associated with an increase in myocardial oxygen consumption (MO2) compared with an intravenous infusion of dobutamine that improved left ventricular performance to a similar extent, but was associated with a significant increase in MO2 requirements.[61]
Overall, these data confirm that selective inhibition of 3-KAT represents a new therapeutic window in the treatment of patients with heart failure of different aetiologies.
Combined Metabolic Action of Beta-blockers and Trimetazidine
ACE inhibitors and beta-blockers remain the clinical mainstay of the treatment of heart failure. It is interesting to note that beta-blockers may yield an ancillary metabolic effect. Their principal mechanism of action is based on reduction of oxygen consumption by reduced heart rate and inotropism. However, a direct complementary metabolic effect could be exerted by beta-blockers themselves, by reducing peripheral lipolysis and determining reduction of FFA availability. There is indeed evidence that beta-blockade can reduce FFA use in favour of greater glucose use in patients with cardiac disease.[64] This change in myocardial energetics could provide a potential mechanism for the decreased MO2 and improved energy efficiency seen with beta-adrenoreceptor blockade in the treatment of ischaemic heart disease and heart failure.[65]
The issue of whether non-selective, compared with selective beta-adrenoreceptor blockers are more efficient in shifting total body substrate use from lipid to glucose oxidation[66] remains controversial.[67] Nevertheless, a better metabolic disposition of non-selective beta-blockers may contribute to improved survival rates observed with their use.[68] In addition, central inhibition of sympathetic nervous activity with moxonidine has been associated with increased mortality rates in patients with chronic heart failure.[69] In fact, despite a significant reduction of cathecolamine spillover and, consequently, heart rate, moxonidine has been shown to increase FFA use and increase MO2 consumption.[70] This could be the reason for the failure of central sympathetic inhibition in preventing death in long-term studies in patients with chronic heart failure. It also indicates that the predominant mechanism of action of beta-blockers in cardiac syndromes is likely related to mechanisms of action other than simple heart rate reduction. In patients with heart failure the magnitude of heart rate reduction may therefore be a marker of improved functional response following beta-blockade administration, a consequent effect rather than a mechanism. Nonetheless, a clinical trial in which the cardiac ‘funny’ (If) channel inhibitor ivabradine (a pure heart rate-lowering agent) was added to beta-blockade (SHIFT [Systolic Heart Failure Treatment With the lf Inhibitor Ivabradine Trial]) clearly demonstrated that the greater the heart rate reduction the greater the reduction of hospitalisation events in patients with heart failure.[71] Therefore, apart from the importance of heart rate lowering per se, a complementary synergistic metabolic action of beta-blockers and trimetazidine can be hypothesised: whereas the former reduce FFA availability, the latter decrease their cardiac use. Overall, this drug-induced metabolic shift could reduce FFA oxidation and increase the flux through pyruvate dehydrogenase with a consequent energy-sparing effect.[54,72]
Additional data also suggest that the metabolic effect of trimetazidine may also take place in other organs and tissues.[72] In fact, apart from a reduction of whole-body energy demand, a trend for a reduction of whole-body lipid oxidation and of fasting plasma FFA concentration has also been observed.[72] This general metabolic shift could reduce the overall metabolic requirements of the body, resulting in an attractive adaptation strategy in the context of coronary and myocardial insufficiencies. Interestingly, beta-blockers have also been shown to exert a direct effect on whole-body metabolism. In trained athletes, beta-adrenergic blockade abolishes the marked increase in plasma glucose levels during intense exercise as a result of enhanced peripheral glucose uptake, with no significant change in glucose production.[73] These effects of adrenergic blockade on glucose kinetics could be mediated by direct effects or indirectly through changes in lipid substrates and/or counter-regulatory hormones.
Other Inhibitors of Fatty Acids Oxidation
Etomoxir, perhexiline and oxfenicine are carnitine palmitoyltransferase I (CPT-I) inhibitors. CPT-I is the key enzyme for mitochondrial FFA uptake; its inhibition, therefore, reduces FFA oxidation and their inhibitory effect on pyruvate dehydrogenase. As a consequence, glucose oxidation is increased.[74,75] Etomoxir, initially developed as an antidiabetic agent, has been observed to improve left ventricular performance of pressure-overloaded rat heart.[76] These effects have been considered due to a selective modification of gene expression of hypertrophic cardiomyocytes.[77] Etomoxir has also been shown to increase phosphatase activation, have a direct effect on peroxisome proliferator-activated receptor-alpha and upregulate the expression of various enzymes involved in beta-oxidation.[77] The first clinical trial employing etomoxir in patients with heart failure showed a significant clinical and cardiac function improvement.[78] In experimental animal studies, etomoxir has also been shown to improve glucose metabolism.[79] However, the use of etomoxir may be limited by the observations that it may cause cardiac hypertrophy[80] and oxidative stress.[81]
Analogous to etomoxir, perhexiline and oxfenicine, originally classified as calcium antagonists, reduce cardiac use of long-chain fatty acids by inhibiting CPT-I.[82–84] They were initially developed as antianginal agents.[85,86] However, they have since been employed in patients with heart failure. In a previous study, metabolic modulation with perhexiline improved maximal oxygen consumption at the cardiopulmonary exercise test, left ventricular ejection fraction, symptoms, resting and peak stress myocardial function, and skeletal muscle energetics.[87] More recently, and similarly to trimetazidine, perhexiline has been shown to improve cardiac energetics and symptom status with no evidence of altered cardiac substrate use, further supporting the hypothesis of energy deficiency in heart failure and further consideration of metabolic therapies in its management.[88] Therefore, similarly to 3-KAT inhibitors, CPT-I inhibitors may represent a novel treatment in patients with heart failure with a good safety profile, provided that the dosage is adjusted according to plasma levels.
FFA Inhibition in Older Patients
Age-related changes of mitochondria impair the human host cells homeostasis and contribute to the development of most common ageing diseases. Older subjects without overt cardiac diseases are prone to develop heart failure with preserved ejection fraction. Risk factors do not fully account for the aged heart functional loss that might be underlined by a common pathogenic denominator (i.e. cell energy alteration at mitochondrial level in organs requiring high energy). In older men without overt cardiovascular disease, the presence of pre-pathologic conditions (pre-hypertension, reduced insulin sensitivity, impaired myocardial contractile reserve, inadequate vasodilation due to endothelial dysfunction, reduced cardiomyocytes renewal, systemic inflammation and raised coagulation capacity) are possibly related to reduced mitochondrial function and density. Several studies have indeed shown reduced mitochondrial content and function with ageing, leading to the theory that decreased mitochondrial content and increased uncoupling with age compromises the energy state of the cell.[89] Indeed, altered beta-oxidation increases the reliance on long-chain fatty acids relative to glucose with subsequent decrease of cellular metabolic efficiency at any given level of tissue activity. On this basis, in older patients with coronary artery disease, partial fatty acid oxidation inhibition by trimetazidine added to standard optimal medical therapy has been shown to improve reverse remodelling of chronically dysfunctional myocardium[90] and improve cardiac symptoms and quality of life.[91] The observed improvement could be related to increased cellular energy reserve,[54] which could be pivotal in a context of ageing-induced reduction of mitochondrial efficiency.
The Importance of a Correct Metabolic Substrate Availability
It remains questionable whether metabolic substrate availability rather than pharmacological shift from fatty acids to glucose oxidation may be appropriate in patients with long-lasting heart failure.[92] In fact, the anti-lipolytic drug acipimox, which reduces substrate availability and impairs fatty acid oxidation, has been shown to worsen left ventricular function in patients with idiopathic dilated cardiomyopathy.[93] In addition, when substrate availability is acutely modulated during exercise testing in patients with stable coronary artery disease and preserved left ventricular function using a high-carbohydrate meal versus a high-fat meal, a lower ischaemic threshold and greater ischaemia magnitude is observed following the high-carbohydrate meal.[94] Reduced lipid uptake and disposal in the setting of heart failure may represent therefore a maladaptive response. A recent study evaluated the metabolic and functional effects of high- and low-serum FFA availability in the presence of normal fasting serum glucose and insulin concentrations.[95] In patients with chronic heart failure, short-term reduction in serum FFA concentration, while serum glucose and insulin concentrations remained closed to the fasting levels, induced an impairment of left ventricular energy metabolism and left ventricular function at rest. A two-fold serum FFA increment in the same experimental conditions did not induce any detectable change.[95] Although in previous studies pharmacological manipulation of the heart substrates preferences has been shown to exert beneficial effects, these data demonstrate that direct deprivation of energy substrates is detrimental for cardiac metabolism and function. The clinical fallout is that metabolic manipulation at the systemic level may be considered in order to optimise the treatment of these patients; we also believe that special cautions should be considered when iatrogenic and acute changes of substrates and regulatory hormones of glucose and FFA metabolism are to be performed in these extremely vulnerable patients.
Conclusion
All cardiac syndromes may induce and be maintained by modifications of cardiac metabolism. Heart failure may be dependent on and promote metabolic changes in part through neurohormonal activation determining an increased use of non-carbohydrate substrates for energy production. This metabolic adaptation yields a state of metabolic inefficiency. The net result is depletion of myocardial ATP, phosphocreatine and creatine kinase levels with decreased efficiency of mechanical work. Different therapeutic approaches have been developed to manipulate cardiac energy metabolism. However, the most effective approach consists in modifying substrate use by the failing heart and includes several pharmacological agents. These agents have been originally adopted to increase the ischaemic threshold in patients with effort angina. However, the results of current research support the concept that shifting the energy substrate preference away from fatty acid metabolism and glucose metabolism could be an effective adjunctive treatment in patients with heart failure. Nevertheless, the exact role of metabolic therapy in heart failure is yet to be established, and a large multicentre randomised trial is necessary.
References
- 1.Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Progr Cardiovasc Dis. 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 2.Lopaschuk GD, Ussher JR, Folmes CD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. DOI: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 3.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;281:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 4.Lommi J, Kupari M, Koskinen P, et al. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. doi: 10.1016/0735-1097(96)00214-8. [DOI] [PubMed] [Google Scholar]
- 5.Riley M, Bell N, Elborn JS, et al. Metabolic response to graded exercise in chronic heart failure. Eur Heart J. 1993;14:1484–1488. doi: 10.1093/eurheartj/14.11.1484. [DOI] [PubMed] [Google Scholar]
- 6.Paolisso G, De Riu S, Marrazzo G, et al. Insulin resistance and hyperinsulinemia in patients with chronic heart failure. Metabolism. 1991;40:972–977. doi: 10.1016/0026-0495(91)90075-8. [DOI] [PubMed] [Google Scholar]
- 7.Mori J, Basu R, McLean BA, et al. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail. 2012;5:493–503. doi: 10.1161/CIRCHEARTFAILURE.112.966705. DOI: 10.1161/CIRCHEARTFAILURE.112.966705. [DOI] [PubMed] [Google Scholar]
- 8.Dai DF, Johnson SC, Villarin JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. DOI: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellieux C, Aasum E, Larsen TS, et al. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J Mol Cell Cardiol. 2006;41:459–466. doi: 10.1016/j.yjmcc.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Pellieux C, Montessuit C, Papageorgiou I, Lerch R. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-alpha. Cardiovasc Res. 2009;82:341–350. doi: 10.1093/cvr/cvp004. DOI: 10.1093/cvr/cvp004. [DOI] [PubMed] [Google Scholar]
- 11.Mori J, Alrob OA, Wagg CS, et al. Ang II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol. 2013;304:H1103–13. doi: 10.1152/ajpheart.00636.2012. DOI: 10.1152/ajpheart.00636.2012. [DOI] [PubMed] [Google Scholar]
- 12.Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. 2014;171:2080–2090. doi: 10.1111/bph.12475. DOI: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vermes E, Ducharme A, Bourassa MG, et al. Tardif, for the studies of left ventricular dysfunction. enalapril reduces the incidence of diabetes in patients with chronic heart failure: insight from the Studies Of Left Ventricular Dysfunction (SOLVD). Circulation. 2003;107:1291–1296. doi: 10.1161/01.cir.0000054611.89228.92. [DOI] [PubMed] [Google Scholar]
- 14.Yusuf S, Ostergren JB, Gerstein HC, et al. Effects of candesartan on the development of a new diagnosis of diabetes mellitus in patients with heart failure. Circulation. 2005;112:48–53. doi: 10.1161/CIRCULATIONAHA.104.528166. [DOI] [PubMed] [Google Scholar]
- 15.Drexler H, Hayoz D, Munzel T, et al. Endothelial function in chronic congestive heart failure. Am J Cardiol. 1992;69:1596–1601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- 16.Parodi O, De Maria R, Roubina E. Redox state, oxidative stress and endothelial dysfunction in heart failure: the puzzle of nitrate-thiol interaction. J Cardiovasc Med. 2007;8:765–774. doi: 10.2459/JCM.0b013e32801194d4. [DOI] [PubMed] [Google Scholar]
- 17.Traverse JH, Chen Y, Hou M, Bache RJ. Inhibition of NO production increases myocardial blood flow and oxygen consumption in congestive heart failure. Am J Physiol Heart Circ Physiol. 2002;282:H2278–83. doi: 10.1152/ajpheart.00504.2001. [DOI] [PubMed] [Google Scholar]
- 18.Kiowski W, Sutsch G, Hunziker P, et al. Evidence for endothelin-1-mediated vasoconstriction in severe chronic heart failure. Lancet. 1995;346:732–736. doi: 10.1016/s0140-6736(95)91504-4. [DOI] [PubMed] [Google Scholar]
- 19.Yamauchi-Kohno R, Miyauchi T, Hoshino T, et al. Role of endothelin in deterioration of heart failure due to cardiomyopathy in hamsters: increase in endothelin production in the heart and beneficial effect of endothelin A antagonist on survival and cardiac function. Circulation. 1999;99:2171–2176. doi: 10.1161/01.cir.99.16.2171. [DOI] [PubMed] [Google Scholar]
- 20.Fragasso G, Piatti P, Monti L, et al. Acute effects of heparin administration on the ischemic threshold of patients with coronary artery disease: evaluation of the protective role of the metabolic modulator trimetazidine. J Am Coll Cardiol. 2002;39:413–419. doi: 10.1016/s0735-1097(01)01768-5. [DOI] [PubMed] [Google Scholar]
- 21.Monti LD, Setola E, Fragasso G, et al. Metabolic and endothelial effects of trimetazidine on forearm skeletal muscle in patients with type 2 diabetes and ischemic cardiomyopathy. Am J Physiol Endocrinol Metab. 2006;290:E54–9. doi: 10.1152/ajpendo.00083.2005. [DOI] [PubMed] [Google Scholar]
- 22.Maridonneau-Parini I, Harpey C. Effects of trimetazidine on membrane damage induced by oxygen free radicals in human red cells. Br J Clin Pharmacol. 1985;20:148–151. doi: 10.1111/j.1365-2125.1985.tb05047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doutreleau S1, Mettauer B, Piquard F, et al. Chronic L-arginine supplementation enhances endurance exercise tolerance in heart failure patients. Int J Sports Med. 2006;27:567–572. doi: 10.1055/s-2005-865847. [DOI] [PubMed] [Google Scholar]
- 24.Lopaschuck GD, Stanley WC. Glucose metabolism in the ischemic heart. Circulation. 1997;95:313–315. doi: 10.1161/01.cir.95.2.313. [DOI] [PubMed] [Google Scholar]
- 25.Nuutila P, Knuuti MJ, Raitakari M, et al. Effect of antilipolysis on heart and skeletal muscle glucose uptake in overnight fasted humans. Am J Physiol. 1994;267:E941–6. doi: 10.1152/ajpendo.1994.267.6.E941. [DOI] [PubMed] [Google Scholar]
- 26.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23:201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 27.Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634. doi: 10.1007/s00125-002-0800-2. [DOI] [PubMed] [Google Scholar]
- 28.Peabody FW, Meyer AL, Du Bois EF. The basal metabolism of patients with cardiac and renal disease. Arch Int Med. 1916;17:980–1009. [Google Scholar]
- 29.Riley M, Elborn JS, McKane WR, et al. Resting energy expenditure in chronic cardiac failure. Clin Sci. 1991;80:633–639. doi: 10.1042/cs0800633. [DOI] [PubMed] [Google Scholar]
- 30.Poehlman ET, Scheffers J, Gottlieb SS, et al. Increased resting metabolic rate in patients with congestive heart failure. Ann Intern Med. 1994;121:860–862. doi: 10.7326/0003-4819-121-11-199412010-00006. [DOI] [PubMed] [Google Scholar]
- 31.Lommi J, Kupari M, Yki-Järvinen H. Free fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol. 1998;81:45–50. doi: 10.1016/s0002-9149(97)00804-7. [DOI] [PubMed] [Google Scholar]
- 32.Steinberg D, Nestel PJ, Buskirk ER, Thompson RH. Calorigenic effect of norepinephrine correlated with plasma free fatty acid turnover and oxidation. J Clin Invest. 1964;43:167–176. doi: 10.1172/JCI104901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landsberg L, Saville ME, Young JB. Sympathoadrenal system and regulation of thermogenesis. Am J Physiol. 1984;247:E181–9. doi: 10.1152/ajpendo.1984.247.2.E181. [DOI] [PubMed] [Google Scholar]
- 34.Mc Veigh JJ, Lopaschuck GD. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am J Physiol. 1990;259:H1070–85. doi: 10.1152/ajpheart.1990.259.4.H1079. [DOI] [PubMed] [Google Scholar]
- 35.Nicholl TA, Lopaschuck GD, McNeill GH. Effects of free fatty acids and dichloroacetate on isolated working diabetic rat hearts. Am J Physiol. 1991;261:H1053–9. doi: 10.1152/ajpheart.1991.261.4.H1053. [DOI] [PubMed] [Google Scholar]
- 36.Bersin RM, Wolfe C, Kwasman M, et al. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol. 1994;23:1617–1624. doi: 10.1016/0735-1097(94)90665-3. [DOI] [PubMed] [Google Scholar]
- 37.Kantor PF, Lucien A, Kozak R, Lopashuck GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 38.Lopaschuk GD, Barr R, Thomas PD, Dyck JR. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2003;93:e33–7. doi: 10.1161/01.RES.0000086964.07404.A5. [DOI] [PubMed] [Google Scholar]
- 39.MacInness A, Fairman DA, Binding P, et al. The antianginal trimetazidine does not exert its functional benefit via inhibition of mithochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2003;93:e26–32. doi: 10.1161/01.RES.0000086943.72932.71. [DOI] [PubMed] [Google Scholar]
- 40.Fantini E, Demaison L, Sentex E, et al. Some biochemical aspects of the protective effect of trimetazidine on rat cardiomyocytes during hypoxia and reoxygenation. J Mol Cell Cardiol. 1994;26:949–958. doi: 10.1006/jmcc.1994.1116. [DOI] [PubMed] [Google Scholar]
- 41.Brottier L, Barat JL, Combe C, et al. Therapeutic value of a cardioprotective agent in patients with severe ischaemic cardiomyopathy. Eur Heart J. 1990;11:207–212. doi: 10.1093/oxfordjournals.eurheartj.a059685. [DOI] [PubMed] [Google Scholar]
- 42.Lu C, Dabrowski P, Fragasso G, Chierchia SL. Effects of trimetazidine on ischemic left ventricular dysfunction in patients with coronary artery disease. Am J Cardiol. 1998;82:898–901. doi: 10.1016/s0002-9149(98)00500-1. [DOI] [PubMed] [Google Scholar]
- 43.Belardinelli R, Purcaro A. Effects of trimetazidine on the contractile response of chronically dysfunctional myocardium to low-dose dobutamine in ischaemic cardiomyopathy. Eur Heart J. 2001;22:2164–2170. doi: 10.1053/euhj.2001.2653. [DOI] [PubMed] [Google Scholar]
- 44.Fragasso G, Piatti PM, Monti L, et al. Short- and long-term beneficial effects of partial free fatty acid inhibition in diabetic patients with ischemic dilated cardiomyopathy. Am Heart J. 2003;146:E18. doi: 10.1016/S0002-8703(03)00415-0. [DOI] [PubMed] [Google Scholar]
- 45.Rosano GMC, Vitale C, Sposato B, et al. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: a double-blind placebo-controlled study. Cardiovasc Diabetol. 2003;2:16. doi: 10.1186/1475-2840-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Napoli P, Taccardi AA, Barsotti A. Long term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart. 2005;91:161–165. doi: 10.1136/hrt.2003.031310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fragasso G, Palloshi A, Puccetti P, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. 2006;48:992–998. doi: 10.1016/j.jacc.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 48.Sisakian H, Torgomyan A, Barkhudaryan A. The effect of trimetazidine on left ventricular systolic function and physical tolerance in patients with ischaemic cardiomyopathy. Acta Cardiol. 2007;62:493–499. doi: 10.2143/AC.62.5.2023413. [DOI] [PubMed] [Google Scholar]
- 49.Di Napoli P, Di Giovanni P, Gaeta MA, et al. Trimetazidine and reduction in mortality and hospitalization in patients with ischemic dilated cardiomyopathy: a post hoc analysis of the Villa Pini d’Abruzzo Trimetazidine Trial. J Cardiovasc Pharmacol. 2007;50:585–589. doi: 10.1097/FJC.0b013e31814fa9cb. [DOI] [PubMed] [Google Scholar]
- 50.Lavanchy N, Martin J, Rossi A. Anti-ischemia effects of trimetazidine: 31P-NMR spectroscopy in the isolated rat heart. Arch Int Pharmacodyn Ther. 1987;286:97–110. [PubMed] [Google Scholar]
- 51.Conway MA, Allis J, Ouwerkerk R, et al. Detection of low PCr to ATP ratio in failing hypertrophied myocardium by 31P magnetic resonance spectroscopy. Lancet. 1991;338:973–976. doi: 10.1016/0140-6736(91)91838-l. [DOI] [PubMed] [Google Scholar]
- 52.Yabe T, Mitsunami K, Inubushi T, Kinoshita M. Quantitative measurements of cardiac phosphorus metabolites in coronary artery disease by 31P magnetic resonance spectroscopy. Circulation. 1995;92:15–23. doi: 10.1161/01.cir.92.1.15. [DOI] [PubMed] [Google Scholar]
- 53.Nascimben L, Ingwall JS, Pauletto P, et al. The creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94:1894–1901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 54.Fragasso G, De Cobelli F, Perseghin G, et al. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J. 2006;27:942–948. doi: 10.1093/eurheartj/ehi816. [DOI] [PubMed] [Google Scholar]
- 55.Neubauer S, Horn M, Cramer M, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 56.Fragasso G, Rosano G, Baek SH, et al. Effect of partial fatty acid oxidation inhibition with trimetazidine on mortality and morbidity in heart failure: Results from an international multicentre retrospective cohort study. Int J Cardiol. 2013;163:320–325. doi: 10.1016/j.ijcard.2012.09.123. DOI: 10.1016/j.ijcard.2012.09.123. [DOI] [PubMed] [Google Scholar]
- 57.Gao D, Ning N, Niu X, et al. Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart. 2011;97:278–286. doi: 10.1136/hrt.2010.208751. DOI: 10.1136/hrt.2010.208751. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Lu Y, Jiang H, et al. Additional use of trimetazidine in patients with chronic heart failure: a meta-analysis. J Am Coll Cardiol. 2012;59:913–922. doi: 10.1016/j.jacc.2011.11.027. DOI: 10.1016/j.jacc.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 59.Lopatin YM, Rosano GM, Fragasso G, et al. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int J Cardiol. 2015;203:909–915. doi: 10.1016/j.ijcard.2015.11.060. DOI: 10.1016/j. ijcard.2015.11.060. [DOI] [PubMed] [Google Scholar]
- 60.Sabbah HN, Chandler MP, Mishima T, et al. Ranolazine, a partial fatty acid oxidation (pFOX) inhibitor, improves left ventricular function in dogs with chronic heart failure. J Card Fail. 2002;8:416–422. doi: 10.1054/jcaf.2002.129232. [DOI] [PubMed] [Google Scholar]
- 61.Chandler MP, Stanley WC, Morita H, et al. Short-term treatment with ranolazine improves mechanical efficacy in dogs with chronic heart failure. Circ Res. 2002;91:278–280. doi: 10.1161/01.res.0000031151.21145.59. [DOI] [PubMed] [Google Scholar]
- 62.Hayashida W, van Eyll C, Rousseau MF, Pouleur H. Effects of ranolazine on left ventricular regional diastolic function in patients with ischemic heart disease. Cardiovasc Drugs Ther. 1994;5:741–747. doi: 10.1007/BF00877121. [DOI] [PubMed] [Google Scholar]
- 63.Aaker A, McCormack JG, Hirai T, Musch TI. Effects of ranolazine on the exercise capacity of rats with chronic heart failure induced by myocardial infarction. J Cardiovasc Pharmacol. 1996;28:353–362. doi: 10.1097/00005344-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 64.Wallhaus TR, Taylor M, DeGrado TR, Russell DC. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441. [DOI] [PubMed] [Google Scholar]
- 65.Spoladore R, Fragasso G, Perseghin G, et al. Beneficial effects of beta-blockers on left ventricular function and cellular energy reserve in patients with heart failure. Fundam Clin Pharmacol. 2013;27:455–464. doi: 10.1111/j.1472-8206.2012.01029.x. DOI: 10.1111/j.1472-8206.2012.01029.x. [DOI] [PubMed] [Google Scholar]
- 66.Podbregar M, Voga G. Effect of selective and nonselective beta-blockers on resting energy production rate and total body substrate utilization in chronic heart failure. J Cardiac Fail. 2002;8:369–378. doi: 10.1054/jcaf.2002.130238. [DOI] [PubMed] [Google Scholar]
- 67.Sharma V, Dhillon P, Wambolt R, et al. Metoprolol improves cardiac function and modulates cardiac metabolism in the streptozotocin (STZ) diabetic rat. Am J Physiol Heart Circ Physiol. 2008;294:H1609–20. doi: 10.1152/ajpheart.00949.2007. DOI: 10.1152/ajpheart.00949.2007. [DOI] [PubMed] [Google Scholar]
- 68.Poole-Wilson P, Swedberg K, Cleland J, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 69.Cohn JN1, Pfeffer MA, Rouleau J, et al. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). Eur J Heart Fail. 2003;5:659–666. doi: 10.1016/s1388-9842(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 70.Mobini R, Jansson PA, Bergh CH, et al. Influence of central inhibition of sympathetic nervous activity on myocardial metabolism in chronic heart failure: acute effects of the imidazoline I1-receptor agonist moxonidine. Clin Sci. 2006;110:329–336. doi: 10.1042/CS20050037. [DOI] [PubMed] [Google Scholar]
- 71.Swedberg K, Komajda M, Böhm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61198-1. DOI: 10.1016/S0140-6736(10)61198-1. [DOI] [PubMed] [Google Scholar]
- 72.Fragasso G, Salerno A, Lattuada G, et al. Effect of partial inhibition of fatty acid oxidation by trimetazidine on whole body energy metabolism in patients with chronic heart failure. Heart. 2011;97:1495–1500. doi: 10.1136/hrt.2011.226332. DOI: 10.1136/hrt.2011.226332. [DOI] [PubMed] [Google Scholar]
- 73.Howlett KF, Watt MJ, Hargreaves M, Febbraio MA. Regulation of glucose kinetics during intense exercise in humans: effects of alpha- and beta-adrenergic blockade. Metabolism. 2003;52:1615–1620. doi: 10.1016/s0026-0495(03)00330-5. [DOI] [PubMed] [Google Scholar]
- 74.Lopashuck GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036–1043. doi: 10.1161/01.res.63.6.1036. [DOI] [PubMed] [Google Scholar]
- 75.Ratheiser K, Schneeweiss B, Waldhäusl W, et al. Inhibition by etomoxir of carnitine palmitoyltransferase I reduces hepatic glucose production and plasma lipids in non-insulin-dependent diabetes mellitus. Metabolism. 1991;40:1185–1190. doi: 10.1016/0026-0495(91)90214-h. [DOI] [PubMed] [Google Scholar]
- 76.Turcani M, Rupp H. Etomoxir improves left ventricular performance of pressure-overloaded rat heart. Circulation. 1997;96:3681–3686. doi: 10.1161/01.cir.96.10.3681. [DOI] [PubMed] [Google Scholar]
- 77.Zarain-Herzberg A, Rupp H. Therapeutic potential of CPT I inhibitors: cardiac gene transcription as a target. Expert Opin Investig Drugs. 2002;11:345–356. doi: 10.1517/13543784.11.3.345. [DOI] [PubMed] [Google Scholar]
- 78.Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci. 2000;99:27–35. [PubMed] [Google Scholar]
- 79.Schmitz FJ, Rösen P, Reinauer H. Improvement of myocardial function and metabolism in diabetic rats by the carnitine palmitoyl transferase inhibitor etomoxir. Horm Metab Res. 1995;27:515–522. doi: 10.1055/s-2007-980016. [DOI] [PubMed] [Google Scholar]
- 80.Cabreros A, Merlos M, Laguna JC, Carrera MV. Down-regulation of acyl-CoA oxidase gene expression and increased NF-kappaB activity in etomoxir-induced cardiac hypertrophy. J Lipid Res. 2003;44:388–398. doi: 10.1194/jlr.M200294-JLR200. [DOI] [PubMed] [Google Scholar]
- 81.Merrill CL, Ni H, Yoon LW, et al. Etomoxir-induced oxidative stress in HepG2 cells detected by differential gene expression is confirmed biochemically. Toxicol Sci. 2002;68:93–101. doi: 10.1093/toxsci/68.1.93. [DOI] [PubMed] [Google Scholar]
- 82.Kennedy JA, Kiosoglus AJ, Murphy GA, et al. Effect of perhexilline and oxfenicine on myocardial function and metabolism during low flow ischemia/reperfusion in the isolated rat heart. J Cardiovasc Pharmacol. 2000;36:794–801. doi: 10.1097/00005344-200012000-00016. [DOI] [PubMed] [Google Scholar]
- 83.Jeffrey FM, Alvarez L, Diczku V, et al. Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate. J Cardiovasc Pharmacol. 1995;25:469–472. doi: 10.1097/00005344-199503000-00018. [DOI] [PubMed] [Google Scholar]
- 84.Stephens TW, Higgins AJ, Cook GA, Harris RA. Two mechanisms produce tissue-specific inhibition of fatty acid oxidation by oxfenicine. Biochem J. 1985;227:651–660. doi: 10.1042/bj2270651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bergman G, Atkinson L, Metcalfe J, et al. Beneficial effect of enhanced myocardial carbohydrate utilisation after oxfenicine (L-hydroxyphenylglycine) in angina pectoris. Eur Heart J. 1980;1:247–253. doi: 10.1093/oxfordjournals.eurheartj.a061126. [DOI] [PubMed] [Google Scholar]
- 86.Cole PL, Beamer AD, McGowan N, et al. Efficacy and safety of perhexiline maleate in refractory angina: a double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990;81:1260–1270. doi: 10.1161/01.cir.81.4.1260. [DOI] [PubMed] [Google Scholar]
- 87.Lee L, Campbell R, Scheuermann-Freestone M, et al. Metabolic modulation with perhexiline in chronic heart failure. A randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112:3280–3288. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 88.Beadle RM, Williams LK, Kuehl M, et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015;3:202–211. doi: 10.1016/j.jchf.2014.09.009. DOI: 10.1016/j.jchf.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 89.Johannsen DL, Conley KE, Bajpeyi S, et al. Ectopic lipid accumulation and reduced glucose tolerance in elderly adults are accompanied by altered skeletal muscle mitochondrial activity. J Clin Endocrinol Metab. 2012;97:242–250. doi: 10.1210/jc.2011-1798. DOI: 10.1210/jc.2011-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vitale C, Wajngaten M, Sposato B, et al. Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur Heart J. 2004;25:1814–1821. doi: 10.1016/j.ehj.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 91.Marazzi G, Gebara O, Vitale C, et al. Effect of trimetazidine on quality of life in elderly patients with ischemic dilated cardiomyopathy. Adv Ther. 2009;26:455–461. doi: 10.1007/s12325-009-0024-7. DOI: 10.1007/s12325-009-0024-7. [DOI] [PubMed] [Google Scholar]
- 92.Neubauer S. The failing heart. An engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 93.Tuunanen H, Engblom E, Naum A, et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation. 2006;114:2130–2137. doi: 10.1161/CIRCULATIONAHA.106.645184. [DOI] [PubMed] [Google Scholar]
- 94.Fragasso G, Montano C, Lattuada G, et al. A high carbohydrate meal yields a lower ischemic threshold than a high fat meal in patients with stable coronary disease. Int J Cardiol. 2011;147:209–213. doi: 10.1016/j.ijcard.2009.08.023. DOI: 10.1016/j.ijcard.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 95.Salerno A, Fragasso G, Esposito A, et al. Effects of short-term manipulation of serum FFA concentrations on left ventricular energy metabolism and function in patients with heart failure: no association with circulating bio-markers of inflammation. Acta Diabetol. 2015;52:753–761. doi: 10.1007/s00592-014-0695-7. DOI: 10.1007/s00592-014-0695-7. [DOI] [PubMed] [Google Scholar]