

Abstract
Neuronal apoptosis is a crucial pathological process in early brain injury after subarachnoid hemorrhage (SAH). The effective therapeutic strategies to ameliorate neuronal apoptosis are still absent. We intended to determine whether intranasal administration of exogenous Netrin-1 (NTN-1) could attenuate neuronal apoptosis after experimental SAH, specifically via activating DCC-dependent APPL-1/AKT signaling cascade. Two hundred twenty-five male Sprague-Dawley rats were subjected to the endovascular perforation model of SAH. Recombinant human NTN-1 (rNTN-1) was administered intranasally. NTN-1 small interfering RNA (siRNA), APPL-1 siRNA, and AKT inhibitor MK2206 were administered through intracerebroventricular (i.c.v.) injection. SAH grade, neurological score, neuronal apoptosis assessed by cleaved caspase-3 (CC-3) expression and Fluoro-Jade C (FJC) staining, double immunofluorescence staining, and Western blot were examined. Our results revealed that endogenous NTN-1 level was increased after SAH. Administration of rNTN-1 improved neurological outcomes at 24 h and 72 h after SAH, while knockdown of endogenous NTN-1 worsened neurological impairments. Furthermore, exogenous rNTN-1 treatment promoted APPL-1 activation, increased phosphorylated-AKT and Bcl-2 expression, as well as decreased apoptotic marker CC-3 expression and the number of FJC-positive neurons, thereby alleviated neuronal apoptosis. Conversely, APPL-1 siRNA and MK2206 abolished the anti-apoptotic effect of exogenous rNTN-1 at 24 h after SAH. Collectively, intranasal administration of exogenous rNTN-1 attenuated neuronal apoptosis and improved neurological function in SAH rats, at least in apart via activating DCC/APPL-1/AKT signaling pathway.
Keywords: Subarachnoid hemorrhage, Early brain injury, Apoptosis, Netrin-1, APPL-1
1. Introduction
Aneurysmal subarachnoid hemorrhage (SAH) remains the most devastating form of stroke with high mortality and disability rates throughout the world (Hemphill et al., 2015). Increasing evidence has suggested that early brain injury (EBI) is a crucial factor that determines the prognosis of SAH, although the definitive mechanisms contributing to EBI remain elusive (Chen et al., 2014; Pang et al., 2016). Neuronal apoptosis is the most significant pathological process in EBI. Thus, anti-apoptosis is a promising therapeutic strategy in management of EBI (Suzuki, 2015; Xie et al., 2012; Yin et al., 2016).
Netrin-1 (NTN-1), a laminin-related protein, is highly expressed in neurons, as well as mediate axon outgrowth and neuronal migration during embryonic development by interacting with its receptors (Rajasekharan and Kennedy, 2009). Moreover, it is now well established that NTN-1 exerts versatile functions in diverse biological processes, such as cell survival, differentiation, apoptosis, angiogenesis and tissue morphogenesis (Lai Wing Sun et al., 2011; Lu et al., 2012; Mehlen and Furne, 2005; Wilson et al., 2006). Specifically, NTN-1 has been regarded as an anti-apoptotic molecule in various tissues, including brain, heart, kidney, and cancer (Layne et al., 2015; Liao et al., 2013; Llambi et al., 2001; Mehlen and Guenebeaud, 2010; Wang et al., 2009; Wu et al., 2008). In the adult brain, previous studies showed that NTN-1 could attenuate ischemia-induced neuronal apoptosis and reduce infarct size, as well as improve neurological function after ischemic stroke (Liao et al., 2013; Wu et al., 2008). However, the anti-apoptotic property of NTN-1 by interacting with deleted in colorectal cancer (DCC) receptor has not been reported in the setting of SAH.
APPL-1 (adaptor protein containing PH domain, PTB domain, and leucine zipper motif-1), is a multifunctional endosomal adaptor protein. By binding with the transmembrane receptors, such as DCC, EGF, adiponectin receptors, APPL-1 serves as a key regulator for the fundamental cellular processes including survival, proliferation, and metabolism (Holmes et al., 2011; Lin et al., 2006; Liu et al., 2002; Miaczynska et al., 2004; Tan et al., 2010). APPL-1 has been shown to regulate the activity of AKT via its interacting PTB domain (Schenck et al., 2008).
In this study, we hypothesized that intranasal administration of rNTN-1 could attenuate neuronal apoptosis after SAH. The anti-apoptotic property of rNTN-1 is mediated by DCC-dependent APPL-1 signaling pathway, which promotes phosphorylated-AKT (p-AKT) and Bcl-2 activity, and thereby inhibits cleaved caspase-3 (CC-3) expression.
2. Materials and Methods
2.1. Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Loma Linda University. They were in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines, and complied with the NIH guidelines for the use of Animals in Neuroscience Research. Two hundred twenty-five male Sprague-Dawley (SD) rats (Indianapolis, IN) weighing 280 to 330 g were used. All rats were housed in a controlled humidity and temperature room with a 12-h light/dark cycle and were raised with free access to water and food.
2.2. SAH Model
The endovascular perforation model of SAH was induced as previously published (Guo et al., 2016). Briefly, rats were intubated and mechanically ventilated throughout the operation with 3% isoflurane anesthesia. The left carotid artery and its branches were separated. A sharpened, 4-0 monofilament nylon suture was inserted into the left internal carotid artery from the external carotid artery, and then was pushed further to perforate the bifurcation of the anterior and middle cerebral arteries. Rats in the sham group were subjected to the same procedures without puncture. After the surgical procedure was completed, the rats were allowed to recover on a heated blanket.
2.3. Experimental Design
Three separate experiments were conducted as follows (Fig. 1).
Fig. 1. Experimental design and animal groups.
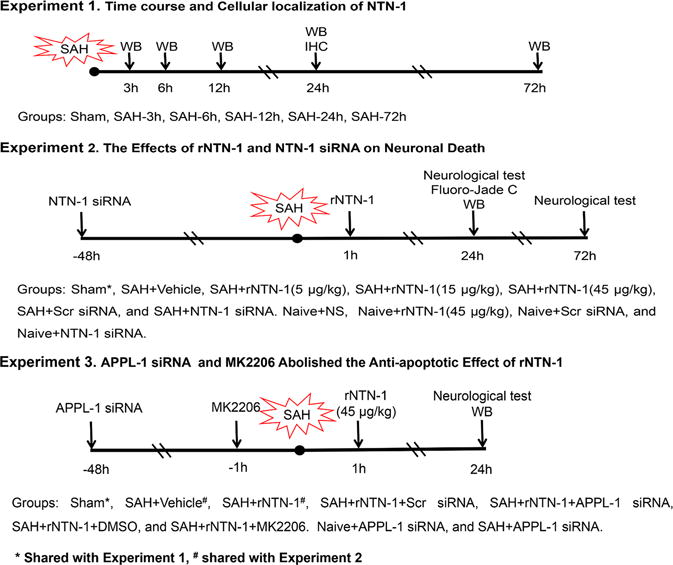
DMSO, Dimethyl Sulfoxide; IHC, immunohistochemistry; MK2206, AKT inhibitor; NTN-1, Netrin-1; NS, normal saline; SAH, Subarachnoid hemorrhage; Scr siRNA, scramble siRNA; WB, Western blot
Experiment 1
To determine the time course of endogenous NTN-1 expression after SAH. Western blots analysis for its expression level was performed using the ipsilateral/left cerebral cortex tissues. The additional four rats in the sham and SAH (24 h) groups were used for double immunohistochemistry staining.
Experiment 2
To evaluate the effect of recombinant human NTN-1 (rNTN-1) treatment and in vivo knockdown of endogenous NTN-1 on neuronal apoptosis. rNTN-1 or NTN-1 small interfering RNA (NTN-1 siRNA) was administered intranasally or intracerebroventricularly (i.c.v.), respectively. Neurobehavioral tests were assessed at 24 h and 72 h after SAH. Neuronal degeneration and Western blot were measured at 24 h post SAH. Rats were randomly divided into seven groups: sham, SAH + vehicle (normal saline, NS), SAH + rNTN-1 (5 μg/kg), SAH + rNTN-1 (15 μg/kg), SAH + rNTN-1 (45 μg/kg), SAH + scramble siRNA (Scr siRNA), and SAH + NTN-1 siRNA. The sample for sham group was shared from Experiment 1. Additionally, to validate the delivery efficiency of intranasal administration of rNTN-1 (45 μg/kg), and knockdown efficiency of NTN-1 siRNA, rats were randomly divided into four groups: Naive + NS, Naive + rNTN-1(45 μg/kg), Naive + Scr siRNA, Naive + NTN-1 siRNA. The expression of NTN-1 in the ipsilateral cortex was analyzed by Western blot.
Experiment 3
To explore the underlying mechanisms of rNTN-1-mediated neuroprotective effects. APPL-1 siRNA and AKT inhibitor MK2206 were administered i.c.v. before SAH induction and then followed with rNTN-1 (45 μg/kg) treatment. Neurobehavioral tests and Western blot were examined at 24 h after SAH. Rats were randomly divided into seven groups: sham, SAH + vehicle (NS), SAH + rNTN-1, SAH + rNTN-1 + Scr siRNA, SAH + rNTN-1 + APPL-1 siRNA, SAH + rNTN-1 + Dimethyl Sulfoxide (DMSO), and SAH + rNTN-1 + MK2206. The samples for sham, SAH + vehicle (NS), and SAH + rNTN-1 groups were shared from Experiment 1 and 2. In addition, to test knockdown efficiency of APPL-1 siRNA, rats were randomly divided into two groups: Naive + APPL-1 siRNA, and SAH + APPL-1 siRNA. The expression of APPL-1 in the ipsilateral cortex were analyzed by Western blot.
2.4. Intranasal Administration of rNTN-1
Intranasal administration was performed at 1 h after SAH induction as previously described (Topkoru et al., 2013). Rats were placed in a supine position under 2% isoflurane anesthesia. Sterile normal saline (NS) or rNTN-1 (5 μg/kg, 15 μg/kg or 45 μg/kg; R&D Systems, USA) dissolved in sterile NS was administered intranasally. A total volume of 30 μL was delivered alternately into the left and right nares, 5 μL one naris every 5 minutes for a period of 25 minutes.
2.5. Intracerebroventricular Drug Administration
Intracerebroventricular drug administration was performed as previously described (He et al., 2015). Briefly, rat was placed in a stereotaxic apparatus under 2.5% isoflurane anesthesia. The needle of a 10-μL Hamilton syringe (Microliter 701; Hamilton Company, USA) was inserted through a burr hole into the right lateral ventricle at the following coordinates relative to bregma: 1.5 mm posterior, 1.0 mm lateral, and 3.2 mm beneath the dural surface. siRNA was administered at 48 h prior to SAH induction. In order to enhance the knockdown efficiency, three different rat NTN-1 siRNA duplexes (Thermo Fisher Scientific), or APPL-1 siRNA duplexes (Thermo Fisher Scientific) was mixed and dissolved in sterile NS with a total volume 5 μL (500 pmol), and then injected into the right lateral ventricle by a pump at the rate of 0.5 μL/min. The same volume of Scr siRNA (Thermo Fisher Scientific) was used as a negative control. AKT inhibitor MK2206 (100 μg; Selleck Chemicals, USA) was dissolved in DMSO and further diluted in NS to a final DMSO concentration of 0.5 % (Yan et al., 2016). Five microliters of MK2206 (100 μg), or vehicle (DMSO in NS) was injected at a rate of 0.5 μl/min at 1 h before SAH induction. The needle was kept in situ for an additional 5 min and then withdrawn slowly over 5 min. Finally, the incision was closed with sutures and rats were allowed to recover.
2.6. Short-Term Neurological Score Evaluation
Neurological score was evaluated at 1 h before euthanized in a blinded fashion according to the modified Garcia and Beam balance tests as previously described (Yin et al., 2016). The Garcia neurological assessment included rodent’s spontaneous activity, spontaneous movement of all limbs, forelimbs outstretching, climbing wall of the cage, touch of trunk, and vibrissae touch. Each part of the test was scored from 0 to 3 (maximum score = 18). After this composite sensorimotor assessment, rats were evaluated for walking distances on a wooden beam for 1 minute. The scores for this test ranged from 0 to 4. Higher scores represented better neurological function.
2.7. SAH Grade
The severity of SAH was blindly assessed through a grading system immediately after euthanasia as previously described (Sugawara et al., 2008). The base of the brain was divided into six parts, each part was scored (0–3) according to the amount of subarachnoid blood. The total score was calculated as the total SAH grade (maximum SAH grade = 18). Fourteen mild SAH rats with a score of 8 or less were excluded from this study.
2.8. Fluoro-Jade C Staining
Degenerating neurons were evaluated by Fluoro-Jade C (FJC) staining as previously described (Shi et al., 2016). Briefly, rats were transcardially perfused under deep anesthesia with cold 60 mL of PBS (0.1M, pH 7.4) followed by 60 mL 10% paraformaldehyde solution at 24 h after SAH. Brains were removed and fixed in 10% paraformaldehyde at 4°C for 24 h, and then transferred into 30% sucrose solution for 72 h. Frozen coronal slices (10 μm) were sectioned in cryostat (CM3050S; Leica Microsystems). According to the manufacturer’s instructions, a modified FJC ready-to-dilute staining kit was used (Biosensis, USA). Slides were immersed in 1% sodium hydroxide solution for 5 min, followed by being rinsed for 2 min in 70% ethanol, then 2 min in distilled water. Slides were incubated in 0.06% potassium permanganate solution for 10 min, after 2 min in distilled water, transferred into 0.0001% solution of FJC (Millipore, USA) which was dissolved in 0.1% acetic acid. Slides were rinsed with distilled water for 1 min, three times. Slides were dried for 5 min, and immersed in xylene for 1 min, then added cover slipped with DPX mounting media (Sigma-Aldrich, USA). The sections were visualized by a blinded investigator using a fluorescence microscope Leica DMi8 (Leica Microsystems, Germany). The extent of neuronal damage was evaluated by the average number of FJC-positive neurons in six sections per brain with ImageJ software (ImageJ 1.5, NIH, USA). The data were expressed as cells/mm2.
2.9. Immunofluorescence Staining
Double fluorescence staining was performed as described previously (Huang et al., 2015). Frozen coronal sections (10 μm) were blocked with 5% donkey serum for 1 h and incubated at 4°C overnight with primary antibodies: rabbit anti-NTN-1 (1:500, Abcam), and mouse anti-NeuN (1:500, Abcam) followed by incubation with appropriate fluorescence-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) for 2 h at room temperature. Negative control staining was conducted by omitting the primary antibody. Finally, slides were covered with DAPI (Vector Laboratories, Inc.). The sections were visualized under a fluorescence microscope Leica DMi8. Microphotographs were analyzed with LASX software.
2.10. Western Blot Analysis
Rats were anesthetized with isoflurane at 24 h post SAH, and perfused with cold 120 mL of PBS (0.1M, pH 7.4). After removal of brain, the ipsilateral/left cerebral cortex tissues that facing blood clots were instantly collected and stored in −80 °C freezer until use. Western blot was performed as described previously (Tong et al., 2016). After protein samples preparation, equal amounts of protein (50 μg) were separated by SDS-PAGE gel electrophoresis, and then transferred onto nitrocellulose membranes. Membranes were blocked and incubated with the following primary antibodies overnight at 4 °C: rabbit anti-NTN-1 (1:800, Abcam), rabbit anti-APPL-1 (1:2000, Cell Signaling), rabbit anti-phospho-AKT (Ser473) (1:1000, Cell Signaling), rabbit anti-AKT (1:1000, Cell Signaling), rabbit anti-Bcl-2 (1:500, Santa Cruz Biotechnology), and rabbit anti-CC-3 (1:1000, Abcam). β-actin was used as an internal loading control. Then, membranes were processed with horseradish-peroxidase conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Immunoblots were probed with an ECL Plus chemiluminescence reagent kit (Amersham Biosciences). The relative density of protein was analyzed by ImageJ software.
2.11. Statistical Analysis
Data were expressed as a mean ± SD. All analyses were performed using GraphPad Prism 6 (GraphPad software). Statistical difference between groups were analyzed using one-way ANOVA, followed by Turkey multiple comparison post hoc test. Statistical significance was defined as p < 0.05.
3. Results
3.1. Mortality Rates and SAH Grade
No rats died in the sham group. There was no significant difference in mortality rates among all SAH groups (p > 0.05, Fig. 2A). The overall mortality of SAH was 19.1% (34/178). At 24 h after SAH, subarachnoid blood clots were obviously present around the Circle of Willis (Fig. 2B). The SAH grade scores were not significantly different among all SAH groups (p > 0.05, Fig. 2C).
Fig. 2. Mortality rate and subarachnoid hemorrhage (SAH) grade.
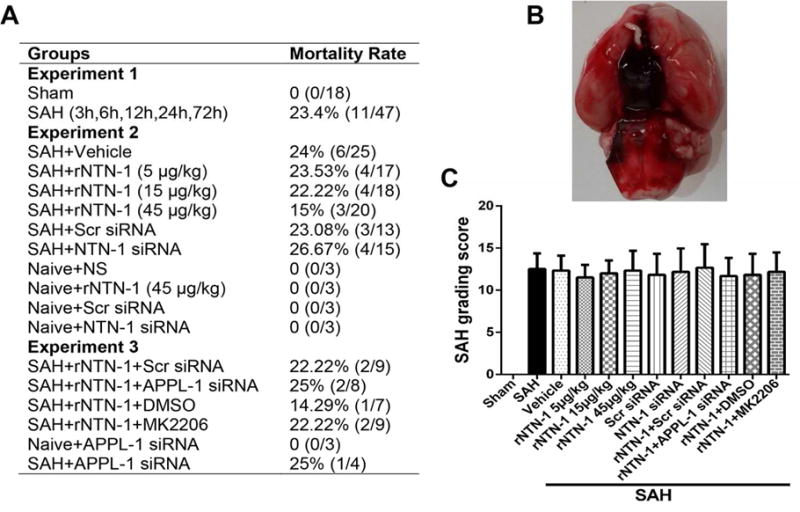
(A) Mortality rates of all SAH groups. (B) Subarachnoid blood clots were obviously present around the Circle of Willis at 24 h after SAH. (C) SAH grade scores of all SAH groups. n = 6 for each group. DMSO, Dimethyl Sulfoxide; IHC, immunohistochemistry; MK2206, AKT inhibitor; NTN-1, Netrin-1; NS, normal saline; Scr siRNA, scramble siRNA
3.2. Expression of NTN-1 and DCC after SAH
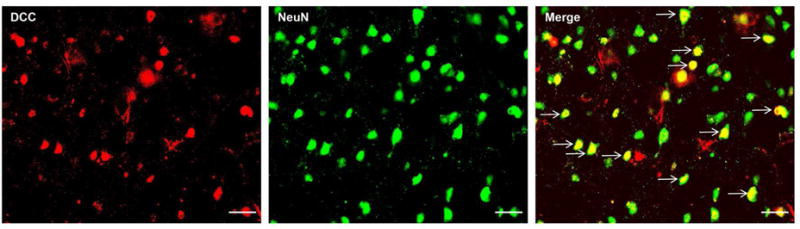
Western blot result showed that elevation of endogenous NTN-1 in the left cerebral cortex started from 12 h, and peaked at 72 h after SAH (p < 0.05, Fig. 3A). Double immunohistochemistry staining revealed that the NTN-1 was mainly expressed in neurons in the cerebral cortex, and the number of NTN-1 positive neurons in the SAH (24 h) group significantly increased compared with that in sham group (Fig. 3B). DCC was also expressed in neurons (Fig. 10) in the cerebral cortex at 24 hours after SAH.
Fig. 3. Expression of endogenous Netrin-1 (NTN-1) after subarachnoid hemorrhage (SAH).
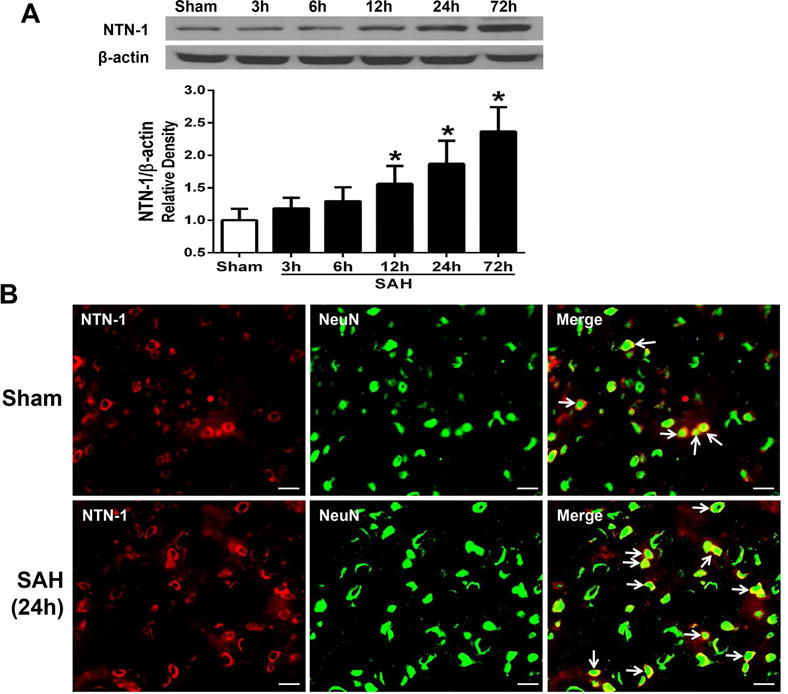
(A) Representative western blot band and quantitative analysis of NTN-1 time course from the ipsilateral cerebral cortex after SAH. The level of endogenous NTN-1 expression increased from 12 h, and peaked at 72 h after SAH. Relative density of NTN-1 protein has been normalized against the sham group. n=6 for each group per time point. *p < 0.05 vs sham. (B) Representative microphotographs of double immunofluorescence staining showed the localization of NTN-1 (red) with NeuN (green) in sham and SAH (24 h) groups. The number of NTN-1 positive neurons significantly increased at 24 h after SAH. n=2 for each group. Scale bar=50 μm
Figure 10.
Cellular localization of DCC receptor after subarachnoid hemorrhage (SAH). Representative microphotographs of double immunofluorescence staining showed that DCC was expressed in neurons. Scale bar=50 μm.
3.3. Effects of rNTN-1 Treatment on Short-Term Neurobehavioral Functions after SAH
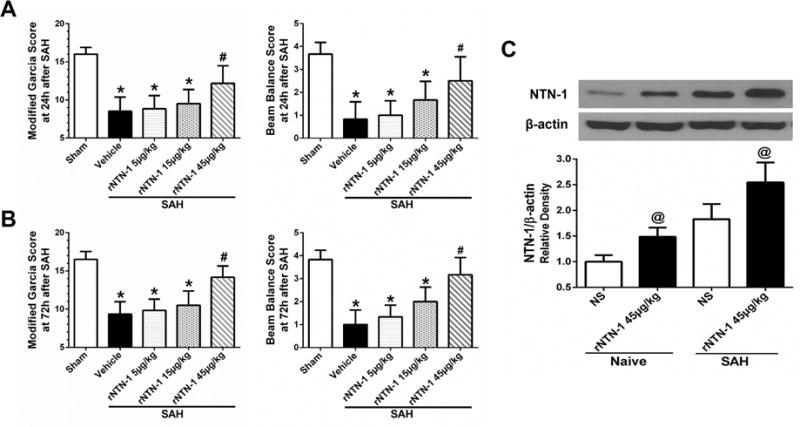
Neurological impairments were evident in vehicle group compared with sham group both at 24 h and 72h after SAH (p < 0.05, Fig. 4A, B). However, post-SAH administration of high dose of rNTN-1 (45 μg/kg) significantly attenuated neurobehavioral deficits both at 24 h and 72 h after SAH when compared with vehicle group and rNTN-1 groups at dose of 5 or 15 μg/kg (p < 0.05, Fig. 4A, B). Based on this finding, we used rNTN-1 (45 μg/kg) for the following studies. In addition, the delivery efficiency of intranasal administration of rNTN-1 (45 μg/kg) was validated by Western blot analysis. Following intranasal administration of rNTN-1, NTN-1 expression in the ipsilateral cortex was significantly increased in the rNTN-1 treatment group compared with NS group at 23 h after rNTN-1 administration (p < 0.05, Fig. 4C), which indicated that intranasal administration of rNTN-1 (45 μg/kg) was successfully delivered into the brain.
Fig. 4. The neuroprotective effects of recombinant Netrin-1 (rNTN-1) at 24 h after subarachnoid hemorrhage (SAH).
(A, B) High dose of rNTN-1 treatment improved neurological deficits both at 24 h and 72h after SAH. n=6 for each group. *p < 0.05 vs sham; #p < 0.05 vs Vehicle, rNTN-1 (5 μg/kg), and rNTN-1 (15 μg/kg). (C) NTN-1 expression in the ipsilateral cortex was significantly increased in the rNTN-1 treatment group, which indicated that intranasal administration of rNTN-1 (45 μg/kg) was successfully delivered into the brain. n=3 for each group. @p < 0.05 vs NS. NS, normal saline
3.4. Effects of NTN-1 siRNA on Short-Term Neurobehavioral Functions after SAH
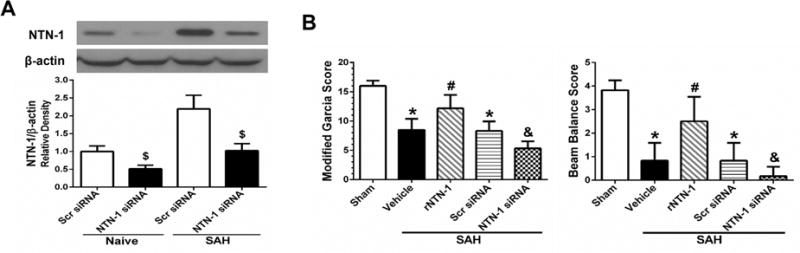
The silencing efficiency of NTN-1 siRNA was confirmed by Western blot. NTN-1 siRNA pretreatment markedly reduced the NTN-1 expression in the ipsilateral cortex, compared with Scr siRNA group at 72 h after injection (p < 0.05, Fig. 5A). In vivo knockdown of endogenous NTN-1 using NTN-1 siRNA remarkably exacerbated neurological impairments at 24 h after SAH, compared with rNTN-1 treatment and Scr siRNA groups (p < 0.05, Fig. 5B).
Fig. 5. The effect of Netrin-1 (NTN-1) siRNA on neurobehavioral functions at 24 h after subarachnoid hemorrhage (SAH).
(A) NTN-1 expression in the cortex was significantly inhibited by NTN-1 siRNA. n=3 for each group. $p < 0.05 vs Scr siRNA. (B) While recombinant Netrin-1 (rNTN-1) treatment improved the neurological outcomes, silencing of endogenous NTN-1 exacerbated neurological impairments. n=6 for each group. *p < 0.05 vs sham, #p < 0.05 vs Vehicle, &p < 0.05 vs rNTN-1 and Scr siRNA. Scr siRNA, scramble siRNA
3.5. Effects of rNTN-1 and NTN-1 siRNA on Neuronal Death after SAH
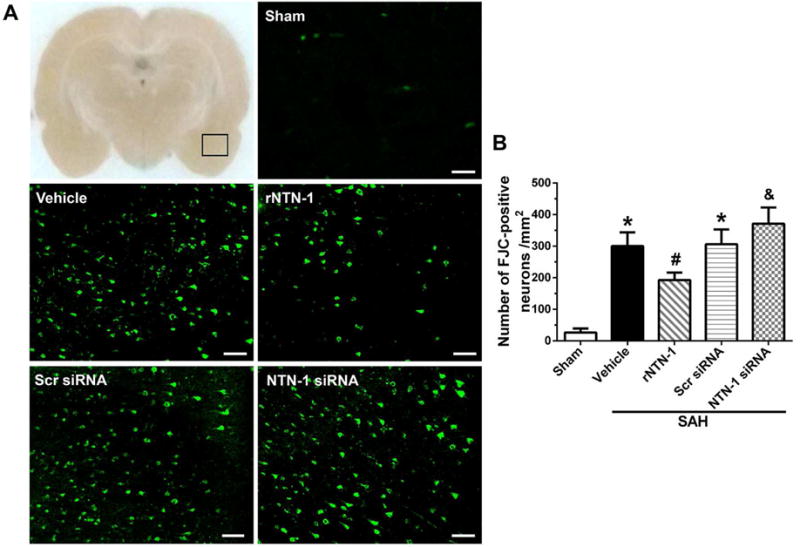
Since SAH results in neuronal degeneration and apoptosis, to test whether rNTN-1 treatment can attenuate those adverse effects, FJC staining was used in this study. As shown in Fig. 6, the numbers of FJC-positive neurons in the ipsilateral cortex increased in vehicle group at 24 h post SAH, when compared to sham group (p < 0.05). However, administration of exogenous rNTN-1 reduced the numbers of FJC-positive neurons in the ipsilateral cortex, when compared to vehicle group at 24h after SAH (p < 0.05, Fig. 6A, B). Conversely, knockdown of endogenous NTN-1 by NTN-1 siRNA significantly aggravated degenerating neurons, when compared with the rNTN-1 treatment and Scr siRNA groups (p < 0.05, Fig. 6A, B).
Fig. 6. The effects of recombinant Netrin-1 (rNTN-1) and NTN-1 siRNA on neuronal death at 24 h after subarachnoid hemorrhage (SAH).
(A) Representative microphotographs of Fluoro-Jade C (FJC)-positive neurons in the ipsilateral cortex. (B) Quantitative analysis of FJC-positive neurons showed that the numbers of FJC-positive neurons remarkably increased at 24 h after SAH. Whereas intranasal administration of exogenous rNTN-1 significantly decreased the numbers of FJC-positive neurons. In contrast, the numbers of FJC-positive neurons markedly increased after NTN-1 siRNA pretreatment. n=4 for each group. *p < 0.05 vs sham, #p < 0.05 vs Vehicle, &p < 0.05 vs rNTN-1 and Scr siRNA. Scr siRNA, scramble siRNA. Scale bar=80 μm
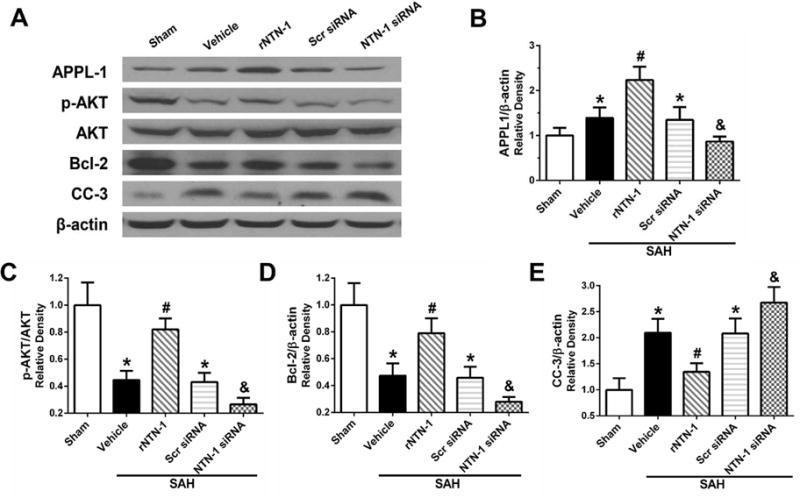
3.6. Effects of rNTN-1 and NTN-1 siRNA on APPL-1, AKT, Bcl-2, and CC-3 Expression after SAH
At 24 h after SAH, the expression of phosphorylated-AKT (p-AKT) and Bcl-2 were remarkably decreased, whereas APPL-1 and CC-3 were increased, when compared to the sham group (p < 0.05, Fig. 7A to E). However, rNTN-1 (45 μg/kg) treatment further enhanced APPL-1 expression, increased p-AKT and Bcl-2 expression, and inhibited CC-3 expression, when compared to vehicle group (p < 0.05, Fig. 7A to E). Silencing of endogenous NTN-1 evidently suppressed APPL-1, p-AKT and Bcl-2 expression, but increased CC-3 expression, when compared with the rNTN-1 treatment and Scr siRNA groups (p < 0.05, Fig. 7A to E).
Fig. 7. Changes in expression of downstream signaling molecules of APPL-1/AKT after recombinant Netrin-1 (rNTN-1) and NTN-1 siRNA at 24 h post subarachnoid hemorrhage (SAH).
Exogenous NTN-1 significantly further augmented APPL-1 expression which contributed to the increase in AKT phosphorylation and Bcl-2 expression (A to D). However, when silenced the endogenous NTN-1 using NTN-1 siRNA, the expression of APPL-1, p-AKT, and Bcl-2 were decreased (A to D). The Bcl-2 level negatively regulated to the apoptotic marker cleaved caspase-3 (CC-3) expression (E) in the ipsilateral cortex. Relative densities of each protein have been normalized against the sham group. n=6 for each group. *p < 0.05 vs sham, #p < 0.05 vs Vehicle, and &p < 0.05 vs rNTN-1 and Scr siRNA. Scr siRNA, scramble siRNA
3.7. Knockdown of APPL-1 Reversed the Anti-apoptotic Property of rNTN-1 after SAH
The knockdown efficacy of APPL-1 siRNA was demonstrated by Western blot. APPL-1 siRNA significantly decreased APPL-1 expression in the ipsilateral cortex (p < 0.05, Fig. 8A). APPL-1 in vivo knockdown markedly aggravated neurological impairments at 24 h post SAH, compared with exogenous rNTN-1 treatment group (p < 0.05, Fig. 8B). Consistently, APPL-1 siRNA pretreatment significantly inhibited expression of p-AKT and Bcl-2, whereas increased CC-3 expression, when compared with rNTN-1 treatment, and rNTN-1 + Scr siRNA groups (p < 0.05, Fig. 8C).
Fig. 8. Knockdown APPL-1 using APPL-1 siRNA abrogated the anti-apoptotic effect of recombinant Netrin-1 (rNTN-1) at 24 h after subarachnoid hemorrhage (SAH).
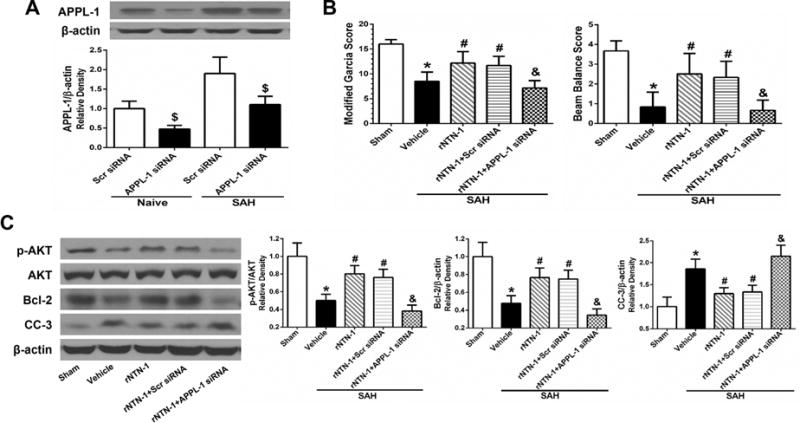
(A) APPL-1 siRNA efficiently knocked down APPL-1 expression in the ipsilateral cortex. n=3 for each group. $P<0.05 vs Scr siRNA. (B) Knockdown of APPL-1 markedly worsened neurological impairments. (C) APPL-1 siRAN abrogated the effects of rNTN-1 on the increase in AKT phosphorylation and Bcl-2 expression, inhibition of CC-3 expression in the ipsilateral cortex. Relative densities of each protein have been normalized against the sham group. n=6 for each group. *p < 0.05 vs sham, #p < 0.05 vs Vehicle, and &p < 0.05 vs rNTN-1 and rNTN-1+Scr siRNA. Scr siRNA, scramble siRNA
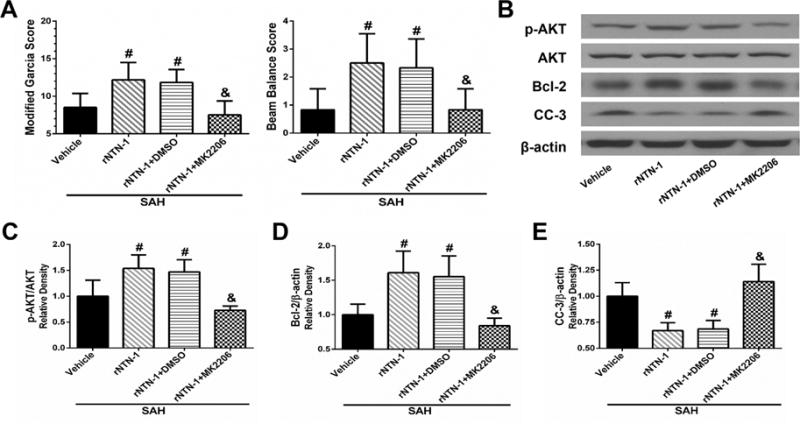
3.8. Inhibition of AKT Abolished rNTN-1-mediated Anti-apoptotic Effect after SAH
To further investigate whether the rNTN-1-mediated anti-apoptotic effect interacts with AKT, AKT inhibitor MK2206 was administered i.c.v. at 1 h before SAH induction followed by rNTN-1 treatment in SAH rats. Pretreatment with MK2206 significantly abolished the protective effect of exogenous rNTN-1 on neurological functions, when compared with rNTN-1 treatment group (p < 0.05, Fig. 9A). Moreover, MK2206 sufficiently decreased p-AKT and Bcl-2 expression, while increased CC-3 expression, when compared with rNTN-1 treatment, and rNTN-1 + DMSO groups (p < 0.05, Fig. 9B to E).
Fig. 9. Inhibition of AKT by MK2206 abolished the protective effects of exogenous recombinant Netrin-1 (rNTN-1) at 24 h after subarachnoid hemorrhage (SAH).
(A) MK2206 significantly aggravated neurological deficits. The effects of rNTN-1 on p-AKT, Bcl-2 protein increase, and CC-3 suppression were abolished by MK2206 (B to E). Relative densities of each protein have been normalized against the sham group. n=6 for each group. *p < 0.05 vs sham, #p < 0.05 vs Vehicle, and &p < 0.05 vs rNTN-1 and rNTN-1+DMSO
4. Discussion
In the present study, we first identified the treatment effect of exogenous rNTN-1 against neuronal apoptosis after SAH, which was at least in part mediated by DCC/APPL-1/AKT signaling pathway. First, we observed that the level of endogenous NTN-1 increased in the left cerebral cortex in the early phase after SAH, mainly expressed in neurons. Second, administration of exogenous rNTN-1 significantly improved neurological functions, concomitant with upregulating APPL-1, p-AKT and Bcl-2 expression and downregulating apoptotic marker CC-3 expression with less FJC-positive neurons. Conversely, knockdown of NTN-1 using NTN-1 siRNA increased neuronal apoptosis, and worsened neurological impairments. Finally, silencing APPL-1 by specific siRNA or inhibiting AKT by MK2206 abolished the protective effects exogenous rNTN-1 on neurological outcome, and neuronal apoptosis by decreasing p-AKT and Bcl-2, and increasing apoptotic marker CC-3.
The importance of EBI in SAH has been well established (Chen et al., 2014). Neuronal apoptosis is characterized as one of the major pathological changes of EBI (Chen et al., 2014; Yin et al., 2016). NTN-1 has been shown as a critical regulator involved in cell apoptosis, morphogenesis, tissue development, and tumorigenesis (Lai Wing Sun et al., 2011; Lu et al., 2012; Mehlen and Furne, 2005; Wilson et al., 2006). With regard to apoptosis, previous studies demonstrated that deletion of NTN-1 significantly contributed to substantial neuronal apoptosis and cell death in NTN-1 knockout mice during brainstem development (Llambi et al., 2001). In contrast, NTN-1 treatment prevented ischemia-induced apoptosis in the brain, heart and kidney via different signaling pathways (Layne et al., 2015; Liao et al., 2013; Wang et al., 2009; Wu et al., 2008). In the present study, we observed that the expression of endogenous NTN-1 was upregulated as early as 12h post SAH. The result was different from the previous observations in the cerebral ischemia model. After ischemia, endogenous NTN-1 expression level did not changed in the infarct zone (Liao et al., 2013; Wu et al., 2008), but it increased from 7 days, and peaked at 14 days in the peri-infarct area (Tsuchiya et al., 2007). We postulated that such discrepancy might be due to differences in animal models, time points, and focus areas, or experimental conditions. As neuronal apoptosis is one of the important pathological process in EBI after SAH, our observations showed that administration of exogenous rNTN-1 significantly increased the anti-apoptotic markers such as p-AKT and Bcl-2 expression, and decreased apoptotic marker CC-3 expression and the number for FJC-positive neurons, and improved the neurological outcomes, while silencing endogenous NTN-1 by specific siRNA increased neuronal apoptosis and aggravated neurological deficits. It is consistent with the findings in ischemic stroke model in which exogenous NTN-1 treatment attenuated neuronal apoptosis (Liao et al., 2013; Wu et al., 2008).
The exact mechanisms of NTN-1- mediated neuroprotection have been remained unclear. DCC, a main receptor of NTN-1, is referred to as dependence receptors which induce apoptosis and cell death if the ligand is absent, but mediate cell survival if the ligand is bound (Bagri and Ashkenazi, 2010; Bredesen et al., 2004). A most recent study revealed that receptor DCC-dependent signaling cascade was required for NTN-1-activated anti-apoptotic function in mesenchymal stem cells (Son et al., 2013). With respect to downstream mediators of DCC, a previous study by Liu et al. (Liu et al., 2002) has revealed that knockdown of endogenous APPL-1 by specific siRNA inhibited DCC-induced apoptosis in colon adenocarcinoma cell, which suggested that APPL-1 was a downstream effector of DCC apoptotic pathway. Additionally, several studies have provided clear evidence that APPL-1 interacts with, and regulates the activity of AKT, which is a central molecule in multiple anti-apoptotic signaling pathways (Lin et al., 2006; Mao et al., 2006; Schenck et al., 2008; Shan et al., 2016; Wang et al., 2012). However, so far little is known about the exact downstream signaling cascade initiated by DCC/APPL-1 in the neurons. In the current study, Western blot analysis showed that following administration of exogenous rNTN-1, DCC-dependent APPL-1/AKT signaling molecule complex were activated, and effectively protected neurons from SAH-induced apoptosis through promoting APPL-1, p-AKT and Bcl-2, as well as inhibiting apoptotic marker CC-3 expression, thereby improved neurological function. In contrast, when APPL-1 were silenced with specific siRNA, anti-apoptotic effects of exogenous rNTN-1 were blocked, due to suppressing APPL-1, AKT phosphorylation and Bcl-2, subsequently increasing CC-3 expression. These results echoed a previous study that NTN-1 stimulated DCC-dependent APPL-1/AKT signaling complex formation, and activated AKT phosphorylation in mesenchymal stem cells (Son et al., 2013). Therefore, our results suggest that activation of receptor DCC-dependent APPL-1/AKT signaling cascade underlies the anti-neuronal apoptotic function of NTN-1 after SAH.
This present study has some limitations. In addition to anti-apoptotic effect, our study cannot rule out the possibility that NTN-1-mediated anti-inflammation and blood-brain barrier preserving may be involved in the neuroprotective role of NTN-1 in EBI after SAH (Podjaski et al., 2015). Additionally, future studies are needed to further explore the long-term outcome of NTN-1 in brain injury after SAH and its possible underlying signaling mechanisms.
In summary, our findings indicated that exogenous recombinant NTN-1 could attenuate neuronal apoptosis and improve neurological outcome after SAH, which was probably mediated by activation of its receptor DCC-dependent APPL-1/AKT signaling cascade and subsequent suppression of apoptotic proteins expression. Thus, NTN-1 may be an attractive candidate for anti-apoptosis treatment in EBI after SAH.
Highlights.
Subarachnoid hemorrhage (SAH) resulted in neuronal apoptosis and neurological deficits in a rat model.
Intranasal administration of exogenous recombinant Netrin-1 (rNTN-1) significantly attenuated neuronal apoptosis and improved neurological functions.
The mechanisms of rNTN-1-mediated anti-apoptosis may be mediated by activating DCC-dependent APPL-1 which is a key regulator for the fundamental cellular processes.
The downstream signaling of NTN-1/DCC/APPL-1 in subarachnoid hemorrhage may be related to AKT, Bcl-2 and Caspase-3.
rNTN-1 may be an attractive candidate for anti-apoptosis treatment in early brain injury after SAH.
Acknowledgments
This study was partially supported by a grant from National Institutes of Health NS081740 and NS082184 to Dr J.H. Zhang, and a grant from Chongqing Natural Science Foundation Project (CSTC2013jcyjA10054), Medical Research Projects of Chongqing Municipal Health Bureau (2013-1-018) to Dr Z.Y. Xie.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- Bagri A, Ashkenazi A. UNCovering the molecular machinery of dependence receptor signaling. Mol Cell. 2010;40:851–853. doi: 10.1016/j.molcel.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Mehlen P, Rabizadeh S. Apoptosis and dependence receptors: a molecular basis for cellular addiction. Physiol Rev. 2004;84:411–430. doi: 10.1152/physrev.00027.2003. [DOI] [PubMed] [Google Scholar]
- Chen S, Feng H, Sherchan P, Klebe D, Zhao G, Sun X, Zhang J, Tang J, Zhang JH. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol. 2014;115:64–91. doi: 10.1016/j.pneurobio.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Hu Q, Xu L, Guo ZN, Ou Y, He Y, Yin C, Sun X, Tang J, Zhang JH. Lipoxin A4 Reduces Inflammation Through Formyl Peptide Receptor 2/p38 MAPK Signaling Pathway in Subarachnoid Hemorrhage Rats. Stroke. 2016;47:490–497. doi: 10.1161/STROKEAHA.115.011223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Xu L, Li B, Guo ZN, Hu Q, Guo Z, Tang J, Chen Y, Zhang Y, Tang J, Zhang JH. Macrophage-Inducible C-Type Lectin/Spleen Tyrosine Kinase Signaling Pathway Contributes to Neuroinflammation After Subarachnoid Hemorrhage in Rats. Stroke. 2015;46:2277–2286. doi: 10.1161/STROKEAHA.115.010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill JC, 3rd, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, Fung GL, Goldstein JN, Macdonald RL, Mitchell PH, Scott PA, Selim MH, Woo D, American Heart Association Stroke, C., Council on, C., Stroke N., Council on Clinical, C. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2015;46:2032–2060. doi: 10.1161/STR.0000000000000069. [DOI] [PubMed] [Google Scholar]
- Holmes RM, Yi Z, De Filippis E, Berria R, Shahani S, Sathyanarayana P, Sherman V, Fujiwara K, Meyer C, Christ-Roberts C, Hwang H, Finlayson J, Dong LQ, Mandarino LJ, Bajaj M. Increased abundance of the adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif (APPL1) in patients with obesity and type 2 diabetes: evidence for altered adiponectin signalling. Diabetologia. 2011;54:2122–2131. doi: 10.1007/s00125-011-2173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Sherchan P, Wang Y, Reis C, Applegate RL, 2nd, Tang J, Zhang JH. Phosphoinositide 3-Kinase Gamma Contributes to Neuroinflammation in a Rat Model of Surgical Brain Injury. J Neurosci. 2015;35:10390–10401. doi: 10.1523/JNEUROSCI.0546-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Wing Sun K, Correia JP, Kennedy TE. Netrins: versatile extracellular cues with diverse functions. Development. 2011;138:2153–2169. doi: 10.1242/dev.044529. [DOI] [PubMed] [Google Scholar]
- Layne K, Ferro A, Passacquale G. Netrin-1 as a novel therapeutic target in cardiovascular disease: to activate or inhibit? Cardiovasc Res. 2015;107:410–419. doi: 10.1093/cvr/cvv201. [DOI] [PubMed] [Google Scholar]
- Liao SJ, Gong Q, Chen XR, Ye LX, Ding Q, Zeng JS, Yu J. Netrin-1 rescues neuron loss by attenuating secondary apoptosis in ipsilateral thalamic nucleus following focal cerebral infarction in hypertensive rats. Neuroscience. 2013;231:225–232. doi: 10.1016/j.neuroscience.2012.11.059. [DOI] [PubMed] [Google Scholar]
- Lin DC, Quevedo C, Brewer NE, Bell A, Testa JR, Grimes ML, Miller FD, Kaplan DR. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol Cell Biol. 2006;26:8928–8941. doi: 10.1128/MCB.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yao F, Wu R, Morgan M, Thorburn A, Finley RL, Jr, Chen YQ. Mediation of the DCC apoptotic signal by DIP13 alpha. J Biol Chem. 2002;277:26281–26285. doi: 10.1074/jbc.M204679200. [DOI] [PubMed] [Google Scholar]
- Llambi F, Causeret F, Bloch-Gallego E, Mehlen P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 2001;20:2715–2722. doi: 10.1093/emboj/20.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wang Y, He X, Yuan F, Lin X, Xie B, Tang G, Huang J, Tang Y, Jin K, Chen S, Yang GY. Netrin-1 hyperexpression in mouse brain promotes angiogenesis and long-term neurological recovery after transient focal ischemia. Stroke. 2012;43:838–843. doi: 10.1161/STROKEAHA.111.635235. [DOI] [PubMed] [Google Scholar]
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- Mehlen P, Furne C. Netrin-1: when a neuronal guidance cue turns out to be a regulator of tumorigenesis. Cell Mol Life Sci. 2005;62:2599–2616. doi: 10.1007/s00018-005-5191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlen P, Guenebeaud C. Netrin-1 and its dependence receptors as original targets for cancer therapy. Curr Opin Oncol. 2010;22:46–54. doi: 10.1097/CCO.0b013e328333dcd1. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Pang J, Chen Y, Kuai L, Yang P, Peng J, Wu Y, Chen Y, Vitek MP, Chen L, Sun X, Jiang Y. Inhibition of Blood-Brain Barrier Disruption by an Apolipoprotein E-Mimetic Peptide Ameliorates Early Brain Injury in Experimental Subarachnoid Hemorrhage. Transl Stroke Res. 2016 doi: 10.1007/s12975-016-0507-1. [DOI] [PubMed] [Google Scholar]
- Podjaski C, Alvarez JI, Bourbonniere L, Larouche S, Terouz S, Bin JM, Lecuyer MA, Saint-Laurent O, Larochelle C, Darlington PJ, Arbour N, Antel JP, Kennedy TE, Prat A. Netrin 1 regulates blood-brain barrier function and neuroinflammation. Brain. 2015;138:1598–1612. doi: 10.1093/brain/awv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan S, Kennedy TE. The netrin protein family. Genome Biol. 2009;10:239. doi: 10.1186/gb-2009-10-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]
- Shan H, Bian Y, Shu Z, Zhang L, Zhu J, Ding J, Lu M, Xiao M, Hu G. Fluoxetine protects against IL-1beta-induced neuronal apoptosis via downregulation of p53. Neuropharmacology. 2016;107:68–78. doi: 10.1016/j.neuropharm.2016.03.019. [DOI] [PubMed] [Google Scholar]
- Shi X, Xu L, Doycheva DM, Tang J, Yan M, Zhang JH. Sestrin2, as a negative feedback regulator of mTOR, provides neuroprotection by activation AMPK phosphorylation in neonatal hypoxic-ischemic encephalopathy in rat pups. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16656201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son TW, Yun SP, Yong MS, Seo BN, Ryu JM, Youn HY, Oh YM, Han HJ. Netrin-1 protects hypoxia-induced mitochondrial apoptosis through HSP27 expression via DCC- and integrin alpha6beta4-dependent Akt, GSK-3beta, and HSF-1 in mesenchymal stem cells. Cell Death Dis. 2013;4:e563. doi: 10.1038/cddis.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H. What is early brain injury? Transl Stroke Res. 2015;6:1–3. doi: 10.1007/s12975-014-0380-8. [DOI] [PubMed] [Google Scholar]
- Tan Y, You H, Wu C, Altomare DA, Testa JR. Appl1 is dispensable for mouse development, and loss of Appl1 has growth factor-selective effects on Akt signaling in murine embryonic fibroblasts. J Biol Chem. 2010;285:6377–6389. doi: 10.1074/jbc.M109.068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong LS, Shao AW, Ou YB, Guo ZN, Manaenko A, Dixon BJ, Tang J, Lou M, Zhang JH. Recombinant Gas6 augments Axl and facilitates immune restoration in an intracerebral hemorrhage mouse model. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16658490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topkoru BC, Altay O, Duris K, Krafft PR, Yan J, Zhang JH. Nasal administration of recombinant osteopontin attenuates early brain injury after subarachnoid hemorrhage. Stroke. 2013;44:3189–3194. doi: 10.1161/STROKEAHA.113.001574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya A, Hayashi T, Deguchi K, Sehara Y, Yamashita T, Zhang H, Lukic V, Nagai M, Kamiya T, Abe K. Expression of netrin-1 and its receptors DCC and neogenin in rat brain after ischemia. Brain Res. 2007;1159:1–7. doi: 10.1016/j.brainres.2006.12.096. [DOI] [PubMed] [Google Scholar]
- Wang W, Reeves WB, Pays L, Mehlen P, Ramesh G. Netrin-1 overexpression protects kidney from ischemia reperfusion injury by suppressing apoptosis. Am J Pathol. 2009;175:1010–1018. doi: 10.2353/ajpath.2009.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YB, Wang JJ, Wang SH, Liu SS, Cao JY, Li XM, Qiu S, Luo JH. Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J Neurosci. 2012;32:11919–11929. doi: 10.1523/JNEUROSCI.3852-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BD, Ii M, Park KW, Suli A, Sorensen LK, Larrieu-Lahargue F, Urness LD, Suh W, Asai J, Kock GA, Thorne T, Silver M, Thomas KR, Chien CB, Losordo DW, Li DY. Netrins promote developmental and therapeutic angiogenesis. Science. 2006;313:640–644. doi: 10.1126/science.1124704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TW, Li WW, Li H. Netrin-1 attenuates ischemic stroke-induced apoptosis. Neuroscience. 2008;156:475–482. doi: 10.1016/j.neuroscience.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Xie Z, Lei B, Huang Q, Deng J, Wu M, Shen W, Cheng Y. Neuroprotective effect of Cyclosporin A on the development of early brain injury in a subarachnoid hemorrhage model: a pilot study. Brain Res. 2012;1472:113–123. doi: 10.1016/j.brainres.2012.06.053. [DOI] [PubMed] [Google Scholar]
- Yan F, Cao S, Li J, Dixon B, Yu X, Chen J, Gu C, Lin W, Chen G. Pharmacological Inhibition of PERK Attenuates Early Brain Injury After Subarachnoid Hemorrhage in Rats Through the Activation of Akt. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-9790-9. [DOI] [PubMed] [Google Scholar]
- Yin C, Huang GF, Sun XC, Guo Z, Zhang JH. Tozasertib attenuates neuronal apoptosis via DLK/JIP3/MA2K7/JNK pathway in early brain injury after SAH in rats. Neuropharmacology. 2016;108:316–323. doi: 10.1016/j.neuropharm.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]