

Abstract
Osteocalcin is an important extracellular matrix bone protein that contributes to the structural properties of bone through its interactions with hydroxyapatite mineral and with collagen I. This role may be affected by glycation, a labile modification the levels of which has been shown to correlate with bone fragility. Glycation starts with the spontaneous addition of a sugar onto a free amine group on a protein, forming an Amadori product, and then proceeds through several environment-dependent stages resulting in the formation of an advanced glycation end product. Here, we induce the first step of this modification on synthetic osteocalcin, and then use multiple mass spectrometry fragmentation techniques to determine the location of this modification. Collision-induced dissociation resulted in spectra dominated by neutral loss, and was unable to identify Amadori products. Electron-transfer dissociation showed that the Amadori product formed solely on osteocalcin’s N-terminus. This suggests that the glycation of osteocalcin is unlikely to interfere with osteocalcin’s interaction with hydroxyapatite. Instead, glycation may interfere with its interaction with collagen I or another bone protein, osteopontin. Potentially, the levels of glycated osteocalcin fragments released from bone during bone resorption could be used to assess bone quality, should the N-terminal fragments be targeted.
Keywords: osteocalcin, bone, glycation, bone matrix, non-collagenous protein
1. Introduction
Osteocalcin (OC) is the most abundant non-collagenous protein found in the extracellular matrix of bone [1]. In humans, it is a 49 amino acid protein with three carboxylated residues (Figure 1) that enable it to bind calcium, giving it a strong affinity for the hydroxyapatite in bone [2]. Osteocalcin is known to influence the morphology of bone mineral and contribute to bone fracture resistance [3; 4; 5]. Studies have shown that osteocalcin localizes in areas where microcracks form in bone, suggesting it is one of the first elements to fail in the production of a bone fracture [4; 5; 6]. Because of the implied physical interaction between the osteocalcin molecule and bone mineral, alterations to the structure of the molecule may interfere with the molecule’s ability to dissipate energy during microcracking and therefore contribute to the deterioration of bone quality. When bone is resorbed, the osteocalcin embedded in bone is released primarily as fragments into circulation [7; 8].
Figure 1.

Osteocalcin sequence annotating areas of interest to this study, highlighting in boxes the potential glycation sites, the trypsin digestion sites by arrows. A shaded “E” signifies a carboxylation site, or a “Gla” residue.
In many long-lived proteins, exposure to sugar results in a non-enzymatic glycation. This process occurs when the open chain form of various sugars (e.g., glucose, ribose) interact with a free amino group on the protein, such as those on lysines, arginines, and the protein n-terminus [9]. The interaction between the amino group and the carboxyl group of the sugar links the two, losing a water molecule and forming a Schiff base. The complex then undergoes a rearrangement to form an Amadori product (Figure 2). From this point, multiple different reactions can occur to the attached sugar to form an advanced glycation end-product (AGE), such as pentosidine, glucosepane, or carboxymethyl lysine [10]. AGEs have been implicated in the deterioration of tissue properties in many different tissues, including bone.[11] AGE crosslinking in collagen has been demonstrated to increase bone fragility [12; 13; 14; 15; 16; 17].
Figure 2.
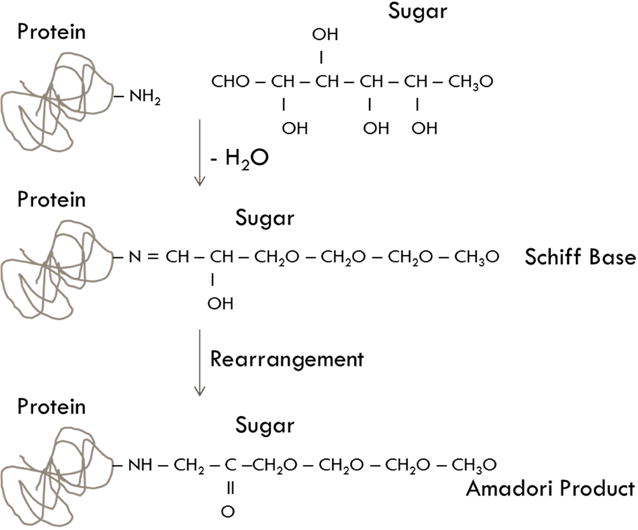
Schematic of the formation of Amadori Products on a protein
OC from both humans and cows has been shown to be glycated in bone, with increases noted in the amount of glycated protein with age [18]. There are potentially five sites that can be glycated on osteocalcin, including any of four arginines and the n-terminus of the protein. Gundberg et al [18]. hypothesized that osteocalcin was glycated at the N-terminus of the protein because the Edman degradation reaction was inhibited for glycated osteocalcin. This result implied that the N-terminal end of glycated osteocalcin was chemically modified, preventing the reaction from occurring. The Edman degradation proceeded uninhibited after glycated osteocalcin was treated with periodate to remove the sugar. Since this landmark study, there have been no further efforts to characterize the modification of OC and determine the potential glycation sites.
Recent advances in mass spectrometry have led to more definitive identifications of AGEs associated with proteins [19; 20; 21; 22; 23; 24]. However, limited application of mass spectrometry to localize the precursors to AGEs, the Amadori products, have been reported [22; 25]. In this study, we glycated osteocalcin in vitro to localize the modification and evaluated the potential sites of glycation.
2. Materials and Methods
2.1 Glycation
To glycate osteocalcin in vitro, synthetic intact human osteocalcin 1–49 (Sigma Aldrich, O5761) was incubated either with Hanks buffered saline (HBS) alone or HBS with 1.43 M ribose or glucose at 60°C for 2 or 4 hours. After incubation, osteocalcin was buffer exchanged to 50 mM ammonium bicarbonate using 5,000 MWCO spin columns (3× washes with 13000 rpm centrifugation) to remove excess sugar and HBS.
1.2 Mass Spectrometry
After incubation and buffer exchange, an aliquot was removed for analysis of intact protein and the remainder was subjected to trypsin digestion at a ratio of 1:100. The intact samples were defrosted and all of the samples were reduced with 10 mM dithiothreitol thenalkylated with 30 mM iodoacetamide for 45 minutes each. For the digested peptides, ribosylated and control osteocalcin was digested overnight at 37°C using Promega Trypsin Gold at a ratio of 1:100.
In preparation for analysis by mass spectrometry, all intact and digested OC were desalted by stage tip [26]. Using a pipet tip loaded with C18 media from a 3M Empore Extraction disk, after binding, protein or peptides were washed with 0.1% formic acid, and then eluted with 80% acetonitrile and 0.1% formic acid.
Intact mass spectrometry analysis was performed on a Thermo Scientific LTQ Orbitrap XL in positive mode with a spray voltage of 4.5 kV and a source temperature of 275 °C. The sheath and auxiliary gas were set to 45 and 20, the FTMS Full AGC target was 200,000, and the Ion Trap MSn AGC target was 10,000 for CID and the FTMS MSn AGC Target for HCD was 100,000. The samples were separated using an Agilent 1200 Series HPLC (high-performance liquid chromatography) system with a BioBasic-18 150 × 2.1 mm column with a particle size of 5 μm (Thermo Scientific). Using a flow rate of 250 uL/min, the samples were eluted using the gradient detailed in Table 1. The instrument was set to fragment the top five most abundant precursor ions using CID for the intact peptides and the top three most abundant precursor ions using HCD for the digested peptides, both using 35% normalized collision energy. Both MS1 and MS2 data had a resolution of 30,000.
Table 1.
Elution through LC column over time, using a flow rate of 250 μL/min through the column.
| Time (minutes) | Percent 0.01% Formic Acid | Percent 70% Acetonitrile + 0.01% Formic Acid |
|---|---|---|
| 0–5 | 98 | 2 |
| 5–35 | 70 | 30 |
| 35–60 | 40 | 60 |
| 60–64 | 5 | 95 |
| 64–75 | 98 | 2 |
The spectra were analyzed manually using Thermo Xcalibur 2.2.38 software specifically for evidence of glycation. Mass shifts of 132.11 for ribose and 162.14 for glucose were used to confirm the presence of in vitro formed glycation. Higher-energy collisional dissociation (HCD) fragmentation was performed concurrently with the above protocol.
Electron-transfer dissociation (ETD) mass spectrometry was performed at the Cornell Biotechnology Resource Center (BRC). The nanoLC-MS/MS analysis was carried out using an Orbitrap Fusion (Thermo-Fisher Scientific, San Jose, CA) mass spectrometer containing a nanospray Flex Ion Source, which is coupled with the UltiMate3000 RSLCnano (Dionex, Sunnyvale, CA). Each reconstituted sample was injected onto a PepMap C-18 RP nano trap column (3 μm, 75 μm × 20 mm, Dionex) with nanoViper Fittings at 20 uL/min flow rate for on-line desalting and then separated on a PepMap C-18 RP nano column (2 μm, 75μm × 15cm), and eluted in a 30 min gradient of 5% to 35% acetonitrile (ACN) in 0.1% formic acid at 300 nL/min, followed by a 5-min ramping to 95% ACN-0.1% FA and a 5-min hold at 95% ACN-0.1% FA. The column was re-equilibrated with 2% ACN-0.1% FA for 20 min prior to the next run.
The Orbitrap Fusion is operated in positive ion mode with nano spray voltage set at 1.6 kV and source temperature at 275 °C. All data are acquired under Xcalibur 3.0 operation software (Thermo-Fisher Scientific) in data-dependent acquisition (DDA) mode using FT mass analyzer for one survey MS scan for selecting precursor ions followed by Top 3 second data-dependent CID-MS/MS and ETD-MS/MS toggle scans for precursor peptides with multiple charged ions (>2 for CID and >3 for ETD) above a threshold ion count of 5000 with normalized collision energy of 30% for CID and 150ms reaction time for ETD.
MS survey scans at a resolution of 120,000 (fwhm at m/z 200), for the mass range of m/z 375–1575, and Q isolation window (m/z) at 1.6 was used for CID-based MS/MS scans in ion trap. Dynamic exclusion parameters were set at 1 and a 40 s exclusion duration with ±10 ppm exclusion mass tolerance. The rapid ion trap scan with 70 ms for maximal injection time and 10,000 AGC target was applied for CID and ETD analysis. Fragmentation data was adapted into graphical form using ProSight Lite.
3. Results
3.1 In vitro Glycation of Osteocalcin
Preliminary studies attempting to replicate in vivo glycation in bone used longer incubation times of up to three weeks at 37°C, but using this technique on osteocalcin alone showed that osteocalcin in solution quickly degrades, making it unusable for mass spectrometry analysis (data not shown). Thus, a shorter incubation time at a higher temperature was chosen. After incubation at 60°C for both two and four hours, a measureable increase in glycated protein was detected although the unmodified protein still dominated the signal from both intact and digested proteins (Figures 3 and 4).
Figure 3.
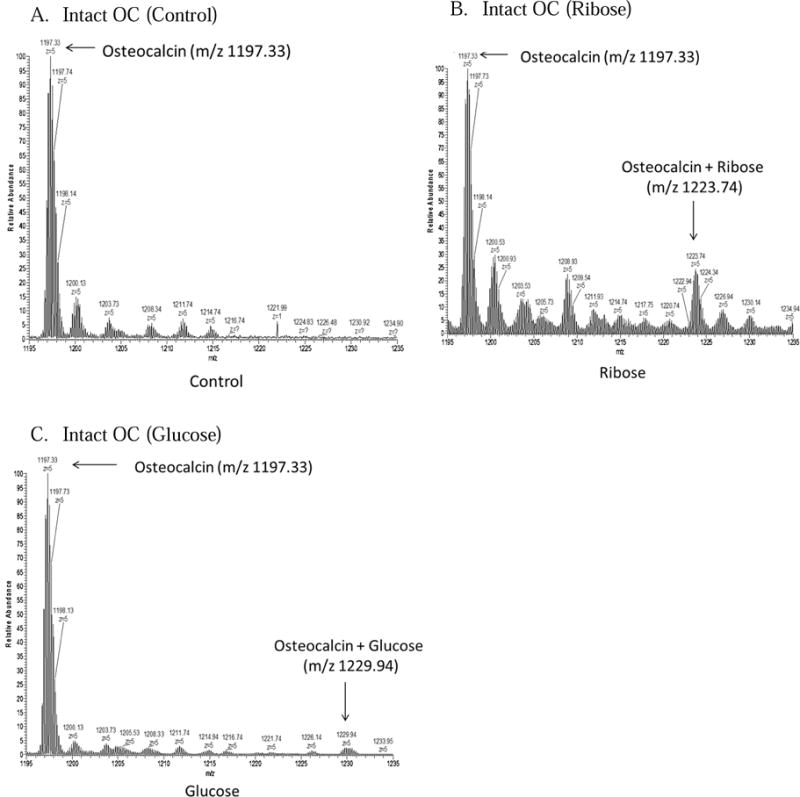
Spectra 3A–C show the areas of interest from LC/MS runs of osteocalcin incubated with buffer alone, glucose, and ribose.
Figure 4.
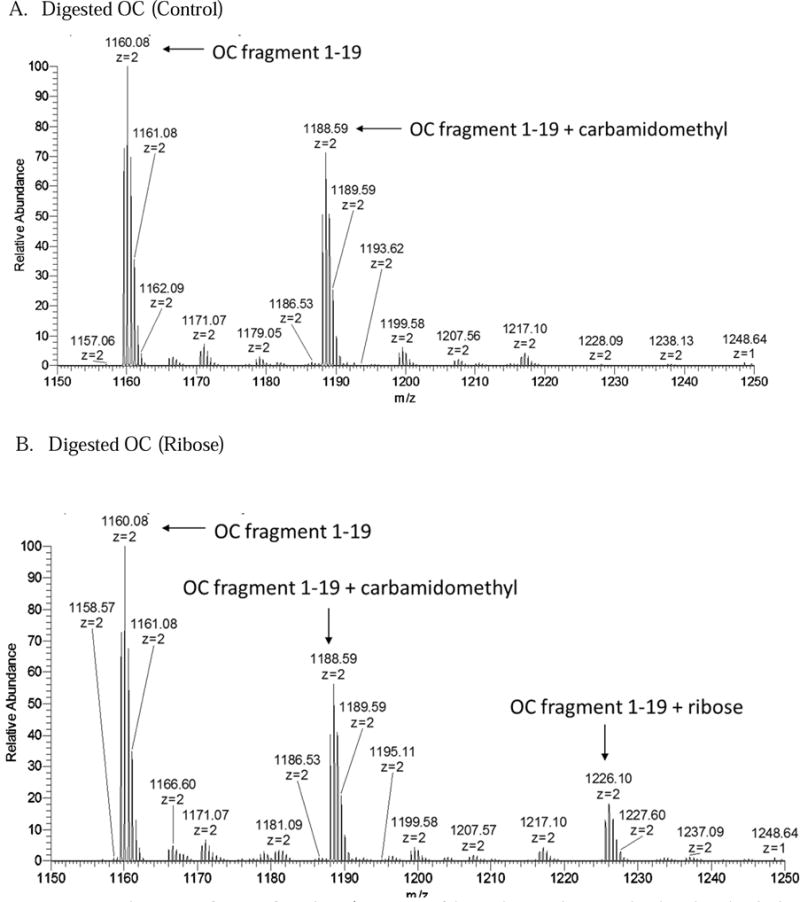
Spectra 4A–B show areas of interest from the LC/MS spectra of digested osteocalcin control and incubated with ribose.
3.2 Intact Synthetic Osteocalcin
Analysis of intact unmodified osteocalcin identified the synthetic osteocalcin as having a mass of 5978.63, which corresponds to the mass of the unmodified protein plus three gamma-carboxylated Glu residues. After incubation with ribose or glucose, we found a distinct mass shift of 132.05 Da for ribose and 163.05 Da for glucose showing formation of the Amadori products (Figure 3). Ribose had a greater relative amount of Amadori formation (20% ribosylated) compared to glucose (2% glycated) (Figure 3). The shifts observed for ribose and glucose corresponded to the weight of the additional sugar, less a water molecule.
3.3 Digested Synthetic Osteocalcin
Digested OC shows similar shifts as the intact protein. Upon digestion, osteocalcin was digested into two pieces: fragment 1–19, or the N-terminal peptide (YLYQWLGAPVPYPDPLEPR), and fragment 20–43 (REVCELNPDCDELADHIGFQEAYR). The fragments that might form from residues 44–49 is are too small to be observed within the range of the instrument. OC 20–43 contains two gamma-carboxylated residues at amino acids 21 and 24, making it difficult to analyze due to its acidity.[27] Spectra from digested control osteocalcin showed a dominant peak of the N-terminal peptide that was carboxylated but unglycated. Furthermore, smaller peaks, corresponding to the peptide with the added carbamidomethyl group on the n-terminus from over-alkylation with iodoacetamide, were also present (Figure 4). The second peptide, OC 20–43, appeared in very low abundance in both singly carboxylated and doubly carboxylated forms; this peptide also retains one of the arginine residues that are typically cleaved by trypsin. The fragmentation of this peptide showed a sequence consistent with a missed trypsin cleavage at arginine 20, a pattern that is sometimes observed in cases of paired arginines and is also shown in the digestion of bovine osteocalcin.[27;28] There was no mass shift on either the singly or doubly carboxylated peptide, suggesting that ribosylation does not occur on any amino acid within the peptide (Figure 5).
Figure 5.
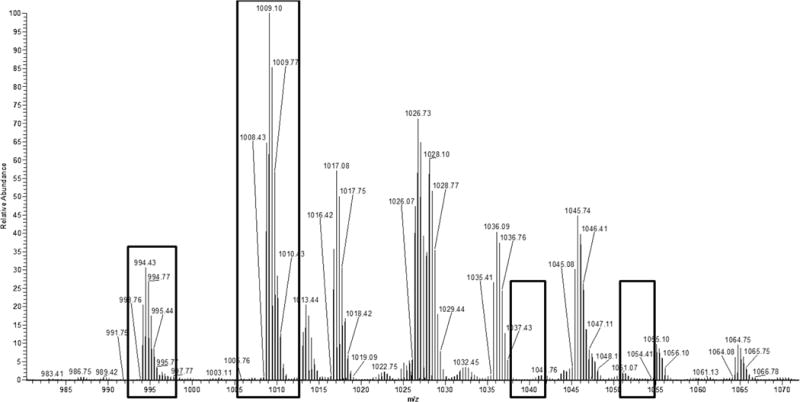
Spectrum showing the presence of the OC 20–42 peptide present in both single and double carboxylations. Empty boxes show areas in the spectrum where peaks would be expected should the peptide possess an Amadori product, which are absent.
The spectrum from the digested ribosylated sample showed that, similar to the intact protein, an N-terminal peptide peak appeared on the spectrum with a shift corresponding to the weight of a ribosylation (Figure 4). HCD MS/MS analysis of the digested sample peaks, corresponding to the added sugar, were attempted to further localize the Amadori modification on the N-terminal peptide. However, the resulting spectra showed that before the peptide itself was fragmented, the Amadori product was removed by the high energy collision. ETD analysis of the same peak was successful in keeping the modification intact. The associated fragmentation pattern showed an added mass of 132.04 on all c-series ions, including the peak corresponding to the n-terminal tyrosine alone (Figure 6). The mass shifted tyrosine peak was absent in control osteocalcin and fragmentations of non-ribosylated peptides in the same sample.
Figure 6.
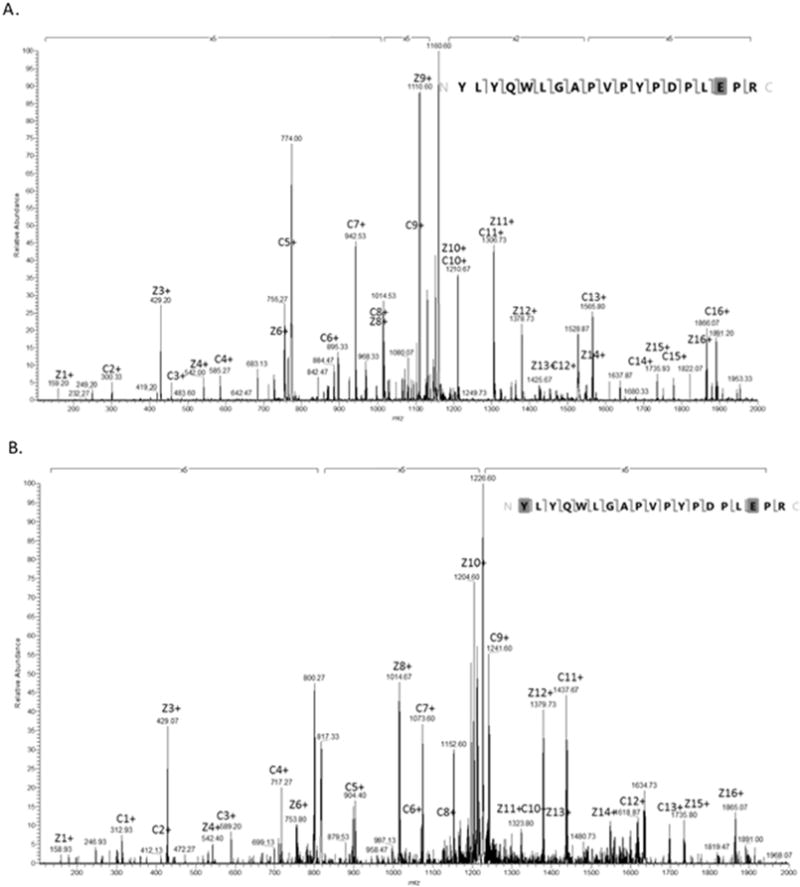
Spectrum (a) shows the fragmentation of the 773.7233 peak, which corresponds to the unmodified OC N-terminal peptide. (b) Shows the annotated spectrum of the 817.7371 peak fragmentation, which corresponds to the weight of the OC N-terminal peptide with a ribosylation. The fragmentation shows c- and z- ions indicating the presence of the ribosylation on the n-terminus of the peptide. Both spectra shown are derived from the ribosylated OC sample.
4. Discussion
Here we show directly the location of glycation on osteocalcin through in vitro processes. In particular, we saw mass shifts on all c-series fragments including C1, placing the glycation at the n-terminus of the peptide. Additionally, ETD allows for localization of the carboxyglutamic acid without neutral loss [27].
Results from the intact and digested ribosylated and glycated OC showed the occurrence of a smaller percent glycation than ribosylation. Thus, to strike a balance between the time spent in incubation to obtain a higher yield but minimizing the amount of osteocalcin permitted to fragment in solution, ribose was chosen for final experiments because of its higher reactivity.
While the LC/MS spectrum without fragmentation cannot show the exact location of a modification, it is capable of identifying that a glycation is present (Figure 4). In this study, the presence of the shifted N-terminal peptide in the digested ribose spectrum demonstrates that the Amadori product must be located on this peptide, on either the n-terminal tyrosine or the arginine residue at position 19. In particular our data alone imply that the protein was glycated at the n-terminus; because it has been shown that glycation can prevent the action of trypsin, the presence of glycated arginine would have prevented the action of the trypsin, preventing the formation of that peptide [23]. The HCD fragmentation of the N-terminal peptide failed to localize the modification; the sugar contains labile bonds that break preferentially to the amino acid backbone, leading to a spectrum dominated by peaks reflecting multiple instances of neutral water losses and making it difficult to obtain site specific glycation information [23; 29; 30]. These results confirm the unsuitability of collision-based fragmentation techniques for localization of Amadori products. While alternative methods like ETD are not necessary to identify the presence of a modified peptide, they are indeed necessary to show a modification’s precise location.
The relative abundances of the peaks for ribosylated, glycated, and control peptides suggest that identification of this modification from a more complex sample would most likely be unfeasible without further purification of the Amadori products (e.g., through boronate chromatography) [18; 31]. Even with in vitro glycation in a simple system with no competing interactions, we found that the relative abundances of the modified peptides are small relative to the amount of unmodified peptide. The in vitro modification results in glycation at the same location as observed in situ by Gundberg et al [18]. Thus, our method is applicable for modeling the glycation of other proteins, and evaluating how their function is impaired over time. The yield of glycated osteocalcin is limited by osteocalcin’s poor stability in solution; however, proteins that are more stable in solution may be capable of longer incubation periods, increasing the relative amount of glycated protein to be analyzed. Previous assessments on the role of glycation on bone health have tended to focus on AGEs rather than Amadori products. Measuring the levels of early glycation/Amadori products, in addition to AGEs, may more comprehensively explain bone fragility associated with this common non-enzymatic process.
Information regarding the site of Amadori product addition to osteocalcin can be used to understand which functions of osteocalcin that are susceptible to modification. Because the N-terminus is distant from the carboxylated residues that make up the binding site of osteocalcin to hydroxyapatite, alteration of the N-terminus is unlikely to interfere with the osteocalcin’s binding affinity. However, the N-terminal end of osteocalcin may factor in its interactions with collagen I or with osteopontin, both of which are involved in the formation of dilatational bands in fracture as shown in Poundarik et al.’s proposed model of the role of osteocalcin in bone fragility fractures [5].
Osteocalcin released from bone during resorption has been shown to exist in intact and fragmented form in circulation [7; 8; 32; 33]. It is present solely in fragmented form in urine, typically consisting of the middle section with truncated C- and N- termini, although some of these fragments do include the N-terminus that was identified as a glycation target in this study. Small fragments containing the N-terminus would also be present that would contain a percentage of glycated peptide [8; 32]. Should N-terminal OC fragments be purified from serum or urine, the level of glycation in those fragments may be useful as an indication of bone fragility.
In conclusion, this study has shown the exact location of the post-translational modification of osteocalcin by glucose and ribose. These results were produced under controlled conditions in vitro and successfully replicated in vivo results. Because osteocalcin has been shown to have a structural role in bone, the glycation of osteocalcin found here is important to characterize and localize.
Acknowledgments
Funding: This work was supported by the National Institute of Health, AR 49635.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg CM, Bradley A, Karsenty G. Increased bone formation in osteocalcin-deficient mice. Nature Letters. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- 2.Hauschka PV, Carr SA. Calcium-Dependent a-Helical Structure in Osteocalcin. Biochemistry. 1982;21:2538–2547. doi: 10.1021/bi00539a038. [DOI] [PubMed] [Google Scholar]
- 3.Boskey AL, Gadaleta S, Gundberg CM, Doty SB, Ducy P, Karsenty G. Fourier Transform Infrared Microspectroscopic Analysis of Bones of Osteocalcin-Deficient Mice Provides Insight Into the Function of Osteocalcin. Bone. 1998;23:187–196. doi: 10.1016/s8756-3282(98)00092-1. [DOI] [PubMed] [Google Scholar]
- 4.Poundarik AA, Gundberg CM, Vashishth D. Non-collageneous proteins influence bone crystal size and morphology: A SAXS study. IEEE. 2011 [Google Scholar]
- 5.Poundarik AA, Diab T, Sroga GE, Ural A, Boskey AL, Gundberg CM, Vashishth D. Dilatational band formation in bone. PNAS. 2012;109:19178–19183. doi: 10.1073/pnas.1201513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikel O, Laurencin D, McCallum SA, Gundberg CM, Vashishth D. NMR investigation of the role of osteocalcin and osteopontin at the organic-inorganic interface in bone. Langmuir. 2013;29:13873–82. doi: 10.1021/la403203w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivaska KK, Hentunen TA, Vaaraniemi J, Ylipahkala H, Pettersson K, Vaananen HK. Release of intact and fragmented osteocalcin molecules from bone matrix during bone resorption in vitro. J Biol Chem. 2004;279:18361–9. doi: 10.1074/jbc.M314324200. [DOI] [PubMed] [Google Scholar]
- 8.Ivaska KK, Kakonen SM, Gerdhem P, Obrant KJ, Pettersson K, Vaananen HK. Urinary osteocalcin as a marker of bone metabolism. Clin Chem. 2005;51:618–28. doi: 10.1373/clinchem.2004.043901. [DOI] [PubMed] [Google Scholar]
- 9.Lapolla A, Traldi P, Fedele D. Importance of measuring products of non-enzymatic glycation of proteins. Clin Biochem. 2005;38:103–15. doi: 10.1016/j.clinbiochem.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Q, Ames JM, Smith RD, Baynes JW, Metz TO. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease. J Proteome Res. 2009;8:754–69. doi: 10.1021/pr800858h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sroga GE, Vashishth D. Effects of bone matrix proteins on fracture and fragility in osteoporosis. Curr Osteoporos Rep. 2012;10:141–50. doi: 10.1007/s11914-012-0103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karim L, Vashishth D. Heterogeneous glycation of cancellous bone and its association with bone quality and fragility. PLoS One. 2012;7:e35047. doi: 10.1371/journal.pone.0035047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ural A, Janeiro C, Karim L, Diab T, Vashishth D. Association between non-enzymatic glycation, resorption, and microdamage in human tibial cortices. Osteoporos Int. 2014 doi: 10.1007/s00198-014-2938-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valcourt U, Merle B, Gineyts E, Viguet-Carrin S, Delmas PD, Garnero P. Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. J Biol Chem. 2007;282:5691–703. doi: 10.1074/jbc.M610536200. [DOI] [PubMed] [Google Scholar]
- 15.Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of Nonenzymatic Glycation on Biomechanical Properties of Cortical Bone. Bone. 2001;28:195–201. doi: 10.1016/s8756-3282(00)00434-8. [DOI] [PubMed] [Google Scholar]
- 16.Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17:1514–23. doi: 10.1007/s00198-006-0155-5. [DOI] [PubMed] [Google Scholar]
- 17.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 18.Gundberg CM, Anderson M, Dickson I, Gallop PM. “Glycated” Osteocalcin in Human and Bovine Bone. The Journal of Biological Chemistry. 1986;261:14557–14561. [PubMed] [Google Scholar]
- 19.Mikulikova K, Eckhardt A, Pataridis S, Miksik I. Study of posttranslational non-enzymatic modifications of collagen using capillary electrophoresis/mass spectrometry and high performance liquid chromatography/mass spectrometry. J Chromatogr A. 2007;1155:125–33. doi: 10.1016/j.chroma.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Bhonsle HS, Korwar A, Kesavan S, Bhosale SD, Bansode S, Kulkarni MJ. “Zoom-In”-A targeted database search for identification of glycation modifications analyzed by untargeted tandem mass spectrometry. European Journal of Mass Spectrometry. 2012;18:475–481. doi: 10.1255/ejms.1203. [DOI] [PubMed] [Google Scholar]
- 21.Brancia FL, Bereszczak JZ, Lapolla A, Fedele D, Baccarin L, Seraglia R, Traldi P. Comprehensive analysis of glycated human serum albumin tryptic peptides by off-line liquid chromatography followed by MALDI analysis on a time-of-flight/curved field reflectron tandem mass spectrometer. J Mass Spectrom. 2006;41:1179–85. doi: 10.1002/jms.1083. [DOI] [PubMed] [Google Scholar]
- 22.Kislinger T, Humeny A, Peich CC, Becker CM, Pischetsrieder M. Analysis of protein glycation products by MALDI-TOF/MS. Ann N Y Acad Sci. 2005;1043:249–59. doi: 10.1196/annals.1333.030. [DOI] [PubMed] [Google Scholar]
- 23.Lapolla A, Fedele D, Reitano R, Arico NC, Seraglia R, Traldi P, Marotta E, Tonani R. Enzymatic digestion and mass spectrometry in the study of advanced glycation end products/peptides. J Am Soc Mass Spectrom. 2004;15:496–509. doi: 10.1016/j.jasms.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 24.Wa C, Cerny RL, Clarke WA, Hage DS. Characterization of glycation adducts on human serum albumin by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chim Acta. 2007;385:48–60. doi: 10.1016/j.cca.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Tang N, Brock JW, Mottaz HM, Ames JM, Baynes JW, Smith RD, Metz TO. Enrichment and analysis of nonenzymatically glycated peptides: boronate affinity chromatography coupled with electron-transfer dissociation mass spectrometry. J Proteome Res. 2007;6:2323–30. doi: 10.1021/pr070112q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 27.Cleland TP, Thomas CJ, Gundberg CM, Vashishth D. Influence of carboxylation on osteocalcin detection by mass spectrometry. Rapid Commun Mass Spectrom. 2016;30:2109–15. doi: 10.1002/rcm.7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thiede B, Lamer S, Mattow J, Siejak F, Dimmler C, Rudel T, Jungblut PR. Analysis of missed cleavage sites, tryptophan oxidation and N-terminal pyroglutamylation after in-gel tryptic digestion. Rapid Commun Mass Spectrom. 2000;14:496–502. doi: 10.1002/(SICI)1097-0231(20000331)14:6<496::AID-RCM899>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q, Frolov A, Tang N, Hoffmann R, van de Goor T, Metz TO, Smith RD. Application of electron transfer dissociation mass spectrometry in analyses of non-enzymatically glycated peptides. Rapid Commun Mass Spectrom. 2007;21:661–6. doi: 10.1002/rcm.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frolov A, Hoffmann P, Hoffmann R. Fragmentation behavior of glycated peptides derived from D-glucose, D-fructose and D-ribose in tandem mass spectrometry. J Mass Spectrom. 2006;41:1459–69. doi: 10.1002/jms.1117. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q, Tang N, Brock JWC, Mottaz HM, Ames JM, Baynes JW, Smith RD, Metz TO. Enrichment and Analysis of Nonenzymatically Glycated Peptides Boronate Affinity Chromatography Coupled with Electron-Transfer Dissociation Mass Spectrometry. Journal of Proteome Research. 2007;6:2323–2330. doi: 10.1021/pr070112q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rehder DS, Gundberg CM, Booth SL, Borges CR. Gamma-Carboxylation and Fragmentation of Osteocalcin in Human Serum Defined by Mass Spectrometry. Molecular and Cellular Proteomics. 2015 doi: 10.1074/mcp.M114.047621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gundberg CM, Weinstein RS. Multiple Immunoreactive Forms of Osteocalcin in Uremic Serum. J Clin Invest. 1986;77:1762–1767. doi: 10.1172/JCI112499. [DOI] [PMC free article] [PubMed] [Google Scholar]