

Abstract
Pulmonary oedema (PO) is a common manifestation of acute heart failure (AHF) and is associated with a high-acuity presentation and with poor in-hospital outcomes. The clinical picture of PO is dominated by signs of pulmonary congestion, and its pathogenesis has been attributed predominantly to an imbalance in Starling forces across the alveolar–capillary barrier. However, recent studies have demonstrated that PO formation and resolution is critically regulated by active endothelial and alveolar signalling. PO represents a medical emergency and treatment should be individually tailored to the urgency of the presentation and acute haemodynamic characteristics. Although, the majority of patients admitted with PO rapidly improve as result of conventional intravenous (IV) therapies, treatment of PO remains largely opinion based as there is a general lack of good evidence to guide therapy. Furthermore, none of these therapies showed simultaneous benefit for symptomatic relief, haemodynamic improvement, increased survival and end-organ protection. Future research is required to develop innovative pharmacotherapies capable of relieving congestion while simultaneously preventing end-organ damage.
Keywords: Pulmonary oedema, afterload, alveolar active signalling, therapies
Acute heart failure (AHF) is a heterogeneous clinical syndrome including diverse phenotypes sharing similar presenting signs and symptoms.[1] The diversity of aetiologies and precipitants of HF and their specific pathophysiologic mechanisms, may result in distinct clinical profiles requiring specific treatment approaches.
Pulmonary oedema (PO) is a common manifestation of AHF associated with a high-acuity presentation and significant haemodynamic abnormalities. PO is defined as alveolar or interstitial oedema verified by chest X-ray and/or with arterial oxygen saturation <90 % on room air accompanied by severe respiratory distress.[2]
Epidemiology
PO is the second most common clinical presentation of AHF syndromes (AHFS), though the prevalence and in-hospital mortality may exhibit substantial geographic variation (see Table 1).[3–10] Moreover, overlapping features among AHF clinical profiles and/or the confounding presence of noncardiac comorbidities may lead to incomplete or inaccurate classification. These patients are often not included in clinical trials due to their disease severity and inability to wait for study medication to be initiated. PO may be a deadly clinical manifestation of HF (see Table 1) and recent experience[11] suggests the vast majority of deaths occurred soon after admission.
Table 1: Prevalence, Demography, Clinical Presentation, In-Hospital Management, and In-Hospital Mortality Rate of Patients Admitted with Pulmonary Oedema in Recent European Heart Failure Registries.
| EHFS II[3] (2004–2005) | HF pilot[4] (2009–2010) | RO-AHFS[5] (2008–2009) | ALARM HF[6] (2006–2007) | AHEAD[7] (2006–2009) | OFICA[8] (2009) | |
| PO prevalence (%) | 16.2 | 13.3 | 28.7 | 37 | 18.4 | 38 |
| Age (years) | 71.2±11 | 72.7±11 | 70.9 ± 10.4 | – | 73.8 | 80 |
| Male (%) | 59.4 | – | 49.7 | – | 55.9 | 48 |
| Ischemic etiology (%) | 55 | 56.5 | 70.8 | 31.8 | 67.4 | 49 |
| LVEF >45 % (%) | 38.4 | 26.7 | 39 | 25.3 | 35 | 42 |
| AFib (%) | 28.1 | 26.3 | 25.1 | 24.7 | 21.4 | – |
| SBP (mmHg) | – | 146±37 | 167.2±43.3 | 138.5+37.1 | 145 (95; 218) | 130 (110–150) |
| HR (beats/minute) | 100 | 97±24 | 103.4±25.3 | 109.5+25.2 | 98 (63; 141) | 88 (73–104) |
| IV diuretics (%) | 94 | 94.5 | 88.6 | 94.9 | – | – |
| IV vasodilators (%) | 72.7 | 62.5 | 73.6 | 50.3 | – | – |
| IV inotropes (%) | 29 | 14.6 | 18.4 | 25 | 20.1 | 8.8 |
| NIMV (%) | 20.5 | – | 1.8 | 10.8 | 9.2 | 15.4 |
| IMV (%) | 11 | – | 7 | 15.8 | 17.1 | – |
| PO–ACM (%) | 9.1 | 5.6 | 7.4 | 7.4 | 7.1 | 7.3 |
ACM = all-cause mortality; AFib = atrial fibrillation; AHEAD = Acute Heart Failure Database; ALARM-HF = Acute Heart Failure Global Survey of Standard Treatment; ESC-HF pilot = European Society of Cardiology – Heart Failure pilot study; EHFS II = European Heart Failure Survey II; HR = heart rate; IV = intravenous; LVEF = left ventricular ejection fraction; NIMV = noninvasive mechanical ventilation; OFICA = French Observational Survey on Acute Heart Failure; PO = pulmonary oedema; RO-AHFS = Romanian Acute Heart Failure Syndromes; SBP = systolic blood pressure.
Pathophysiologic Mechanisms
The pathogenesis of hydrostatic PO has been attributed predominantly to a difference in Starling forces[12,13] (i.e. fluid extravasation attributable to an increased hydrostatic or reduced oncotic pressure gradient across the intact alveolo-capillary barrier). Furthermore, capacity of the lymphatic system to remove fluid from the interstitial space and to drain into the systemic veins is dependent on systemic venous pressure and the integrity of the lymphatics. The clinical picture of PO is dominated by pulmonary congestion due to an acutely increased afterload. Increases in afterload can be the primary mechanism responsible of PO inducing acute cardiac dysfunction and pulmonary congestion, or can be secondary to large cardiac dysfunction (see Figure 1).
Figure 1: Illustration of Pressure-dependent- and Pressure-independent Mechanisms Responsible for Pulmonary Oedema Formation and Resolution.
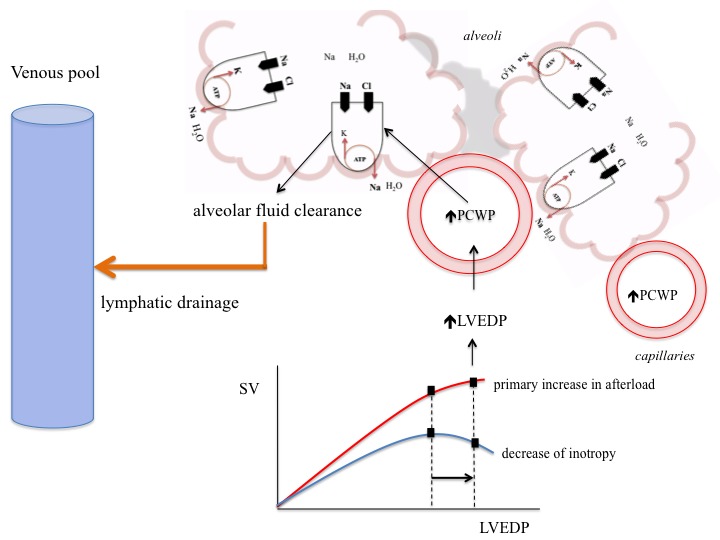
Acute increase in afterload increases fluid transfer across alveolo-capillary membrane. Alveolar epithelial cells are involved in fluid formation and fluid clearance by regulation of sodium and chloride transfer via active signalling processes. Pulmonary oedema resolution depends on active sodium reabsorption as well as on capacity of intact lymphatics to drain fluids out of alveoli into systemic veins. LVEDP = left ventricular end diastolic pressure; PCWP = pulmonary capillary wedge pressure; SV = stroke volume.
In a recent study,[14] the mechanism of PO was described as a consequence of acutely increased afterload in patients with decreased systolic and diastolic capacity to adapt to changes in loading in the presence of maintained right ventricular function. It has been hypothesized that PO patients respond to increased afterload with an increase in heart rate (HR) and peripheral vasoconstriction rather than an acute adaptation in cardiac dimensions.[11] This suggests there are smaller cardiac dimensions and a higher HR and diastolic blood pressure (DBP) in PO patients compared with other profiles.[11]
Peripheral congestion and body weight increases are documented in a relatively small proportion of PO patients, suggesting that relative volume redistribution as opposed to an absolute increase in total body fluid may play a major role in the pathophysiology of the PO phenotype (see Figure 2). The concept of fluid redistribution in PO is further supported by the studies, which have documented that the majority of patients gain little or no weight before heart failure hospitalization, despite the fact that filling pressures rise significantly.[15–17] Sympathetically stimulated reduction in venous capacitance acts to shift volume out of the splanchnic vessels and increase the effective circulating blood volume,[18] leading to increases in preload in the absence of any changes in total body weight.
Figure 2: A. Clinical Signs at Initial Clinical Presentation; B. Time Delay to Presentation in Terms of Symptom Onset.
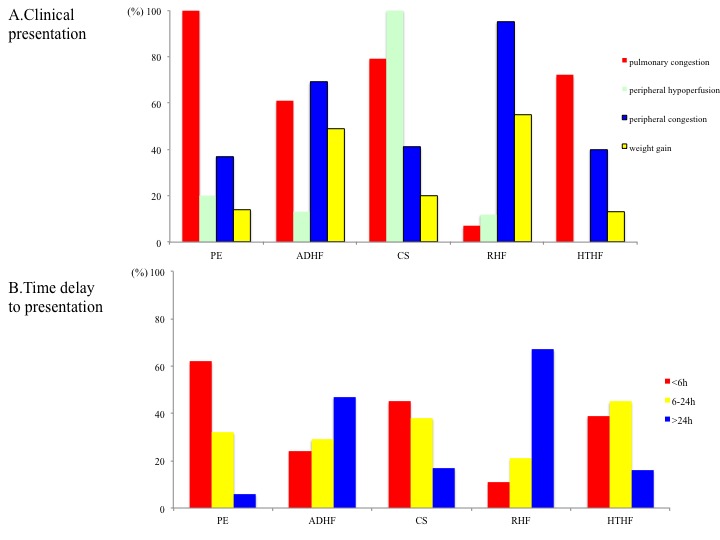
ADHF = acute decompensated heart failure; CS = cardiogenic shock; HTHF = hypertensive heart failure; PO = pulmonary oedema; RHF = right heart failure. Reproduced and modified from Chioncel et al., 2014.[11] Figure 2 was originally published in the Journal of Cardiovascular Medicine as a publish-ahead-of-print version and may be subject to change upon final publication. It is reproduced here with the permission of Wolters Kluwer.
Although pressure-related mechanisms were considered sufficient to explain PO, recent studies show that cardiogenic PO is critically regulated by active signalling processes (see Figure 1), suggesting that endothelial and alveolar responses may contribute critically to the formation of hydrostatic PO. Absorption of excess alveolar fluid is an active process (see Figure 1) that involves transport of sodium out of alveolar air spaces with water following the sodium osmotic gradient. Active transepithelial transport of sodium from the airspaces to the lung interstitial space is a primary mechanism driving alveolar fluid clearance. This mechanism depends on sodium uptake by amiloride-sensitive sodium channels on the apical membrane of alveolar type II cells followed by extrusion of sodium on the basolateral surface by the Na-K-ATPase.[19–22]
A major part of cardiogenic PO formation results from active epithelial secretion of chlorine and secondary fluid flux into the alveolar space.[23] Transepithelial chlorine secretion is triggered by inhibition of epithelial sodium uptake and mediated via cystic fibrosis transmembrane conductance regulator (CFTR) and Na-K-2Cl-co-transporter 1 (NKCC1).[23]
Active sodium transport across the alveolar epithelium is also regulated via basolateral Na-K-ATPase and acute elevation of left atrial pressure inhibits active sodium transport by decreasing the number of Na-K-ATPase at the basolateral membrane of alveolar epithelial cells.[22] Regulation of the Na-K-ATPase is elicited by stimulation of dopaminergic,24 β2 adrenergic receptors,[25,26] and aldosterone, and inhibited by oabain-like compounds.[22,24–26] Furthermore, recent data suggest that lung injury and barrier dysfunction may play a role in the formation and resolution of congestion and pulmonary oedema.[26]
Therapeutic Targets
As PO represents a medical emergency, treatment should be individually tailored to the urgency of the presentation, underlying pathophysiological mechanism, and acute haemodynamic characteristics. Clinical severity must be identified at the time of presentation, since the majority of the deaths occurred on the first day of admission.[11] Systolic blood pressure (SBP) at presentation is one of the most cited predictors of in-hospital mortality in this subset of HF patients (see Table 2), and the recent guidelines recommend that patients with PO can be risk stratified even from the time of diagnosis and subjected to intense monitoring and treatments according to the value of SBP at admission.[27] Beyond decongestion, rapid identification of precipitants and underlying aetiologies is a critical step in management of PO. As reported by registries, clinical scenario (acute myocardial infarction [AMI], cardiomyopathy) is predictive of short-term prognosis (see Table 2).
Table 2: Prognostic Variables for All-cause Mortality in Pulmonary Oedema Patients.
| A. Independent Prognostic Factors Associated with In-hospital Mortality in Observational Studies |
| POPS (n=276 patients)[10] |
| –Low SBP at presentation |
| –WBC count |
| –AMI at presentation |
| –Heart rate |
| ALARM-HF survey (n=1,820 patients)[6] |
| –History of previous cardiovascular event |
| –Cardiomyopathy |
| –Low LVEF |
| –Low SBP |
| –Serum creatinine at presentation |
| –Treatment with diuretics |
| RO–AHFS study (n=924 patients)[11] |
| –Need for inotrope |
| –Need for invasive MV |
| –VFib, sVT during hospitalisation |
| –Acute coronary syndromes at presentation |
| –LBBB at admission |
| –BUN at presentation |
| –Age |
| B. Independent Prognostic Factors Associated with 7–days Mortality in Clinical Trials |
| 3CPO trial (n=1,062 pts)[34] |
| –Ability to obey commands |
| –Low SBP at presentation |
| –Age |
3CPO = 3 Interventions in Cardiogenic Pulmonary Oedema; AMI = acute myocardial infarction; BUN = blood urea nitrogen; LBBB = left bundle brunch block; LVEF = left ventricular ejection fraction; MV = mechanical ventilation; POPS = Pulmonary Oedema Prognostic Score; RBBB = right bundle brunch block; SBP = systolic blood pressure; sVT = sustained ventricular tachycardia; VFib = ventricular fibrillation; WBC = white blood cell count.
The immediate objectives of PO treatment are to improve symptoms, reestablish tissue oxygenation, and stabilize the patient’s haemodynamic condition. Rapid improvement of dyspnea constitutes a major treatment goal and offers an estimate of treatment efficacy. Although dyspnea is directly attributed to pulmonary congestion as result of acute increases in filling pressure, its mechanisms are multifactorial. Overall, imbalance between the neural output of the brain and the resulting work performed by the respiratory muscles may result in dyspnea.[28,29] Sensation of dyspnea can be increased by vascular distension and interstitial oedema, which may directly stimulate pulmonary vascular nerve endings.[30] Furthermore, an abrupt increase in pulmonary congestion directly leads to decreased pulmonary compliance, which further contributes to the tachypnea, tachycardia, and hypoxemia. Although, improved standardisation of dyspnea measurements has been proposed,[31] a large area of uncertainty persists in terms of the factors associated with dyspnea relief.[32] In spite of grading and scoring systems, in current clinical practice, evaluation of symptomatic improvement in the PO clinical profile is largely subjective and often based on clinician judgment.
The clinical picture of PO is dominated by pulmonary congestion along with acutely increased afterload. As relative volume redistribution plays a major role in the pathophysiology of the PO,[13] the therapeutic strategies should focus on transitioning back into the venous reservoir. This aim can be achieved by: 1) reducing pulmonary capillary hydrostatic pressure; 2) increasing lymphatic drainage by reducing systemic venous pressure; or 3) removing or displacing fluid from the alveoli (see Figure 1).
An increase of left ventricular (LV) filling pressure and imbalances in Starling forces may explain transfer of fluids across alveolar-capillary membrane (see Figure 1). Further, the physiologic ability of alveolar epithelial cells to clear alveolar spaces from excess fluid is an active signalling process involving ion channels, mechanosensitive receptors, or adrenoreceptors.[19,20,21,25] Very recent data suggest that targeting mechanisms that prevent barrier dysfunction[26] and augment transepithelial sodium transport and enhance the clearance of alveolar oedema may lead to more effective prevention or treatment for PO.[25,33]
A search for the precipitant and underlying aetiology must be conducted early in the course of hospitalisation. It is important to identify those aetiologies that will not respond to conventional intravenous (IV) therapies, such as mechanical complications of AMI, valvular endocarditis, and prosthetic valve dysfunction. For these patients, mechanical circulatory support or rapid cardiac surgery may be life saving. Precipitant factors responsible for haemodynamic instability and the clinical presentation must also be identified and addressed.
Even if decreasing afterload is the main target for therapeutic intervention in PO patients, in some instances, hypoxemia may be too severe or may not be promptly improved by pharmacotherapies. Mechanical ventilation (MV), either invasive or noninvasive, when appropriately addressed, is an effective measure to correct hypoxemia by displacing fluid out of the alveoli. The need for MV, either invasive or noninvasive, must be anticipated in order to avoid waiting too long or in a place without the proper facilities. Noninvasive MV (NIMV) should be considered as adjunctive therapy in patients with PO who have severe respiratory distress and early initiation of NIMV may potentially correct metabolic abnormalities and reduce the proportion of patients requiring intubation.[34]
In particular circumstances, aggressive treatment of pulmonary congestion may alter the fine balance between end-diastolic pressure and end-diastolic volume, which will negatively impact cardiac output (CO), SBP, and end-organ perfusion (renal, coronary). As a result, some patients may experience worsening of ischemia, life-threatening arrhythmias, or a decline in renal function during hospitalisation. Consequently, in addition to decreasing pulmonary congestion, therapeutic strategies should prevent end-organ damage and not increase the risk for ischemia, propensity for cardiac arrhythmias, and/or permanently alter intrarenal haemodynamics.
Treatment
Although, recent clinical registries[3–8,11] have suggested the majority of patients admitted with PO rapidly improve as a result of conventional IV therapies, treatment of PO remains largely opinion-based as there is a general lack of good evidence to guide therapy.[27] Moreover, most of the trial data are derived from an AHF population that does not resemble PO patients, and thus the data are not entirely generalisable to the PO clinical profile.
Although multicenter, randomized, controlled trials have been conducted in patients with AHF, none of studied agents showed simultaneous benefit for symptomatic relief, haemodynamic improvement, preservation of end-organ function, and increased survival. Furthermore, with the exception of RELAX II[35] and PRONTO,[36] the timing of the interventions was more than 24 hours after presentation, raising relevant questions about the applicability of the results to PO patients.
In clinical practice, acute management of PO is based on IV opiates, diuretics, vasodilators, inotropes, and MV. In approximately 70–80 % of cases, PO patients are treated with combinations of IV therapies.[6,11] Data from recent registries[11] showed there is a specific pattern of utilisation of IV therapies (i.e. high doses for short duration), which may explain the patients’ in-hospital course and adverse outcomes.
Morphine
Morphine is commonly used in the treatment of PO, despite the lack of evidence supporting its efficacy. Morphine is given to cause systemic vasodilatation and alleviate the anxiety related to dyspnea (i.e. ‘air hunger’). In vivo experiments have confirmed that IV morphine results in significant reduction in systemic vascular resistance (SVR) and venous return.[37,38] Morphine may induce respiratory depression[39] that may worsen hypoxemia in a patient already hypoxicemic and altered cerebral perfusion, leading to intubation. Furthermore, data from Acute Coronary Syndrome (ACS) registries[40] and from HF registries[41] suggest that morphine therapy was associated with an increase in mortality, need for intensive care unit (ICU) admission, and intubation. The use of IV diuretics and vasodilators can decrease pulmonary congestion and avoid morphine’s adverse effects on respiratory drive. Morphine may be useful in some patients with PO in the setting of myocardial ischemia when analgesia is required, as morphine reduces anxiety and relieves the pain.
Diuretics
Intravenous loop diuretics are an essential component of PO treatment, and recent guidelines[27] consider IV diuretics as first-line therapy. Analysis of recent registries (see Table 1) shows that IV diuretics are administered to approximately 90 % of patients who are hospitalized for PO. Furosemide was the most commonly used diuretic. Loop diuretics inhibit reabsorption of NaCl and produce natriuresis and diuresis. The diuretic effect occurs 35–45 minutes after IV administration. They act by inhibiting the renal Na/2Cl/K co-transporter in the luminal membrane of the thick ascending limb of loop of Henle, which is responsible for the reabsorption of 35 % of filtered sodium.[42] Of note, furosemide inhibits the same Na/2Cl/K co-transporter in alveolar epithelial cells altering transepithelial chloride secretion and increasing alveolar fluid clearance and oedema resolution.[23]
Furthermore, IV furosemide produces direct venodilation, an effect that can be seen as rapidly as 2–5 minutes after administration. The direct venodilation was inhibited by local indomethacin administration but not by blockade of nitric oxide (NO) synthesis, indicating that the direct vascular venodilation is dependent on local prostaglandin but not on NO production.[43] The rapid reduction in venous return produced by furosemide usually occurs before diuresis and may be therapeutically relevant for obtaining symptomatic improvement in PO. However, the net venodilatory effect of furosemide is difficult to assess, since the decrease of circulating volume produced by furosemide is at the cost of neurohormonal activation.[44] Historically, direct venodilation has been one argument for IV diuretic use in PO since the main pathophysiology of PO is represented by fluid redistribution rather than overall fluid accumulation. The efficacy of loop diuretic use is also balanced by limitations of diuretic resistance, neurohormonal activation, and worsening renal function (WRF).
One major concern with use of IV loop diuretic is the phenomenon of nephron adaptation, which occurs in patients previously exposed to long-term use of oral furosemide. Chronic furosemide treatment may induce ‘nephron adaptation’, respectively a reactive increase in active transcellular sodium transport capacity in the tubular segments situated distally to the site of action of furosemide.[45] This may result in a reduced natriuretic response at conventional IV diuretic doses[45,46] and possibly explain utilisation of higher doses of IV diuretics in PO patients.
There are substantial variations in clinical practice concerning dosing, mode of administration, and duration of therapy, since there is very little Level A, Class I evidence available. A prospective trial comparing four strategies (high versus low dose; bolus versus continuous administration) showed no significant differences in terms of dyspnea improvement or 60-day outcomes.[47] However, the high-dose strategy was associated to a trend with a trend toward greater dyspnea improvement at the cost of transient WRF that did not appear to have long-term consequences. Current guidelines[27] recommend as the first IV furosemide dose to be 2.5 times the existing oral dose in PO patients already taking diuretic.
Vasodilators
According to recent guidelines,[27] vasodilators may be considered as an adjuvant to diuretic therapy for dyspnea relief when SBP remains >110 mmHg. Interestingly, SBP is used as a safety signal and not as marker of efficacy. IV nitroglycerin (NTG) is primarily a venodilator that lowers preload and reduce pulmonary congestion.[48] The hemodynamic benefit of NTG is produced by activation of cyclic guanosine monophosphate (cGMP)-dependent protein kinases.[49]
NTG produces redistribution of blood from the central circulation into larger capacitance veins, decreasing pulmonary venous congestion and decreasing LV impedance, which results in a decrease in left atrial pressure.[50] Concomitant high-dose nitrate therapy and low-dose IV furosemide[51] was associated with a reduced need for intubation and a lower risk of other cardiovascular events compared with high-dose IV furosemide in PO patients. Utilisation of IV nitrates in AHF is limited by reactive neurohormonal activation and tolerance. Nitrate tolerance,[50] defined as a loss of hemodynamic effect despite dose escalation, may develop within hours, and may contribute to the pattern of IV nitrate utilisation observed in registries.
Nesiritide, a recombinant B-type natriuretic peptide (BNP) with vasodilatory properties, is associated to significant decrease of pulmonary capillary wedge pressure (PCWP).[52,53] Compared with NTG, nesiritide resulted in significant decrease of filling pressure but there was no difference between nesiritide and NTG in terms of dyspnea improvement.[54] In the pivotal ASCEND-HF trial,[55] nesiritide showed moderate dyspnea improvement, but neutral effects on mortality and re-hospitalisations and was associated with an increase in rates of hypotension.
Relaxin is a hormone that induces NO activation of guanylate cyclase (GC) and triggers haemodynamic adaptive changes that occur during pregnancy. The recombinant human form, serelaxin, demonstrated potential hemodynamic benefits for AHF patients. In experimental studies,[56,57] serelaxin decreased SVR concomitant to increase (CO) and renal blood flow. In a phase III placebo-controlled trial, which enrolled patients within 16 hours of presentation,[58] serelaxin improved visual analog scale (VAS)-assessed dyspnea, but did not significantly improve dyspnea as measured by a Likert scale. In spite of the lack of benefit on in-hospital mortality, serelaxin significantly improved 180-day all-cause mortality. A recent exploratory analysis of the mortality benefit in RELAX-AHF suggested serelaxin improved markers of cardiac, renal, and hepatic function early during AHF hospitalisation.[59] Hence, improved dyspnea relief, preserved end-organ function, and less use of concomitant IV therapies may support improved long-term outcomes with novel agents in AHF.
Although, some studies have shown that soluble guanylyl cyclase (sGC) activators may offer organ-protective qualities,[60,61] particularly for renal function, a recent study with cinaciguat suggested no clear symptomatic benefit of this therapy.[62] In spite of decreasing PCWP, cinaciguat decreased SBP without improving dyspnea or cardiac index.
Clevipidine, an ultra-short-acting dihydropyridine calcium antagonist with a high degree of vascular selectivity, has been studied in hypertensive AHF in an open-label design study.[36] Clevidipine was initiated very shortly after presentation (i.e. median time 149 minutes) and was associated with marked improvement of dyspnea, lower rates of ICU admissions, and reduced length of stay compared with usual care supporting a new model of intervening within the first few hours after presentation.
Inotropes
The majority of patients admitted with PO have pulmonary congestion related to high LV filling pressures. Although most are hypertensive or normotensive on admission, approximately 10–15 % of PO patients present with low SBP as a result of low CO[6,11] and inotropic agents are required. Other PO patients may experience an unexpected and abrupt decrease of SBP during hospitalisation as result of aggressive treatment of pulmonary congestion or resolution of a reactive stress response. This subset subsequently requires IV inotropes to maintain CO and perfusion pressure. Low SBP at presentation or signs of tissue hypoperfusion, as well as need of inotropic therapies, were all variables associated with short-term mortality in registries enrolling PO patients (see Table 2). The most commonly used inotropes are sympatomimethic agents (i.e. dobutamine and dopamine). These agents have been associated with adverse events such as ischemia, tachyarrhythmias, and hypotension, and may increase in-hospital and postdischarge mortality.[63,64] However, as systemic hypoperfusion in the setting of low CO occurs, sympathomimetic agents remain a mainstay of therapy, despite their associated long-term adverse events, probably mediated through worsening myocardial injury. For sympathomimetic agents, previous treatment with beta-blockers may greatly influence the anticipated clinical response. Other inotropic agents, non-AmpC dependent, have been tested in clinical trials but with unfavourable results. The short-term use of milrinone, an IV inotrope with vasodilatory properties, has been associated with an increase in postdischarge mortality.[65,66] The negative impact on mortality of these agents is considered to be related to myocardial injury as a result of a reduction in SBP and subsequently in coronary perfusion.
Levosimendan exerts positive inotropic effects by enhancing calcium sensitivity of the cardiac contractile elements and exerts direct peripheral vasodilator effects by blocking ATP-dependent potassium channels in vascular smooth muscle.[67] These effects are not attenuated by concomitant treatment with beta-blockers and are sustained beyond the duration of the drug infusion because levosimendan has an active metabolite with a long half-life.[1,63,64,66,67] Levosimendan demonstrated a favorable haemodynamic profile in preclinical and clinical studies[63] as it reduced PCWP and increased CO, suggesting potential benefit for patients with PO with low or normal SBP. However, when tested in rigorous randomized double-blind trials,[68,69] levosimendan showed only modest clinical improvement, and was associated with hypotension, atrial and ventricular arrhythmias, and, in one trial, a trend toward an increase in early mortality.[68]
Istaroxime is a new agent with dual inotropic and lusitropic properties. Istaroxime inhibits Na/K ATPase and stimulates sarcoplasmic reticulum calcium ATPase.[70] In patients hospitalized with HF, istaroxime decreased PCWP and improved diastolic function. Compared with sympathomimetic agents, istaroxime increased SBP and decreased HR and myocardial oxygen demand, characteristics suggesting a possible role in preventing myocardial injury.[71] Although this agent was studied only in chronic HF: reduced ejection fraction (HFrEF), it seems to be an ideal agent for PO patients presenting with low or normal SBP.
Stresscopin, a corticotropin-releasing factor type 2 receptor (CRFR2) selective agonist,[71] has intrinsic inotropic properties via stimulation of G-protein family. In a recent trial,[72] stresscopin increased CO and decreased SVR without significantly affecting HR or SBP, highlighting its potential advantage over standard inotropic agents. However, there was a significant decrease in DBP, which could have an unfathomable effect on coronary perfusion pressure.[72]
Ventilation
The objectives of MV, either invasive or noninvasive, are to improve oxygenation, to reduce work of breathing, to move alveolar and interstitial fluids into capillaries, to reverse respiratory acidosis and hypercapnia, and finally to improve tissue perfusion.
The decision to initiate MV must be anticipated and should be based on clinical judgment, considering the overall clinical picture, but should not be delayed until the patient is in extremis or has an altered level of consciousness.
Even if invasive MV is a life-saving therapy in the care of critically ill patients, it use must be balanced with the potential deleterious cardiac effects.[73] In a PO patient with hypertension, a decrease in preload may be beneficial. However, in a hypotensive patient a decrease in preload may lead to a decrease in CO and SBP. Other potential complications need to be considered, such as barotrauma, and systemic infections.[74] Although the use of invasive MV was predictive of ACM (see Table 2), the prognosis of PO patients treated with MV may depend more on the severity of the hemodynamic perturbation rather than the degree of respiratory failure.[73]
NIMV may be considered as adjunctive therapy in patients with PO who have severe respiratory distress or whose condition does not improve with pharmacologic therapy.[34] In a randomized study[75] early NIMV therapy for PO patients decreased the need for invasive MV and its attendant complications and appeared to augment the response to therapies. PO patients with early NIMV exhibited significantly less respiratory fatigue and translated in lower rate of tracheal intubation. The treatment delay relative to time of hospital presentation may have substantial consequences on patient outcomes such as mortality, need for subsequent tracheal intubation, or subsequent cardiovascular deterioration requiring further medical care. When NIMV fails to improve oxygenation and respiratory acidosis, or encephalopathy worsens, intubation should be considered without delay.
Therapies Targeting Alveolar Ion and Fluid Transport Pathways
Although these agents were studied only in experimental studies, their knowledge may be clinically relevant as some of IV therapies and particular HF background therapies, such as digoxin, mineralocorticoid receptor antagonists, and diuretics may interfere with function of these channels.
Clinical studies show that impaired alveolar ionic and fluid transport mechanisms contribute to the development, severity, and outcome of PO in humans.
β2-adrenergic receptor signalling is required for upregulation of alveolar epithelial active sodium transport in the setting of excess alveolar oedema. The positive, protective effects of β2-adrenergic receptor signalling on alveolar active sodium transport provide substantial support for the use of β2-adrenergic agonists to accelerate alveolar fluid clearance in patients with PO, and some evidence suggests that pharmacologic treatment with β2-adrenergic agonists facilitate recovery from experimental PO.[25,26]
Active sodium transport across the alveolar epithelium is controlled by basolateral Na-K-ATPase.[19,22,25,76] Na-K-ATPase is positively regulated by GCs, aldosterone, catecholamines (β2 and dopaminergic), and growth hormones, and inhibited by oabain-like compounds and mechanical signalling induced by increased left atrial pressure.[19,22,24,25,77] In some experimental studies,[78] aldosterone increased the Na-K-ATPase function and accelerated the clearance of hydrostatic PO, suggesting that aldosterone may be used as a strategy to increase lung oedema clearance.
Transient receptor potential vanilloid receptor 4 (TRPV4) is as one of the most potent agents that inhibit pressure-induced alveolar permeability and it was identified as a promising therapeutic strategy for the treatment of PO.[79,80–82] TRPV4 was linked to elevated pulmonary vascular pressure-mediated Ca2+ uptake by lung endothelium and subsequent acute lung injury.[80,83] TRPV4 is involved in multiple pathways[81,82] including a role in lung vasomotor control, the inflammatory response, and its specific role in resolving PO should be carefully judged.
Conclusions
Patients with PO can be risk stratified from the time of initial presentation and diagnosis, and require intense therapy, which is often dictated by the their initial SBP at admission. However, stratification of patients may improve allocation of resources and focus intensity of care on patients most likely to benefit from early, aggressive therapy. Future research is required to develop innovative pharmacotherapies capable of relieving hemodynamic congestion while simultaneously preserving end-organ function. Targeting the epithelial alveolar cells and active signaling process that produces ion transfer and clearance of fluids from alveolar spaces may accelerate the resolution of PO and/or decrease of the concomitant conventional IV therapies.
References
- 1.Felker GM, Pang PS, Adams KF, et al. Clinical Trials of Pharmacological Therapies in Acute Heart Failure Syndromes. Circulation. 2010;3:314–325. doi: 10.1161/CIRCHEARTFAILURE.109.893222. [DOI] [PubMed] [Google Scholar]
- 2.Dickstein K, Cohen-Solal A, Filippatos G, et al. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM) Eur Heart J. 2008;2008;29:2388–2442. doi: 10.1093/eurheartj/ehn309. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. [DOI] [PubMed] [Google Scholar]
- 3.Nieminen MS, Brutsaert D, Dickstein K, et al. on behalf of the Euro Heart Survey Investigators. Euro Heart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population) Eur Heart J. 2006;27:2725–2736. doi: 10.1093/eurheartj/ehl193. [DOI] [PubMed] [Google Scholar]
- 4.Maggioni AP, Dahlström U, Filippatos G, et al. on behalf of the Heart Failure Association of the ESC (HFA), EURObservational Research Programme: The Heart Failure Pilot Survey (ESC-HF Pilot) Eur J Heart Fail. 2010;12:1076–1084. doi: 10.1093/eurjhf/hfq154. [DOI] [PubMed] [Google Scholar]
- 5.Chioncel O, Vinereanu D, Datcu M, et al. The Romanian Acute Heart Failure Syndromes (RO-AHFS) Registry. Am Heart J. 2011;162:142–153. doi: 10.1016/j.ahj.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 6.Parissis JT, Nikolaou M, Mebazaa A, et al. Acute pulmonary oedema: clinical characteristics, prognostic factors, and in-hospital management. Eur J Heart Fail. 2010;12:1193–1202. doi: 10.1093/eurjhf/hfq138. [DOI] [PubMed] [Google Scholar]
- 7.Spinar J, Parenica J, Vitovec J, et al. Baseline characteristics and hospital mortality in the Acute Heart Failure Database (AHEAD) Main registry. Crit Care. 2011;15:R291. doi: 10.1186/cc10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Logeart D, Isnard R, Resche-Rigon M. on behalf of the working group on Heart Failure of the French Society of Cardiology, Current aspects of the spectrum of acute heart failure syndromes in a real-life setting: the OFICA study. Eur J Heart Fail. 2013;15:465–476. doi: 10.1093/eurjhf/hfs189. [DOI] [PubMed] [Google Scholar]
- 9.Collins SP, Pang PS, Lindsell J, et al. International variations in the clinical, diagnostic, and treatment characteristics of emergency department patients with acute heart failure syndromes. Eur J Heart Fail. 2010;12:1253–1260. doi: 10.1093/eurjhf/hfq133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiutowski M, Waszyrowski T, Krzeminska-Pakula M, et al. Pulmonary edema prognostic score predicts in-hospital mortality risk in patients with acute cardiogenic pulmonary edema. Heart Lung. 2008;37:46–53. doi: 10.1016/j.hrtlng.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Chioncel O, Ambrosy A, Bubenek S, et al. J Cardiovasc Med. (Hagerstown): 2014. Epidemiology, pathophysiology, and in-hospital management of pulmonary edema: data from the Romanian Acute Heart Failure Syndromes registry. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 12.Erdmann AJ 3rd, Vaughan TR, Jr, Brigham KL, et al. Effect of increased vascular pressure on lung fluid balance in unanesthetized sheep. Circ Res. 197;37:271–284. doi: 10.1161/01.res.37.3.271. [DOI] [PubMed] [Google Scholar]
- 13.Clark AL, Cleland JG. Causes and treatment of oedema in patients with heart failure. Nat Rev Cardiol. 2013;10:156–170. doi: 10.1038/nrcardio.2012.191. [DOI] [PubMed] [Google Scholar]
- 14.Margulescu AD, Rimbas RC, Florescu M, et al. Cardiac adaptation in acute hypertensive pulmonary edema. Am J Cardiol. 2012;109:1472–1481. doi: 10.1016/j.amjcard.2012.01.359. [DOI] [PubMed] [Google Scholar]
- 15.Chaudhry SI, Wang Y, Concato J, et al. Patterns of weight change preceding hospitalization for heart failure. Circulation. 2007;116:1549–1554. doi: 10.1161/CIRCULATIONAHA.107.690768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewin J, Ledwidge M, O’Loughlin C, et al. Clinical deterioration in established heart failure: what is the value of BNP and weight gain in aiding diagnosis? Eur J Heart Fail. 2005;7:953–957. doi: 10.1016/j.ejheart.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Zile MR, Bennett TD, St John Sutton M, et al. Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circulation. 2008;118:1433–1441. doi: 10.1161/CIRCULATIONAHA.108.783910. [DOI] [PubMed] [Google Scholar]
- 18.Fallick C, Sobotka PA, Dunlap ME. Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail. 2011;4:669–675. doi: 10.1161/CIRCHEARTFAILURE.111.961789. [DOI] [PubMed] [Google Scholar]
- 19.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82(3):569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 20.Kaestle SM, Reich CA, Yin N, et al. Nitric oxide-dependent inhibition of alveolar fluid clearance in hydrostatic lung edema. Am J Physiol Lung Cell Mol Physiol. 2007;293:L859–L869. doi: 10.1152/ajplung.00008.2007. [DOI] [PubMed] [Google Scholar]
- 21.Filippatos GS, Hughes WF, Qiao R, et al. Mechanisms of liquid flux across pulmonary alveolar epithelial cell monolayers. In Vitro Cell Dev Biol Anim. 1997;33:195–200. doi: 10.1007/s11626-997-0141-z. [DOI] [PubMed] [Google Scholar]
- 22.Azzam ZS, Dumasius V, Saldias FJ, et al. Na,K ATPase overexpression improves alveolar fluid clearance in a rat model of elevated left atrial pressure. Circulation. 2002;105:497–501. doi: 10.1161/hc0402.102848. [DOI] [PubMed] [Google Scholar]
- 23.Solymosi EA, Kaestle-Gembard SM, Vadász I, et al. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci U S A. 2013;110:E2308–16. doi: 10.1073/pnas.1216382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerrero C, Lecuona E, Pesce L, et al. Dopamine regulates Na-K-ATPase in alveolar epithelial cells via MAPKERK-dependent mechanisms. Am J Physiol Lung Cell Mol Physiol. 200;281:L79–L85. doi: 10.1152/ajplung.2001.281.1.L79. [DOI] [PubMed] [Google Scholar]
- 25.Mutlu GM, Snajder JI. Mechanisms of pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:L685–95. doi: 10.1152/ajplung.00247.2005. [DOI] [PubMed] [Google Scholar]
- 26.Alexanian IP, Farmakis D, Korovesi I, et al. Exhaled breath condensate in acute and chronic heart failure: new insights into the role of lung injury and barrier dysfunction. Am J Respir Crit Care Med. 2014;190:342–345. doi: 10.1164/rccm.201402-0272LE. [DOI] [PubMed] [Google Scholar]
- 27.McMurray JJ, Adamopoulos S, Anker G, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J Fail. 2012;14:803–869. doi: 10.1093/eurjhf/hfs105. [DOI] [PubMed] [Google Scholar]
- 28.Manning HL, Schwartzstein RM. Pathophysiology of dyspnea. N Engl J Med. 1995;333:1547–1553. doi: 10.1056/NEJM199512073332307. [DOI] [PubMed] [Google Scholar]
- 29.Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest. 2004;125:669–682. doi: 10.1378/chest.125.2.669. [DOI] [PubMed] [Google Scholar]
- 30.Snashall PD, Chung KF. Airway obstruction and bronchial hyperresponsiveness in left ventricular failure and mitral stenosis. Am Rev Respir Dis. 1991;144:945–956. doi: 10.1164/ajrccm/144.4.945. [DOI] [PubMed] [Google Scholar]
- 31.Mebazaa A, Pang PS, Tavares M, et al. The impact of early standard therapy on dyspnoea in patients with acute heart failure: the URGENT-dyspnoea study. Eur Heart J. 2010;31:832–841. doi: 10.1093/eurheartj/ehp458. [DOI] [PubMed] [Google Scholar]
- 32.Gheorghiade M, Follath F, Ponikowski P, et al. Assessing and grading congestion in acute heart failure: a scientific statement from the acute heart failure committee of the heart failure association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur J Heart Fail. 2010;12:423–433. doi: 10.1093/eurjhf/hfq045. [DOI] [PubMed] [Google Scholar]
- 33.Sartori C, Matthay MA, Scherrer U. Transepithelial sodium and water transport in the lung. Major player and novel therapeutic target in pulmonary edema. Adv Exp Med Biol. 2001;502:315–338. doi: 10.1007/978-1-4757-3401-0_21. [DOI] [PubMed] [Google Scholar]
- 34.Gray A, Goodacre S, Newby DE, et al. Noninvasive ventilation in acute cardiogenic pulmonary edema. N Engl J Med. 2008;359:142–151. doi: 10.1056/NEJMoa0707992. [DOI] [PubMed] [Google Scholar]
- 35.Teerlink JR, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. doi: 10.1016/S0140-6736(12)61855-8. [DOI] [PubMed] [Google Scholar]
- 36.Peacock WF, Chandra A, Collins S, et al. Clevidipine in acute heart failure: Results of the A Study of Blood Pressure Control in Acute Heart Failure–A Pilot Study (PRONTO) Am Heart J. 2014;167:529–536. doi: 10.1016/j.ahj.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 37.Vismara LA, Leaman DM, Zelis R. The effects of morphine on venous tone in patients with acute pulmonary oedema. Circulation. 1976;54:335. doi: 10.1161/01.cir.54.2.335. [DOI] [PubMed] [Google Scholar]
- 38.Grossmann M, Abiose A, Tangphao O, et al. Morphine-induced venodilation in humans. Clin Pharmacol Ther. 1996;60:554–560. doi: 10.1016/S0009-9236(96)90151-4. [DOI] [PubMed] [Google Scholar]
- 39.Dahan A, Aarts L, Smith TW. Incidence, reversal, and prevention of opioid-induced respiratory depression. Anesthesiology. 2010;112:226–238. doi: 10.1097/ALN.0b013e3181c38c25. [DOI] [PubMed] [Google Scholar]
- 40.Meine TJ, Roe MT, Chen AY, et al. The association of intravenous morphine use and outcomes in acute coronary syndromes: Results from the CRUSADE Quality Improvement initiative. Am Heart J. 2005;149:1043–1049. doi: 10.1016/j.ahj.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Peacock WF, Hollander JE, Diercks DB, et al. Morphine and outcomes in acute decompensated heart failure: an ADHERE analysis. Emerg Med J. 2008;25:205–209. doi: 10.1136/emj.2007.050419. [DOI] [PubMed] [Google Scholar]
- 42.Brater DC. Pharmacology of diuretics. Am J Med Sci. 2000;319:38–50. doi: 10.1097/00000441-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 43.Pickkers P1, Dormans TP, Russel FG, et al. Direct vascular effects of furosemide in humans. Circulation. 1997;96:1847–1852. doi: 10.1161/01.cir.96.6.1847. [DOI] [PubMed] [Google Scholar]
- 44.Francis GS, Benedict C, Johnstone DE, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- 45.Kaissling B, Bachmann S, Kriz W. Structural adaptation of the distal convoluted tubule to prolonged furosemide treatment. Am J Physiol. 1985;248:F374–F381. doi: 10.1152/ajprenal.1985.248.3.F374. [DOI] [PubMed] [Google Scholar]
- 46.Loon NR, Wilcox CS, Unwin RJ. Mechanism of impaired natriuretic response to furosemide during prolonged therapy. Kidney Int. 1989;36:682–689. doi: 10.1038/ki.1989.246. [DOI] [PubMed] [Google Scholar]
- 47.Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805. doi: 10.1056/NEJMoa1005419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elkayam U, Akhter MW, Singh H, et al. Comparison of effects on left ventricular filling pressure of intravenous nesiritide and high-dose nitroglycerinin patients with decompensated heart failure. Am J Cardiol. 2004;93:237–240. doi: 10.1016/j.amjcard.2003.09.051. [DOI] [PubMed] [Google Scholar]
- 49.Munzel T, Feil R, Mulsch A, et al. Physiology and pathophysiology of vascular signaling controlled by guanosine 3_,5_-cyclic monophosphate-dependent protein kinase. Circulation. 2003;108:2172–2183. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- 50.Münzel T, Daiber A, Gori T. Nitrate Therapy New Aspects Concerning Molecular Action and Tolerance. Circulation. 2011;123:2132–2144. doi: 10.1161/CIRCULATIONAHA.110.981407. [DOI] [PubMed] [Google Scholar]
- 51.Cotter G, Metzkor E, Kaluski E, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary edema. Lancet. 1998;351:389–393. doi: 10.1016/S0140-6736(97)08417-1. [DOI] [PubMed] [Google Scholar]
- 52.Clarkson PB, Wheeldon NM, Macleod C, et al. Clin Sci. Vol. 88. (Lond): 1995. Brain natriuretic peptide: effect on left ventricular filling patterns in healthy subjects; pp. 159–164. [DOI] [PubMed] [Google Scholar]
- 53.Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. N Engl J Med. 2000;343:246–253. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- 54.Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540. doi: 10.1001/jama.287.12.1531. Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF). [DOI] [PubMed] [Google Scholar]
- 55.O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
- 56.Debrah DO, Conrad KP, Jeyabalan A, et al. Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension. 2005;46(4):745–750. doi: 10.1161/01.HYP.0000184230.52059.33. [DOI] [PubMed] [Google Scholar]
- 57.Conrad KP, Debrah DO, Novak J, et al. Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology. 2004;145:3289–3296. doi: 10.1210/en.2003-1612. [DOI] [PubMed] [Google Scholar]
- 58.Teerlink JR, Metra M, Felker GM, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo controlled, parallel-group, dose-fi nding phase IIb study. Lancet. 2009;373:1429–1439. doi: 10.1016/S0140-6736(09)60622-X. [DOI] [PubMed] [Google Scholar]
- 59.Metra M, Cotter G, Davison BA, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. doi: 10.1016/j.jacc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Tamargo J, Duarte J, Caballero R, Delpon E. Cinaciguat, a soluble guanylate cyclase activator for the potential treatment of acute heart failure. Curr Opin Investig Drugs. 2010;11:1039–1047. [PubMed] [Google Scholar]
- 61.Benz K, Orth SR, Simonaviciene A, et al. Blood pressure-independent effect of long-term treatment with the soluble heme-independent guanylyl cyclase activator HMR1766 on progression in a model of noninflammatory chronic renal damage. Kidney Blood Press Res. 2007;30:224–233. doi: 10.1159/000104091. [DOI] [PubMed] [Google Scholar]
- 62.Gheorghiade M, Greene SJ, Filippatos G, et al. Cinaciguat, a soluble guanylate cyclase activator: results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur J Heart Fail. 2012;14:1056–1066. doi: 10.1093/eurjhf/hfs093. [DOI] [PubMed] [Google Scholar]
- 63.Thackray S, Easthaugh J, Freemantle N, Cleland JG. The effectiveness and relative effectiveness of intravenous inotropic drugs acting through the adrenergic pathway in patients with heart failure—a meta-regression analysis. Eur J Heart Fail. 2002;4:515–529. doi: 10.1016/s1388-9842(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 64.Gheorghiade M, Vaduganathan M, Ambrosy A. Current management and future directions for the treatment of patients hospitalized for heart failure with low blood pressure. Heart Fail Rev. 2013;18:107–122. doi: 10.1007/s10741-012-9315-1. [DOI] [PubMed] [Google Scholar]
- 65.Cuffe MS, Califf RM, Adams KF, Jr, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287:1541–1547. doi: 10.1001/jama.287.12.1541. [DOI] [PubMed] [Google Scholar]
- 66.De Luca L, Mebazaa A, Filippatos G, et al. Overview of emerging pharmacologic agents for acute heart failure syndromes. Eur J Heart Fail. 2008;10:201–213. doi: 10.1016/j.ejheart.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Nieminen MS, Akkila J, Hasenfuss G. Hemodynamic and neurohumoral effects of continuous infusion of levosimendan in patients with congestive heart failure. J Am Coll Cardiol. 2000;36:1903–1912. doi: 10.1016/s0735-1097(00)00961-x. [DOI] [PubMed] [Google Scholar]
- 68.Packer M, Colucci W, Fisher L, et al. Effect of levosimendan on the short-term clinical course of patients with acutely decompensated heart failure. JACC Heart Fail. 2013;1:103–111. doi: 10.1016/j.jchf.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 69.Mebazaa A, Nieminen MS, Packer M, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE randomized trial. JAMA. 2007;297:1883–1891. doi: 10.1001/jama.297.17.1883. [DOI] [PubMed] [Google Scholar]
- 70.Hsu SY, Hsueh AJ. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7:605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- 71.Gheorghiade M, Blair JE, Filippatos GS, et al. Hemodynamic, echocardiographic, and neurohormonal effects of istaroxime, a novel intravenous inotropic and lusitropic agent: a randomized controlled trial in patients hospitalized with heart failure. J Am Coll Cardiol. 2008;51:2276–2285. doi: 10.1016/j.jacc.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 72.Gheorghiade M, Greene SJ, Ponikowski P, et al. Haemodynamic effects, safety, and pharmacokinetics of human stresscopin in heart failure with reduced ejection fraction. Eur J Heart Fail. 2013;15:679–689. doi: 10.1093/eurjhf/hft023. [DOI] [PubMed] [Google Scholar]
- 73.Crane SD, Elliott MW, Gilligan P, et al. Randomised controlled comparison of continuous positive airways pressure, bilevel non-invasive ventilation, and standard treatment in emergency department patients with acute cardiogenic pulmonary oedema. Emerg Med J. 2004;21:155–161. doi: 10.1136/emj.2003.005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cox CE, Martinu T, Sathy SJ, et al. Expectations and outcomes of prolonged mechanical ventilation. Crit Care Med. 2009;37:2888. doi: 10.1097/CCM.0b013e3181ab86ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Plaisance P, Pirracchio R, Berton C, et al. A randomized study of out-of-hospital continuous positive airway pressure for acute cardiogenic pulmonary oedema: physiological and clinical effects. Eur Heart J. 2007;28:2895. doi: 10.1093/eurheartj/ehm502. [DOI] [PubMed] [Google Scholar]
- 76.Factor P. Role and regulation of lung Na,K-ATPase. Cell Mol Biol. 2001;47:347–361. [PubMed] [Google Scholar]
- 77.Pesce L, Comellas A, Sznajder JI. Beta-adrenergic agonists regulate Na-K-ATPase via p70S6k. Am J Physiol Lung Cell Mol Physiol. 2003;285:L802–7. doi: 10.1152/ajplung.00266.2002. [DOI] [PubMed] [Google Scholar]
- 78.Olivera WG1, Ciccolella DE, Barquin N, et al. Aldosterone regulates Na,K-ATPase and increases lung edema clearance in rats. Am J Respir Crit Care Med. 2000;161(2 Pt 1):567–573. doi: 10.1164/ajrccm.161.2.9808050. [DOI] [PubMed] [Google Scholar]
- 79.Vincent F, Duncton AJ. TRPV4 Agonists and Antagonists. Curr Top Med Chem. 2011;1117:2216–2226. doi: 10.2174/156802611796904861. [DOI] [PubMed] [Google Scholar]
- 80.Bakthavatchalam R, Kimball SD. Modulators of Transient Receptor Potential Ion Channels. Annu Rep Med Chem. 2010;45:37–53. [Google Scholar]
- 81.TRPV4: an exciting new target to promote alveolocapillary barrier function. Am J Physiol Lung Cell Mol Physiol. 2014;307:L817–L821. doi: 10.1152/ajplung.00254.2014. [DOI] [PubMed] [Google Scholar]
- 82.Yue Z, Xie J, Yu AS. Role of TRP channels in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2015;308:H157–H182. doi: 10.1152/ajpheart.00457.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]