

Abstract
The past several years have seen dramatic leaps in our understanding of how gene expression is rewired at the translation level during tumorigenesis to support the transformed phenotype. This work has been driven by an explosion in technological advances and is revealing previously unimagined regulatory mechanisms that dictate functional expression of the cancer genome. In this Review we discuss emerging trends and exciting new discoveries that reveal how this translational circuitry contributes to specific aspects of tumorigenesis and cancer cell function, with a particular focus on recent insights into the role of translational control in the adaptive response to oncogenic stress conditions.
Alterations in translational control reflect a powerful means of adjusting the development1–5, maintenance6–9 and overall fitness10–13 of cancer cells. Seminal studies have revealed that nearly all major oncogenic signalling pathways found to be deregulated in human cancers — including RAS–MAPK, PI3K–AKT–mTOR, MYC and WNT–β-catenin — ultimately lead to reprogramming of the genome at the translation level1–5,10 (FIG. 1a). Oncogenic signalling pathways enhance translation initiation largely through stimulation of the eukaryotic translation initiation factor 4F (eIF4F) complex, either directly by altering the expression and phosphorylation of eIF4F complex members3,14–17 or indirectly through regulation of eIF4F complex formation and activity4,9,18–22 (FIG. 1a). In particular, the eIF4E initiation factor has emerged as a crucial node of translational control (BOX 1) that is hyperactivated downstream of oncogenic signalling pathways through enhanced transcription16,17, via phosphorylation downstream of the MAPK-interacting serine/threonine kinases (MNKs)3,14,15 and by mTOR-dependent inactivation of its negative regulators, the tumour-suppressive eIF4E-binding proteins (4EBPs)4,9,18–20,23. Although most translational control is typically attributed to modulations at the level of initiation, substantial regulation also occurs at the stage of translation elongation, and recent work suggests that oncogenic activation of eukaryotic translation elongation factor 2 (eEF2)-dependent translation elongation may be crucial for some cancers5,24 (BOX 1; FIG. 1a).
Figure 1. Oncogenic activation of mRNA translation.

a | A central output of oncogenic MYC, RAS–MAPK, PI3K–AKT–mTOR and WNT–β-catenin signalling pathways is the aberrant activation of mRNA translation at the initiation and elongation steps. Translation initiation, the first step in this process, is considered the primary rate-limiting step of protein synthesis and typically proceeds in a ‘cap-dependent’ manner that relies on the ability of the eukaryotic translation initiation factor 4F (eIF4F) complex to bind to the 5′ 7-methylguanosine cap present on mature mRNAs62. Oncogenic signalling promotes translation initiation predominantly through alterations in the eIF4F complex, which comprises the major cap-binding protein eIF4E, the scaffolding protein eIF4G and the helicase eIF4A. The eIF4F complex drives translation initiation through the ability of eIF4E to bind to the 5′ cap and interact with eIF4G, which recruits the 43S ribosomal pre-initiation complex (comprising a 40S ribosomal subunit, the eIF2–GTP–Met-tRNAiMet ternary complex, eIF3 and several additional accessory factors). Oncogenic signalling can hyperactivate eIF4E through enhanced transcription16,17, through phosphorylation of eIF4E at serine 209 by the MAPK-interacting serine/threonine kinases (MNKs)3,14,15 and through mTOR complex 1 (mTORC1)-dependent phosphorylation and inactivation of the eIF4E inhibitors eIF4E-binding proteins (4EBPs)4,9,18–20,23. Upon recruitment of the 43S complex to the 5′ untranslated region of mRNA, it scans in the 5′ to 3′ direction until reaching a start codon, a process facilitated by eIF4A helicase unwinding of secondary structures and promoted by ribosomal protein S6 kinase (S6K)-dependent stimulation of eIF4A activity through inhibition of programmed cell death protein 4 (PDCD4) and activation of eIF2B21,22. Start codon recognition by the 43S complex is followed by GTP hydrolysis within the ternary complex and joining of the 60S ribosomal subunit to form a translationally competent ribosome. Translation can also be regulated at the elongation stage by oncogenic signalling, largely through S6K-dependent inhibition of eukaryotic translation elongation factor 2 (eEF2) kinase (eEF2K)24,232,233. Phosphorylation of eEF2K by S6K relieves its suppression of eEF2 (REF. 205), promoting the codon by codon translocation of the ribosome along the mRNA. Dashed arrows indicate indirect activation. b | Oncogenic activation of translation initiation and elongation supports tumorigenesis in part by driving selective changes in the translation of specific mRNA transcripts independently of alterations in transcript levels or global increases in protein synthesis. This selective translational control of specific mRNA transcripts underlies the acquisition and execution of distinct cancer cell behaviours central to the transformed phenotype, such as increased cell growth and proliferation5, altered metabolism28, enhanced angiogenesis175, proper reactive oxygen species (ROS) control10, immune cell recruitment3, and invasion and metastasis15,26. CCL, C-C motif chemokine ligand; CCND3, cyclin D3; FTH1, ferritin heavy polypeptide 1; GCLC, glutamate–cysteine ligase catalytic subunit; MTA1, metastasis-associated 1; PRPS2, phosphoribosyl pyrophosphate synthetase 2; RAPTOR, regulatory associated protein of mTORC1; VEGFA, vascular endothelial growth factor A; YB1, Y-box binding protein 1.
Box 1. New insights into translation initiation and elongation in cancer.
Translation initiation is typically considered the primary rate-limiting step of protein synthesis and usually proceeds in a cap-dependent manner that relies on the ability of the eukaryotic initiation factor 4F (eIF4F) initiation complex to bind to the 5′ 7-methylguanosine cap on mature mRNAs62. Historically, eIF4E was identified as the quantitatively limiting factor in the eIF4F complex200,201. As such, even modest increases or decreases in eIF4E expression were expected to profoundly influence mRNA translation and cellular function. In line with this, seminal studies revealed that overexpression of eIF4E is sufficient to drive transformation in cell lines202,203 and spontaneous tumorigenesis in mice204. However, the recent generation of an eIF4E-haploinsufficient mouse has created a paradigm shift in our understanding of the role of eIF4E levels in development and tumorigenesis10. Unexpectedly, eIF4E-haploinsufficient mice were found to be physiologically normal, yet strikingly resistant to tumour formation. This study further revealed that eIF4E is present at levels that exceed those required for normal translational control and development and instead become limiting specifically for expression of the ‘oncogenic translation programme’. These findings challenge the nearly 30-year-old dogma that eIF4E dose is limiting for normal protein synthesis and cellular homeostasis and further delineate how crucial eIF4E expression levels are for cancer development.
Recent studies suggest that translation elongation, like translation initiation, can also be a crucial node of hyperactivation downstream of oncogenic signalling in cancer cells24. Translation elongation is regulated largely at the level of eukaryotic translation elongation factor 2 (eEF2) kinase (eEF2K), which blocks ribosomal translocation along the mRNA by phosphorylating and inhibiting eEF2 (REF. 205). Exciting new work has revealed that inhibition of eEF2K is essential for cellular proliferation in a mouse model of intestinal tumorigenesis driven by oncogenic WNT signalling5. In particular, this study demonstrated that the ability to enhance translation elongation was a crucial output of mTOR complex 1 (mTORC1) signalling, which is predominantly thought to exert translational control at the level of translation initiation. Paradoxically, although this work demonstrates a pro-tumorigenic role for the inhibition of eEF2K activity, certain cancers instead show increased eEF2K activity206,207. This is likely to be due to the crucial role that eEF2K plays in adapting to cellular stress conditions frequently found in tumours (see the section on translational adaption to stress).
This link between translational control and oncogenic signalling has been echoed by an increased appreciation, spanning both basic and translational biology, of a great disconnect between protein abundance and transcriptional activity within cells (reviewed in REF. 25). In this regard, translational control has emerged not merely as a layer of ‘fine-tuning’ but also as a central regulator of gene expression in cancer. However, a crucial question has been to what extent oncogenic signalling pathways modulate protein synthesis in order to cause cellular transformation. Key insights have come from pioneering studies using mice with spontaneous mutations that decrease overall levels of protein synthesis and thus serve as genetic tools to dampen elevated protein synthesis rates in mouse models of cancer development2,4. This work elegantly demonstrated that genetically constraining the ability of oncogenic MYC or AKT signalling to enhance global protein synthesis rates is sufficient to block cell growth, proliferation and tumorigenic potential in mouse models of lymphomagenesis.
Although global translation rates are generally enhanced in cancer cells, it has become increasingly clear that oncogenic signalling also induces transcript-specific changes in translation2,4–7,10,26. For example, the acute inhibition of KRAS and AKT signalling in glioblastoma cells has been shown to cause a rapid decrease in the translation of specific subsets of mRNAs27. Interestingly, no major changes in total transcript levels were found at these early time points, highlighting that altered translational control is a fundamental and immediate response to oncogenic signalling that may precede changes in transcriptional control.
Accumulating data are revealing that selective mRNA translation has a key role in steering distinct aspects of the transformed phenotype. Indeed, recent studies using unbiased translational profiling techniques have identified large groups of functional gene classes that are translationally regulated downstream of oncogenic signalling pathways6,7,10,26. This work has demonstrated that oncogenic regulation of translation supports not only cellular proliferation and growth5, phenotypes strongly associated with increased global protein synthesis rates, but also specific gene expression programmes that drive a diverse array of cancer cell behaviours, including altered cellular metabolism28, metastasis15,26 and resistance to oxidative stress10 (FIG. 1b).
In this Review, we highlight exciting new studies that have begun to unravel the molecular mechanisms underlying translational specificity in expression of the cancer genome and describe an expanding code of cis-regulatory elements that govern post-transcriptional expression of key mRNAs deregulated in cancer. In particular, we discuss regulatory elements embedded in mRNA untranslated regions (UTRs), including both structural and sequence-specific motifs, that have been identified as crucial mediators of translational control in cancer. In addition, we review how codon usage choice and tRNA expression have been shown to be highly biased and distinct for genes with different functions and expression levels in transformed cells. Importantly, although it touches on many different and new aspects of mRNA translation, this Review is not intended to provide a comprehensive or historical account of translational control in cancer, as several excellent reviews have already been published on the subject29–31. Instead, it focuses on recent insights into the mechanistic basis by which cancer cells co-opt specific translational networks that support tumorigenesis, discusses emerging roles for translation in mediating oncogenic cellular stress responses and highlights technological advances that have progressed our understanding of how the landscape of mRNA translation is specifically tuned in cancer cells.
Structural RNA regulatory elements
Some of the first insights into selective mRNA translation came from classic studies demonstrating that the presence of complex secondary structures within the 5′UTR inhibits mRNA translation, which provided a conceptual framework for the mechanistic basis of selective mRNA translation in cancer32,33. Subsequent studies revealed that the overexpression of eIF4E was sufficient to overcome translational limitations imposed by 5′UTR secondary structures34, mainly through the ability of eIF4E to recruit the eIF4A helicase as part of the eIF4F complex and unwind these structures35–38 (FIG. 2a). Indeed, a large body of work has demonstrated a key role for eIF4A in promoting the translation of mRNAs with structured 5′UTRs and uncovered many underlying principles of eIF4A regulation and activity (reviewed in REF. 39). Importantly, complex 5′UTR structures have been identified in several pro-tumorigenic mRNAs that are translationally induced in an eIF4E-dependent manner, including the metabolic enzyme ornithine decarboxylase (ODC), as well as fibroblast growth factor 2 (FGF2) and the pro-angiogenic factor vascular endothelial growth factor A (VEGFA)40–42. In line with this, pharmacological inhibitors of eIF4A, such as silvestrol, display potent antitumour activity in preclinical mouse models of cancer6,43–45. However, the full impact of 5′UTR structures on translation of the cancer genome has remained largely unresolved.
Figure 2. mRNA regulatory elements direct specialized translation of the oncogenic programme.

Structural and sequence-specific regulatory elements underlie the code for selective control of the translation of specific pro-tumorigenic mRNAs. a | General secondary structure in the 5′ untranslated region (5′UTR) can steer selective translation of mRNAs, such as the metabolic enzyme ornithine decarboxylase (ODC)40, by conferring enhanced sensitivity to eukaryotic initiation factor 4F (eIF4F) complex formation, which promotes unwinding of these structures through the eIF4A helicase. b | Likewise, mRNAs with G-quadruplexes in their 5′UTR, such as the super-enhancer-associated transcription factor ETS1, selectively require eIF4A helicase activity for their translation6. c | Transcripts containing a structural element called an internal ribosome entry site (IRES), such as the anti-apoptotic BCL2 (REF. 50), can be selectively translated in a cap-independent manner by directly recruiting the ribosome to the 5′UTR, frequently with the aid of IRES trans-acting factors (ITAFs). d | Many structural elements confer translational specificity through interactions with distinct RNA-binding proteins (RBPs). For example, the epithelial–mesenchymal transition regulator disabled homologue 2 (DAB2) contains a 3′UTR transforming growth factor-β (TGFβ)-activated translation (BAT) element that recruits RBPs to form a complex that blocks translation elongation59,60. e | Structural elements in the 5′UTR have even been shown to interact with eIF3, which non-canonically binds RNA in a manner dependent on the presence of distinct stem–loops. For example, direct interaction of eIF3 with a stem–loop in the 5′UTR of the mRNA encoding the mitotic regulator JUN promotes its translation61. f | One of the shortest sequence-specific elements in the 5′UTR is an alternative translation start site, typically comprising an upstream AUG (uAUG) or, more rarely, a non-AUG codon. These upstream start codons are in-frame with the primary open reading frame (ORF) and can drive the expression of protein isoforms with novel functions, such as the long form of the tumour suppressor PTEN (PTEN-long), the translation of which starts from an upstream CUG codon64. g | Upstream ORFs (uORFs) are discontiguous from the primary ORF and typically act to inhibit translation, as has been shown for the growth factor receptor HER2 (REF. 74). However, under certain conditions uORFs can act to promote translation (see FIG. 4). h | MicroRNAs (miRNAs) recognize complementary sequences in target mRNAs and guide transcript-specific translational repression as part of the miRNA-induced silencing complex (miRISC), as has been shown for let-7 regulation of the RAS oncogenes98. Cancer cells can reduce their 3′UTR length through alternative cleavage and polyadenylation (APA) to avoid miRNA-mediated regulation85. i | Many RBPs recognize sequence-specific RBP-binding domains (RBDs) in the 3′UTR to regulate transcript-specific translation, as has been shown for the RBP Musashi homologue 2-induced translation of the self-renewal factor Ikaros family zinc finger protein 2 (IKZF2)110. j | There is recent and growing appreciation of the presence of numerous sequence-specific elements in the 5′UTR, including the translation inhibitory element (TIE)118, pyrimidine-rich translational element (PRTE)26,28 and cytosine-enriched regulator of translation (CERT)10, all of which can direct selective cap-dependent translation of distinct transcripts that influence tumour development and progression. Although the underlying molecular mechanisms by which these elements exert translational control remains to be elucidated, they may function in part by recruiting specific trans-acting factors (light blue circles labelled ‘?’) that interact directly or indirectly with the translation initiation complex. FTH1, ferritin heavy polypeptide 1; PRPS2, phosphoribosyl pyrophosphate synthetase 2.
Recent progress in understanding how 5′UTR structure influences genome-wide translation in cancer has been made by ribosome-profiling experiments surveying the effects of eIF4A inhibition by silvestrol in tumour cells6,7. Strikingly, this work has not only validated the well-established ability of eIF4A to direct the translation of mRNAs with generally complex 5′UTR secondary structures but has also identified a role for smaller structural domains of 9–12 nucleotides, including the G-quadruplex, in conferring sensitivity to eIF4A helicase activity (FIG. 2b). Intriguingly, eIF4A inhibition in breast cancer cells was largely found to affect the translation of cell cycle, cell morphology and cell survival genes, whereas T cell acute lymphoblastic leukaemia (T-ALL) cells displayed reduced translation of mRNAs functionally enriched for transcription factors, oncogenes and super-enhancer-associated genes6,7. Although the broader functional role of the G-quadruplex and related structural motifs remain to be validated, these findings suggest that smaller structural domains within 5′UTRs may also steer specific cancer cell behaviours.
One of the most extensively studied 5′UTR structural elements able to direct transcript-specific translation is the internal ribosome entry site (IRES). IRES-dependent translation is thought to fine-tune gene expression by promoting selective cap-independent mRNA translation (FIG. 2c), particularly during distinct cellular conditions in which cap-dependent translation is inhibited, such as during mitosis46, megakaryocyte differentiation47 and cellular stresses such as DNA damage48. A role for IRES-dependent translation in cancer has been best characterized for tumour-associated stress conditions such as hypoxia (discussed further below); however, several studies suggest a more complex role for IRES-dependent translation during cancer. For example, IRES-dependent translation has been shown to drive the expression of proteins with contrasting cellular functions, such as the pro-apoptotic protein apoptotic peptidase-activating factor 1 (APAF1)49 and the anti-apoptotic protein BCL-2 (REF. 50), as well as oncoproteins such as MYC51 and tumour suppressors such as p53 and p27 (REFS 52–54). Although approximately 10% of all mRNAs have been predicted to contain IRES elements55, the functional role of IRES elements in many cellular mRNAs remains to be studied. Moreover, the impact of IRES-driven translation on gene expression in cancer is largely unknown. The potential diversity of cellular functions regulated by IRES-dependent translation may reflect a role for distinct structural features of the IRES as well as associated IRES trans-acting factors (ITAFs) in directing the expression of specific mRNA classes. New technologies to study these structural features (for example, selective 2-hydroxyl acylation analysed by primer extension (SHAPE) in the case of IRES and photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) for ITAFs; TABLE 1) are likely to fuel future progress in understanding their role during tumorigenesis.
Table 1.
Technologies for studying post-transcriptional control of the cancer genome
| Technology | Variations | Description |
|---|---|---|
| Translational profiling | Polysome microarray | Translational profiling that uses classic microarray technology to analyse mRNAs associated with actively translating ribosomes isolated by sucrose gradient fraction234 |
| Ribosome profiling | Deep sequencing of ribosome-protected mRNA fragments as a genome-wide measure of translation with subcodon resolution235 | |
| Proximity-specific ribosome profiling | Ribosome profiling using a spatially restricted biotin ligase and biotin-acceptor-tagged ribosomes as a strategy to look at proximity-specific translation236 | |
| RNA–protein interaction network analysis | Crosslinking and immunoprecipitation followed by high-throughput sequencing (CLIP-Seq) | Also known as high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP), a method that uses high-throughput sequencing of RNAs isolated by UV crosslinking and immunoprecipitation to map genome-wide binding sites of a given protein within a 30-nucleotide region of RNA237 |
| Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) | An alternative to CLIP-Seq that uses PAR analogues, such as 4-thiouridine, added to cell culture to enable enhanced crosslinking and precise mapping of RBP-binding sites by scoring for crosslinking-induced thymidine to cytidine transitions238 | |
| Individual-nucleotide-resolution UV crosslinking and immunoprecipitation (iCLIP) | An adaptation of CLIP-Seq that enables improved individual-nucleotide resolution by using an adaptor that exploits the tendency of cDNAs to prematurely truncate before the crosslinked nucleotide239 | |
| RBP interactome capture | A technique using either UV or PAR crosslinking of RNA–protein interactions followed by oligo(dT) purification of mRNAs and mass spectrometry (MS) to identify the full repertoire of RBPs within a cell113 | |
| Comprehensive identification of RBPs by MS (ChIRP-MS) | A method for identifying the specific subset of proteins associated with a particular RNA using formaldehyde crosslinking of protein–RNA interactions, followed by RNA purification using tiled biotinylated probes specific for the RNA of interest, and MS to identify bound proteins240 | |
| RNA antisense purification with MS (RAP-MS) | An approach similar to ChIRP-MS that uses UV crosslinking and stable isotope labelling by amino acids in culture (SILAC) to perform quantitative MS of RNA–protein interactions241 | |
| RNA–RNA interaction network analysis | Crosslinking, ligation and sequencing of hybrids (CLASH) | A high-throughput method to identify RNA–RNA interactions associated with mRNPs based on purification of UV-crosslinked complexes, intra- and intermolecular ligation of RNA–RNA interactions, and sequencing of hybrid RNAs242 |
| RNA structure analysis | Parallel analysis of RNA structure (PARS) | A method for analysing RNA structure genome-wide at single-nucleotide resolution by parallel deep sequencing of RNA fragments generated by treatment with various structure-specific endonucleases243 |
| Fragmentation sequencing (Frag-Seq) | Genome-wide analysis to identify RNA structure based on high-throughput sequencing of RNA fragments produced by the cleavage of single-stranded nucleic acids by nuclease P1 (REF. 244) | |
| Selective 2-hydroxyl acylation analysed by primer extension (SHAPE) | This standard analysis of RNA secondary structure, which uses primer extension to measure 2′-hydroxyl group reactivity with chemical probes, has recently been improved by the development of new probes enabling its application in living cells245 | |
| SHAPE-Seq | A high-throughput version of SHAPE that enables genome-wide analysis of RNA structure by using multiplexed paired-end deep sequencing of primer extension products246 | |
| In vivo click-SHAPE (icSHAPE) | An adaptation of SHAPE that combines genome-wide profiling with in vivo click selective 2′-hydroxyl acylation to overcome previous limitations in chemical probes that precluded analysis of all four nucleotides247 | |
| RNA hybrid and individual-nucleotide-resolution UV CLIP (hiCLIP) | A modified iCLIP procedure that is similar to CLASH but which uses an adaptor in the ligation step enabling it to better resolve RNA–RNA interactions and predict secondary structure248 | |
| RNA modification profiling | m6A profiling | Techniques such as Me-RIPseq134 and m6A-Seq136 measure N6-methyladenosine modifications on RNA by high-throughput sequencing of transcripts captured using an antibody specific for the modification |
| m5C profiling | The recent development of affinity purification-based high-throughput sequencing methods for profiling RNA 5-methylcytosine modifications — such as 5-azacytidine RNA immunoprecipitation (Aza-IP)249 and methylation individual-nucleotide-resolution CLIP (miCLIP)250 — provide some advantages over bisulfite sequencing approaches251, such as the enhanced ability to detect modification on less abundant RNAs | |
| Pseudo-Seq, PSI-Seq and Ψ-Seq | High-throughput sequencing methods for genome-wide identification of RNA pseudouridylation using CMC to modify pseudouridines and generate a block in reverse transcription137,139,140 | |
| N3-CMC-enriched pseudouridine sequencing (Ceu-Seq) | An approach similar to Pseudo-Seq that uses a CMC derivative modified by click chemistry to add biotin, enabling the enrichment of pseudouridine-containing RNAs by biotin pulldown before sequencing138 | |
| Quantitative MS | Optimized MS approaches have been developed for measuring pseudouridine levels at select rRNA sites226, for determining global changes in the full range of tRNA modifications252 and for evaluating global changes in nucleoside modifications across all species of RNA253 | |
| tRNA profiling | tRNA microarray | Chemical ligation-based microarrays have been developed for profiling tRNA isoacceptors at single-nucleotide resolution141, improving on previous hybridization-based microarrays that can only distinguish tRNAs differing by at least eight nucleotides150 |
| tRNA high-throughput sequencing | Recent adaptations of RNA-Seq such as: DM-tRNA-seq254 and ARM-seq162 have been developed to overcome limits in sequencing caused by RNA modifications and structure; these techniques not only enable quantitative analysis of tRNA expression but can also provide dynamic measurements of specific tRNA methylations |
ARM-seq, AlkB-facilitated RNA methylation sequencing; CMC, N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate; DM-tRNA-seq, demethylase-thermostable group II intron RT tRNA sequencing; mRNP, messenger ribonucleoprotein; RBP, RNA-binding protein.
Another major class of UTR structural elements that can direct translational control in cancer comprises motifs recognized by RNA-binding proteins (RBPs)56. Although most RBP recognition motifs are thought to be sequence specific57 (discussed further below), structural RNA elements can also bind to RBPs. For example, the interferon-γ (IFNγ)-activated inhibitor of translation (GAIT) element is a structural RBP recognition motif that directs translational suppression of pro-inflammatory molecules in response to IFNγ signalling58. Another recent example of a structural RBP motif implicated in tumorigenesis is the transforming growth factor-β (TGFβ)-activated translation (BAT) element, a 3′UTR stem–loop region with an asymmetrical bulge that mediates TGFβ-driven translation of select mRNA transcripts59 (FIG. 2d). In the absence of TGFβ, the BAT element inhibits translation elongation by binding to both the eEF1A1 elongation factor and the RBP heterogeneous nuclear ribonucleoprotein E1 (hnRNPE1; also known as PCBP1) to form a messenger ribonucleoprotein (mRNP) complex that blocks translation elongation60. TGFβ signalling relieves this translational suppression by promoting AKT2-dependent phosphorylation of hnRNPE1 on serine 43, which prevents the formation of the BAT–mRNP complex. Importantly, the BAT element seems to be crucial for selective translational control of specific mRNAs involved in TGFβ-induced epithelial–mesenchymal transition (EMT) and metastasis, such as disabled homologue 2 (DAB2) and interleukin-like EMT inducer (ILEI; also known as FAM3C), highlighting a crucial role for RBP-directed translation in steering specific cancer cell behaviours.
A newly expanding appreciation of the role of structural motifs in steering cancer-associated translation stems in part from technological advances in both transcript-specific and genome-wide analysis of mRNA structure as well as unbiased profiling of RNA–protein interactions (TABLE 1). For example, these technologies were recently used to reveal a novel functional role for eIF3 in interacting with structural domains in the 5′UTRs of select cancer-associated mRNAs61. eIF3 is a multimeric complex best known for the scaffolding-like role it plays in connecting the eIF4F and the 43S pre-initiation complexes62; however, this function of eIF3 cannot explain why distinct eIF3 subunits are frequently overexpressed in human cancers29. Using transcriptome-wide PAR-CLIP analysis to identify preferential interactions of eIF3 with specific subsets of mRNAs, this study uncovered eIF3-binding sites in approximately 3% of all expressed transcripts, predominantly in 5′UTRs61. In support of a putative role in cancer, eIF3-bound transcripts included mRNAs associated with cancer-relevant pathways, such as apoptosis, cell cycle and differentiation. SHAPE analysis on two eIF3 target mRNAs, the oncogene JUN and the tumour suppressor B cell translocation gene 1 (BTG1), showed that eIF3-binding sites map to distinct conserved stem–loop regions61 (FIG. 2e). Intriguingly, these two eIF3-binding structures were found to have opposing effects on mRNA translation, suggesting that other context-specific features of the 5′UTR may interact with eIF3 to determine translational outputs that may be important for cancer cell survival and proliferation.
Sequence-specific RNA elements
A growing realization of the discrepancy between mRNA transcript levels and protein expression has kindled interest in translational control mediated by sequence-specific elements in the 5′ and 3′ UTRs. Technological advances, such as genome-wide profiling of actively translating ribosomes at codon-by-codon resolution, are enabling the identification of an increasing number of sequence-specific elements enriched in distinct subsets of transcripts that control important cancer cell behaviours (FIG. 2).
Some of the shortest sequence-specific elements that can affect transcript-specific translation are upstream initiation codons63. These can mark the presence of an alternative translation start (ATS) site that is in-frame and contiguous with the primary open reading frame (ORF) or can comprise an upstream open reading frame (uORF) that is distinct from the primary ORF. ATS sites can enable the expression of a unique protein isoform with different functions from those of the protein encoded by the primary ORF (FIG. 2f). For example, translation from a non-canonical CUG start codon in the PTEN 5′UTR was shown to drive expression of a long form of PTEN (with an additional N-terminal 173 amino acids) that is membrane permeable, can act in a non-cell-autonomous manner to inhibit PI3K signalling and is downregulated in samples from patients with breast cancer compared with matched normal breast tissue64. Although the mechanisms underlying translation from upstream ATS sites remain poorly understood, these elements may be under selective control in cancer cells.
uORFs, instead, are typically thought to inhibit translation of the primary ORF63 (FIG. 2g). Nearly half of all cellular mRNAs are predicted to contain one or more uORFs65. In line with this, recent data from ribosome-profiling experiments have confirmed the presence of translating 80S ribosomes upstream of the primary ORF in a large portion of expressed transcripts66–68. Despite the presence of uORFs across the genome, several studies have reported that genes encoding growth factors and cellular oncogenes are enriched for the presence of uORFs69,70, suggesting that overcoming uORF-mediated translational repression could be important in activating tumorigenic pathways. One prominent mechanism by which cancer cells can overcome uORF-mediated translation suppression is through phosphorylation of the α-subunit of eIF2 during tumour-associated stress conditions (discussed further below). However, cancer cells have been shown to use a diverse array of additional mechanisms to overcome uORF-dependent translational inhibition of specific transcripts. Examples include the transcription of MDM2 from a cryptic promoter to produce a truncated transcript that lacks two inhibitory uORFs that are normally present in the 5′UTR71–73 and the binding of the RBPs Hu antigen R (HUR; also known as ELAVL1) and hnRNPA1 to a translational derepression element (TDE) in the 3′UTR of HER2 (also known as ERBB2) that overrides the inhibitory effect of uORFs74.
Conversely, it has been shown that uORF-mediated translational repression can also positively affect tumorigenesis through gain-of-function mutations, typically in tumour suppressor genes. Indeed, mutations creating a new inhibitory uORF in the 5′UTR of the cyclin-dependent kinase inhibitor 2A (CDKN2A) tumour suppressor gene (which encodes p16INK4A and p14ARF) have been identified in a subset of patients with an inherited predisposition to melanomas75. Interestingly, some mRNAs even depend on uORFs for optimal translation. For example, genetic deletion of a uORF in the 5′UTR of the transcription factor CCAAT/enhancer-binding protein-β (CEBPB) in mice has revealed that this element is required for physiological in vivo expression of the C/EBPβ liver inhibitory protein (LIP) isoform76, which mediates breast cancer cell resistance to the TGFβ cytostatic response77 and increases tumour susceptibility when overexpressed in mice78. Importantly, although these studies demonstrate the potential for uORFs to steer transcript-specific translation in cancer, the vast majority of predicted uORFs remain to be functionally validated.
Most of the sequence-specific elements identified to date that influence translational control of oncogenic gene expression programmes are in the 3′UTR of transcripts, which are typically longer than 5′UTRs and contain a wealth of microRNA (miRNA)-binding sites and RBP motifs that can directly affect mRNA translation79. Importantly, 3′UTRs can be alternatively trimmed or lengthened to exclude or include these specific regulatory elements through a mechanism of alternative cleavage and polyadenylation (APA)80, which is estimated to occur for roughly half of all human genes81,82. Interestingly, proliferative cell states are typically associated with the selective shortening of 3′UTRs through APA83. Moreover, 3′UTR shortening is further selected for in cancer cells and can help to promote the expression of proto-oncogenes such as insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1)84.
mRNA isoforms with shortened 3′UTRs are typically associated with enhanced gene expression that can be attributed, in part, to the loss of miRNA-binding sites80 (FIG. 2h). The relative contribution of translational repression and mRNA decay to miRNA-mediated control of gene expression has been thoroughly debated85,86; however, recent work suggests that altered mRNA translation is one of the primary outputs of miRNA recognition87–93. Multiple mechanisms have been proposed to explain miRNA-mediated translational repression; however, accumulating evidence suggests that inhibition of cap-dependent translation initiation is the major mechanism89,90,94–96.
Interestingly, although numerous studies have demonstrated that specific miRNAs are frequently altered in human cancers, such as the let-7 family that regulates expression of the RAS oncogenes97, it has also been reported that miRNA expression and processing are globally downregulated in human cancer98,99. This raises an intriguing question of what advantage miRNA-mediated suppression through APA-dependent 3′UTR shortening may provide over direct control of miRNA expression and processing. As miRNAs typically recognize multiple mRNA targets within the cell, it is possible that APA-dependent 3′UTR shortening might enable cancer cells to selectively filter miRNA-mediated suppression to target distinct transcripts. Although recent studies have begun to identify specific molecular components required for 3′UTR shortening in cancer cells, such as the cytoplasmic polyadenylation element-binding protein 1 (CPEB1)100,101 and the cleavage factor 1M complex member, cleavage and polyadenylation specificity factor 25 kDa subunit (CFIm25; also known as NUDT21)102, we still have an incomplete understanding of APA regulation and the extent to which it may promote the expression of distinct transcripts that steer cancer-relevant behaviours.
The other major group of sequence-specific elements in the 3′UTR that can direct translational control of oncogenic gene expression programmes comprises a growing number of motifs recognized by RBPs57 (FIG. 2I). Although the selective loss of RBP recognition elements may be another driver of APA-induced 3′UTR shortening in cancer cells, their role in this process remains enigmatic, in part owing to the multifunctional nature of many RBPs, which can influence nearly every aspect of RNA metabolism103. For example, the RBP HUR, which recognizes U- and AU-rich elements in the 3′UTR and has been well-studied in the context of cancer (reviewed in REF. 56), can influence gene expression by promoting mRNA splicing104, stability105 and translation106. At the translational level, RBPs can regulate every step of protein synthesis from initiation to termination56. Indeed, RBPs have even been shown to promote programmed translational readthrough (PTR) of stop codons. For example, the binding of hnRNPA2B1 to a 63-nucleotide element in the 3′UTR of VEGFA mRNA was recently shown to promote PTR and drive expression of a novel antiangiogenic isoform VEGFAx that can inhibit the growth of human colon carcinoma xenografts in nude mice107. Importantly, although RBPs are typically expressed at much higher levels than most cellular proteins, contributing up to 20% of the expressed protein-coding transcriptome108, specific RBPs are frequently altered in human cancers and can associate with poor prognosis109. For instance, the RBP Musashi homologue 2 (MSI2), which promotes the leukaemia stem cell self-renewal programme in part through sustained translation of key self-renewal factors such as Ikaros family zinc finger protein 2 (IKZF2) and MYC110, was recently shown to be upregulated in human samples from patients with chronic myeloid leukaemia (CML) who have progressed from the chronic phase of disease to blast crisis111,112.
Remarkably, several studies suggest that we have only just begun to uncover the full repertoire of cellular RBPs involved in translational control of oncogenic gene expression programmes. Indeed, novel approaches using quantitative mass spectrometry (MS) in combination with ultraviolet (UV) or PAR crosslinking to identify the complete RBP–mRNA interactome have uncovered an unexpected number and diversity of RBPs within the cell, including a large group of non-canonical RBPs not predicted to bind RNA113–115. Moreover, recent work demonstrating that the tumour suppressor adenomatous polyposis coli (APC) can act as an RBP, recognizing mRNAs associated with its primary functions in regulating microtubule organization, motility and cancer116, suggests that even well-known oncogenic players could exert some of their tumorigenic potential through direct mRNA binding and translational control.
Numerous studies are also identifying an expanding list of sequence-specific elements in the 5′UTR of transcripts that are translationally regulated during tumorigenesis, including known RBP-binding motifs and sequence-specific elements for which regulatory mechanisms remain elusive10,26,117,118 (FIG. 2j). One of the oldest-known sequence-specific elements associated with oncogenic signalling pathways is the 5′-terminal oligopyrimidine tract (5′TOP), which consists of a 5′-terminal cytosine residue followed by a stretch of 7–13 pyrimidine nucleotides and was first identified as a translational regulatory motif controlling ribosomal protein expression associated with cellular growth119,120. Subsequent studies demonstrated that rapamycin could selectively repress the translation of 5′TOP-containing transcripts121, linking mTOR signalling to this sequence-specific translational element.
Although 5′TOP-mediated translation was initially attributed to ribosomal protein S6 kinase (S6K)-dependent phosphorylation of 40S ribosomal protein S6 (RPS6), elegant studies genetically inactivating S6K signalling or phosphorylation of RPS6 revealed that perturbing this pathway has no impact on translational control of 5′TOP-containing transcripts122,123. Recent work has now demonstrated that the ability of mTOR to direct the translation of 5′TOP-containing transcripts stems from its capacity to negatively regulate 4EBP-dependent inhibition of eIF4E as part of mTOR complex 1 (mTORC1)26,117. However, evidence exists in some settings, such as during amino acid stimulation, that translational control of 5′TOP-containing transcripts may be independent of 4EBP117,124 and that neither mTORC1 nor mTORC2 signalling alone can fully account for the control of 5′TOP-mediated translation by mTOR125,126.
Interestingly, landmark studies using ribosome profiling to identify the genome-wide repertoire of mRNAs that are translationally sensitive to mTOR ATP-active site inhibitors127,128 found that approximately 60% of mTOR-sensitive mRNAs also contain a 5′TOP-like element termed the pyrimidine-rich translational element (PRTE)26,117. Importantly, the PRTE diverges from the 5′TOP in both sequence and topology, as it consists of an invariant uridine at position 6 flanked by pyrimidines and it does not reside at the +1 position in the 5′UTR. PRTE-mediated translation has been shown to be crucial for expression of a group of pro-invasion genes, including Y-box binding protein 1 (YB1; also known as YBX1) and metastasis-associated 1 (MTA1), which steer cellular metastasis downstream of oncogenic mTOR signalling26, as well as for MYC-driven expression of the rate-limiting nucleotide biosynthesis enzyme phosphoribosyl pyrophosphate synthetase 2 (PRPS2), which supports anabolic metabolism of nucleotides, nucleic acids and proteins necessary for tumorigenesis28. Moreover, it has recently been demonstrated that protein-level expression of the PRTE-containing transcripts YB1 and MTA1 serves as a novel biomarker for prediction of outcomes for patients with prostate cancer, further highlighting the clinical importance of this translational regulatory element129. Importantly, unbiased translational profiling techniques, such as those used to identify the PRTE, could reveal many previously unknown translational elements that guide transcript-specific translation in cancer cells through distinct molecular mechanisms. For example, a functional element termed the cytosine-enriched regulator of translation (CERT) was recently discovered in mRNAs identified by polysome microarray analysis as being translationally sensitive to eIF4E dose during cellular transformation10.
Outstanding questions remain of how these sequence-specific elements, either through formation of higher-order structures or through recruitment of trans-acting factors, steer transcript-specific translation in cancer cells. Although work investigating the underlying mechanism of PRTE-mediated and CERT-mediated translation is currently under way, some progress has already been made with respect to 5′TOP-containing transcripts with the identification of La-related protein 1 (LARP1) as a 5′TOP-binding protein that can mediate mTOR-dependent translational control130. However, LARP1 activity by itself seems insufficient to explain 5′TOP-mediated translation, as a recent study found that LARP1 interacts with a broad class of approximately 3,000 mRNAs that include most but not all 5′TOP-containing transcripts131. Moreover, there is evidence that LARP1 binding to the 5′TOP may actually repress translation rather than activate it132. Further studies using unbiased approaches, such as comprehensive identification of RBPs by MS (CHIRP-MS) to identify physiologically relevant RBPs and in vivo click-SHAPE (icSHAPE) to identify RNA structure, are needed to fully elucidate the underlying mechanisms of this and other translational elements and to move towards a comprehensive understanding of how mRNA structure and sequence synergize to write the translational code. Importantly, this work will need to determine how the expanding list of mRNA modifications (including methylation133–136 and pseudouridylation137–140) and alterations in ribosome biogenesis and function (BOX 2) affect interactions of the translation machinery with regulatory elements in mRNA 5′ and 3′ UTRs to influence translational control during tumorigenesis.
Box 2. The cancer cell ribosome: regulated at its core.
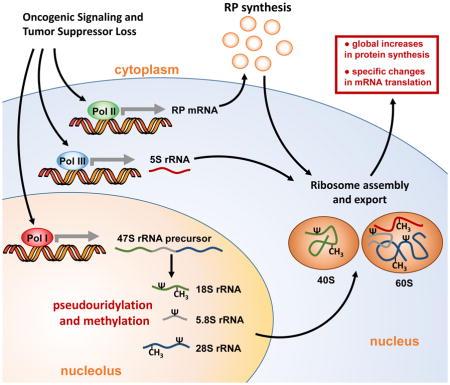
Alterations in ribosome synthesis and activity are a defining feature of cancer cells208. Indeed, a primary output from the activation of oncogenic signalling pathways, such as the MYC, RAS and AKT pathways, and from the loss of tumour suppressors, such as p53 and RB, is to drive increased ribosome biogenesis by enhancing transcription of genes encoding ribosomal proteins and ribosomal RNA (rRNA)145,209–218 (see the figure). Although ribosomes have historically been perceived as ‘housekeeping’ complexes that act as ‘backstage’ participants, supporting protein synthesis without having an active role in directing functional expression of the genome, recent studies have revealed that ribosome biogenesis and activity are in fact highly regulated to steer specific translational outputs219. For example, mutations in the gene encoding ribosomal protein L38 (RPL38) have been shown to cause tissue-specific patterning defects during development as a result of impaired internal ribosome entry site (IRES)-dependent translation of select homeobox (HOX) mRNAs containing an upstream translation inhibitory element118,220.
One of the major mechanisms by which tumour cells alter ribosome biogenesis to drive selective translation of the cancer genome is through rRNA modifications, predominantly methylation and pseudouridylation on nucleotides in functionally important regions. Most rRNA methylation is catalysed by the rRNA 2′-O-methyltransferase fibrillarin as part of a ribonucleoprotein (RNP) complex guided by the C/D class of small nucleolar RNAs (snoRNAs). Fibrillarin overexpression has been linked to p53 loss and can drive specific alterations in mRNA translation, including increased IRES-dependent translation of insulin-like growth factor 1 receptor (IGF1R) and fibroblast growth factor 2 (FGF2)221. rRNA pseudouridylation is catalysed by the pseudouridine synthase dyskerin as part of an RNP complex guided by the H/ACA class of snoRNAs. Dyskerin mutations have been identified in human cancers and are invariably associated with the syndrome X-linked dyskeratosis congenita, an inherited human cancer susceptibility disorder that belongs to a broader class of ribosomopathies208. Decreases in dyskerin function impair IRES-dependent translation of key tumour suppressor genes, such as those encoding p27 and p53, and can cause changes in the expression of distinct subsets of snoRNAs, leading to site-specific alterations in rRNA pseudouridylation222–226. Recently, a growing body of work has also revealed that deregulated expression of C/D and H/ACA snoRNAs is a common feature of human cancers227–231, suggesting a potentially broader role for selective modification of rRNA in cellular transformation and tumour development.
tRNA function and codon usage bias
The human genome contains a total of 61 different sense codons, many of which are synonymous and encode the same amino acid. These 61 codons are translated by 49 different tRNA ‘isoacceptor species’, so-called because they are charged with identical amino acids but have unique anticodon sequences. New unbiased approaches for profiling tRNAs have unexpectedly revealed that the expression of tRNA isoacceptor species is enhanced in several human cancers (including breast cancer, multiple myeloma and ovarian cancers)141–143, raising the question of whether such changes may have functional implications for expression of the cancer genome.
tRNAs have typically been thought to exist in excess of cellular demand; however, exciting new work suggests that this is not always the case. For example, the overexpression of methionine initiator tRNA (tRNAiMet) in human mammary epithelial cells has been shown to drive enhanced proliferation and cellular metabolism144. Unexpectedly, this also caused broad increases in other tRNA levels, calling into question the relative contributions of global and specific changes in tRNA expression to translational control of these cellular processes. Given that tRNA expression can be limiting for cellular growth and proliferation, it is perhaps not surprising that many oncogenic lesions that drive these processes also promote tRNA expression. For example, activation of the AKT–mTOR, RAS–MAPK and MYC oncogenic signalling pathways and loss of the tumour suppressors p53 and RB have been shown to influence RNA polymerase III (Pol III)-dependent transcription of tRNA genes145–148.
How might increases in tRNA expression influence translational control to steer cellular transformation and tumorigenesis? Although it is possible that general increases in tRNA expression are needed simply to meet the enhanced global protein synthesis demands of tumour cells, numerous studies suggest that cancer-associated increases in tRNA expression are not equally distributed across tRNA species141,143. In particular, specific increases in tRNAiMet expression could not only augment global protein synthesis rates by enhancing translation initiation rates through increased ternary complex formation but also simultaneously inhibit the translation of select mRNAs containing multiple uORFs, such as activating transcription factor 4 (ATF4), translation of which is favoured by low levels of the ternary complex (discussed further below).
Additionally, changes in tRNA expression could influence translational outputs by enhancing translation elongation for distinct subsets of mRNAs based on their codon usage. For example, it has been shown that bias in codon usage can contribute significantly to differences in expression between genetic paralogues with similar amino acid identity, such as the KRAS and HRAS oncogenes149. In this scenario, the presence of ‘rare’ codons in KRAS that are generally underrepresented in the genome has been found to limit KRAS expression at the translational level compared with HRAS. Strikingly, replacing rare KRAS codons with more commonly used codons is sufficient not only to enhance KRAS expression but also to promote KRAS-driven tumorigenesis, explaining, at least in part, the inherent differences in tumorigenicity observed between KRAS and HRAS oncogenes.
Although there initially seemed to be a lack of evidence for a broader role of genome-wide codon usage bias in mammalian systems, this has been at least partially reconciled by work demonstrating that codon selection is biased in a highly tissue-specific manner mirrored by tRNA isoacceptor expression150,151. Importantly, these studies suggest that codon usage in mammalian systems could support the translation of specific gene expression programmes (FIG. 3). Indeed, a novel extension of this concept has recently been described, whereby function-related codon usage was found to be hardwired into transcriptional programmes underlying cellular proliferation in both normal cells and cancer cells152. In particular, this study identified selective and divergent expression of tRNA pools between states of cellular proliferation and differentiation that matched well with the respective codon usage bias in cell-autonomous genes driving proliferation and supporting differentiation. Importantly, this work highlights a direct correlation between codon content, tRNA levels and gene expression that comprises a novel mode of translational control underlying transformed cellular phenotypes such as enhanced proliferation. In line with this, the overexpression of specific tRNA isoacceptors in breast cancer cell lines has also been found to correlate positively with codon usage patterns in cancer-related genes involved in the cell cycle, extracellular matrix and transcriptional control141. Collectively, these studies suggest a crucial role for altered tRNA expression in driving specific pro-tumorigenic programmes.
Figure 3. Codon usage: a new layer of control in translation of the cancer genome.

Oncogenic signalling can drive changes in the transcription of tRNA genes and their modifications (denoted here as red asterisks in the anticodon loop), leading to the expression of a specific repertoire of tRNAs in the cancer cell. This can in turn support the translation of pro-tumorigenic mRNAs based on their codon usage bias, whereby mRNAs with codons better matched to the tRNA pool (depicted here by matched codon and tRNA colour) are more likely to be translated. For example, the cancer cell tRNA pool is well matched to the codon usage of transcripts involved in cellular proliferation, such as cell cycle genes, but not to those mRNAs involved in differentiation, such as pattern specification genes141,152. Pol, RNA polymerase.
In addition to being transcriptionally controlled, tRNAs are one of the most heavily modified RNA species in the cell, with more than 90 different modifications that occur after processing and splicing of the initial transcript153. Most of these modifications are thought to have crucial roles in tRNA structural conformation and stability; however, specific modifications in the anticodon loop have been shown to affect tRNA-directed translation153. Recently, several studies on human cancers have identified quantitative changes in tRNA modifications and the enzymes that catalyse them, such as tRNA-isopentenyltransferase154. Although it largely remains to be determined how changes in tRNA modifications contribute functionally towards tumorigenesis, emerging evidence has highlighted a role for tRNA modifications in the regulation of distinct translational outputs in response to stress conditions relevant to the transformed phenotype, including oxidative stress and DNA damage155,156. An interesting example of this comes from the methyltransferase alkylated DNA repair protein alkB homologue 8 (ALKBH8), which is required for specific modifications to the wobble position of select tRNAs, including selenocysteine tRNAs157–159. Selenocysteine is an amino acid uniquely ‘coded’ for by UGA stop codons that lie upstream of a structural motif known as a selenocysteine insertion sequence (SECIS) element, a common feature of several mRNAs that encode antioxidant proteins160. Importantly, the ability of selenocysteine tRNA to recode UGA stop codons requires ALKBH8-mediated tRNA modifications, which are induced downstream of oxidative stress conditions, in order to support the translation of reactive oxygen species (ROS)-detoxifying enzymes, such as glutathione peroxidase 1 (GPX1)161. Importantly, future studies are needed to fully characterize the number and extent of changes in tRNA modifications that occur during tumorigenesis and to elucidate how tumour-associated stress conditions, such as oxidative stress, influence these modifications to control tRNA function. This work will require the continued development of new technologies that enable quantitative transcriptome-wide assessment of tRNA expression and modifications, such as AlkB-facilitated RNA methylation sequencing (ARM-Seq)162 (TABLE 1), and has the potential to provide a new understanding of how tRNA expression, modification and codon usage bias cooperate to drive the translation of gene expression programmes that underlie specific cancer cell behaviours.
Translational adaptation to stress
A key feature of cancer cells that enables them to survive and proliferate is their capacity to adapt to multiple cellular stresses encountered during tumorigenesis, including replicative, oxidative, genotoxic, metabolic and proteotoxic stress, such as endoplasmic reticulum (ER) stress163. These cellular stresses are inherently connected to the activation of oncogenic signalling pathways and can profoundly influence tumour development and progression. For example, the ability of oncogenic signalling to promote enhanced cellular proliferation is directly linked to the induction of replicative stress and genomic instability during early stages of tumorigenesis164. Numerous oncogenes have also been shown to drive altered mitochondrial activity that leads to the generation of ROS, which can be either pro-tumorigenic or antitumorigenic depending on the level and duration of their production165,166. Although ROS can contribute to DNA mutations and act as signalling mediators that support cellular transformation at low levels, high levels of ROS are toxic and invariably associated with decreased cancer cell survival165,166. In addition, the tumour microenvironment can dramatically influence cellular stress states. In particular, insufficient vascularization can contribute to tumour hypoxia and nutrient deprivation, causing metabolic, oxidative and ER stress163. In order to overcome these blocks to tumorigenesis, cancer cells co-opt cytoprotective pathways that enable them to weather and resolve specific stress conditions. Importantly, an increasing number of studies are demonstrating a vital role for translational control in establishing such adaptive stress responses, whereby cancer cells redirect translation initiation and elongation to drive the synthesis of specific proteins that fuel their survival (FIG. 4).
Figure 4. Translational responses and cancer cell adaptation to tumour-associated stress.

The ability of cancer cells to adapt to stresses encountered during tumorigenesis is fundamental for tumour growth and survival163. One of the major cellular responses to stress conditions is global inhibition of protein synthesis, which acts to conserve energy and prevent the accumulation of damaged proteins. This is mediated largely at the level of translation initiation through decreases in eukaryotic initiation factor 4E (eIF4E) activity downstream of mTOR–eIF4E-binding protein (4EBP) signalling168 and inhibition of eIF2A by the eIF2A kinase family, which includes PRKR-like endoplasmic reticulum (ER) kinase (PERK) and general control non-derepressible 2 (GCN2)171. Stress conditions can also block translation elongation through AMP-activated protein kinase (AMPK)-dependent activation of eukaryotic translation elongation factor 2 (eEF2) kinase (eEF2K), which inhibits eEF2 (REF. 11). Although oncogenic signalling frequently suppresses the AMPK–eEF2K pathway, cancer cells selectively reactivate this pathway to promote survival during nutrient deprivation. Cancer cells can also adapt to stress by increasing the translation of selective mRNA transcripts that promote cell survival and resolve stress conditions. This adaptive stress response is driven in part by internal ribosome entry site (IRES)- and upstream open reading frame (uORF)-dependent translational mechanisms that can be maintained or even favoured under conditions in which global protein synthesis is inhibited and can promote the translation of pro-survival genes, such as BCL2 (REF. 50) and X-linked inhibitor of apoptosis protein (XIAP)177, and stress response genes such as excision repair cross-complementation group 5 (ERCC5)181. Likewise, it has been revealed that under hypoxic conditions that inhibit global protein synthesis the eIF4E homologue 4EHP can promote the translation of select mRNAs involved in the adaptive response to hypoxia, including epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor-α (PDGFRA)191. Recently, exciting new studies have revealed that even eIF4E can act to promote the adaptive response to stress, for example, through enhanced translation of reactive oxygen species (ROS) regulators such as ferritin heavy polypeptide 1 (encoded by FTH1) and glutamate–cysteine ligase catalytic subunit (encoded by GCLC)10. As cancer cells do not always globally downregulate protein synthesis in response to stress, this new-found role for eIF4E may be especially relevant for tumorigenesis.
Translational control provides an unparalleled mechanism for cells to rapidly and precisely regulate gene expression during stress conditions167. One of the major cellular responses to a wide range of stress conditions is the global inhibition of protein synthesis, which not only acts as a mechanism of energy conservation but can also prevent the accumulation of damaged and misfolded proteins during specific stress conditions, such as oxidative or ER stress167. Global inhibition of protein synthesis is thought to occur largely at the level of translation initiation through modulation of eIF4E and eIF2 activity. Most cellular stress conditions block eIF4E activity by suppressing mTOR-dependent phosphorylation of 4EBP1, which is thought to be mediated by AMP-activated protein kinase (AMPK)-dependent activation of the mTOR inhibitor TSC2 (also known as tuberin)168, although REDD1 (also known as DDIT4) and RAS GTPases, respectively, have also been implicated in mTOR control during hypoxia and amino acid deprivation169,170.
eIF2, on the other hand, is regulated by a family of kinases that are activated under various stress conditions to phosphorylate serine 51 of the eIF2A subunit, thus blocking the recycling of eIF2 for delivery of tRNAiMet to the 40S ribosome. Perhaps the best characterized eIF2A kinase is PRKR-like ER kinase (PERK; also known as eIF2AK3), which regulates one of the three effector arms of the unfolded protein response (UPR) during ER stress, oxidative stress and hypoxia171. Three other eIF2A kinases — general control non-derepressible 2 (GCN2; also known as eIF2AK4), IFN-induced double-stranded RNA-activated protein kinase (PKR; also known as eIF2AK2) and haem-regulated inhibitor (HRI; also known as eIF2AK1) — have also been identified, although these are typically thought to be more restricted to specific cell types or certain stress conditions. In particular, GCN2 is activated by amino acid deprivation directly in response to the accumulation of uncharged tRNAs172, PKR is classically activated in response to double-stranded RNA during antiviral immune responses173 and HRI expression is restricted to erythroid cells, where it regulates protein synthesis in a haem-dependent manner174.
Traditionally, the inhibition of eIF4E and eIF2 under stress conditions is thought to cause global decreases in protein synthesis while simultaneously enabling selective translation of mRNAs essential to the adaptive stress response167. These mRNAs possess distinct cis-regulatory elements in their 5′UTRs, such as IRES elements and uORFs, that direct transcript-specific translation in a manner compatible with reduced eIF4E and eIF2 activity. For example, hypoxia has been shown to induce a switch from cap-dependent to IRES-dependent translation that promotes tumour survival and angiogenesis through the ability of eIF4G to engage with IRES elements in specific mRNAs175, and IRES-mediated translation of select transcripts (for example, those encoding anti-apoptotic factors such as X-linked inhibitor of apoptosis protein (XIAP) and pro-angiogenic factors such as VEGFC) has been shown to support tumour growth and survival under stress conditions such as DNA damage and hypoxia176–179.
Transcripts containing uORFs can also be selectively translated under stress conditions. For example, a recent study found that nearly all transcripts efficiently translated under the acute induction of oxidative stress contain at least one uORF180. Normally, the presence of one or more uORFs inhibits downstream translation at the major protein-encoding ORF; however, stress-induced phosphorylation of eIF2A can relieve this suppression. For example, DNA damage-induced phosphorylation of eIF2A has been shown to promote translation of a common polymorphic variant of the ERCC5 nucleotide excision repair gene that contains a new uORF in the 5′UTR, and, strikingly, the presence of this uORF polymorphism in paediatric patients with ependymoma was found to confer resistance to platinum-based DNA-damaging agents in association with significantly reduced progression-free survival181. One way that eIF2A phosphorylation can promote such uORF-dependent translation is through a unique ribosome scanning and re-initiation mechanism that relies on a specific configuration of two uORFs, as occurs for the transcript encoding ATF4 (REF. 182). In this scenario, after translating the first uORF, the 40S ribosome remains associated with the mRNA and continues scanning for additional start codons. Normally, eIF2 efficiently delivers tRNAiMet to the scanning 40S ribosome so that it re-initiates translation at the second uORF, which overlaps with the major ORF and blocks its translation. However, under stress conditions in which eIF2 is inhibited, the 40S ribosome bypasses the second uORF before tRNAiMet is delivered, enabling translation re-initiation at the major ORF to occur182. This mechanism promotes the selective translation of ATF4 under stress conditions in order to coordinate the transcription of numerous stress response genes, including genes involved in the production of glutathione, which detoxifies ROS, and autophagy genes such as autophagy related 5 (ATG5) and microtubule-associated protein 1 light chain 3β (MAP1LC3B) that promote cell survival183.
Although translational control during stress has largely been attributed to enhanced IRES- and uORF-dependent translation that is coupled with decreased global protein synthesis, accumulating evidence suggests that this paradigm is insufficient to explain translational regulation of the adaptive stress response during tumorigenesis. For example, several studies have demonstrated that not all cancer cells globally downregulate protein synthesis in response to stress conditions184–186. In fact, ER-stress-dependent activation of the pro-survival PERK–eIF2A pathway in MYC-driven lymphomas is inherently linked to the ability of MYC to enhance global protein synthesis186. Moreover, it is apparent that our understanding of the regulation of IRES- and uORF-containing transcripts and their contribution to the stress response is incomplete. For example, although the ability of eIF4G to promote selective translation under stress conditions has largely been ascribed to its capacity to influence IRES-dependent translation, many eIF4G-dependent transcripts seem to lack functional IRES elements12.
Recently, unanticipated layers of translational control, beyond IRES and uORFs, have been revealed as integral to the adaptive stress response in cancer cells. One unexpected finding came when 4EHP (also known as eIF4E2), a cap-binding homologue of eIF4E best characterized for its role as a translational suppressor crucial for embryonic development and stem cell function187–189, was implicated in the hypoxic stress response. Although eIF4E is typically thought to be the primary cap-binding protein under hypoxic conditions190, 4EHP was recently identified as a central component of a hypoxia-induced translation initiation complex that includes oxygen-regulated hypoxia-inducible factor 2α (HIF2α) and the RBP RBM4191. PAR-CLIP analysis revealed that under hypoxic conditions RBM4 recruits HIF2α to a specific RNA element in the 3′UTR of select mRNAs involved in the adaptive response to hypoxia, including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor-α (PDGFRA) and insulin-like growth factor 1 receptor (IGF1R), promoting their translation through interaction with cap-bound 4EHP. Although this work describes a novel role for 4EHP in promoting translation under hypoxic stress conditions, the full scope of mRNAs regulated by the 4EHP hypoxia complex and the underlying mechanism by which it promotes translation remain outstanding questions. Interestingly, even the major cap-binding protein eIF4E, which is typically thought to be inhibited under stress conditions, has emerged as a surprise player in the response to cellular stress during tumorigenesis. Indeed, eIF4E dose was recently identified as a crucial determinant of the translation of oxidative stress response genes essential for cellular transformation and in vivo lung tumorigenesis, such as ferritin heavy polypeptide 1 (FTH1) and the rate-limiting enzyme for glutathione synthesis glutamate–cysteine ligase catalytic subunit (GCLC)10. Similarly, increased eIF4E dose was shown to rescue human mammary epithelial cells from oncogene-induced replication stress through the activation of a pro-survival DNA damage response192. In line with these studies, eIF4E phosphorylation has also been implicated in resistance to oxidative stress, starvation and DNA-damaging agents in cancer cells193.
Although most work has focused on translation initiation, translation elongation can also be a major node in regulation of the adaptive stress response in cancer cells. Normally, translation elongation is blocked under metabolic stress conditions by AMPK-dependent activation of eEF2 kinase (eEF2K), which phosphorylates and inhibits eEF2 to promote cell survival194. However, oncogenic signalling can suppress eEF2K activity and sensitize cells to nutrient deprivation-induced apoptosis11. Therefore, in order to adapt to metabolic stress, cancer cells selectively reactivate AMPK–eEF2K signalling to inhibit translation elongation and exploit this survival pathway11. Remarkably, eEF2K-dependent inhibition of translation elongation does not seem to influence global protein synthesis rates during nutrient deprivation, suggesting that eEF2 blockade may instead promote cell survival through selective effects on mRNA translation.
Collectively, these findings highlight a newly uncovered vulnerability of cancers that can be exploited therapeutically. Indeed, as tumours face numerous stresses during their development and progression, targeting translational control of the adaptive stress response provides a unique strategy by which to selectively kill cancer cells. Moreover, translation inhibitors have the potential to potently synergize with pharmacological inducers of cellular stress in novel combinatorial approaches. In this regard, it has recently been shown that genetically targeting eIF4E-dependent control of the oxidative stress response sensitizes tumours to small-molecule inducers of ROS10. Similarly, the inhibition of MNK-dependent eIF4E phosphorylation in cancer cells has been found to cooperate with both ROS induction and DNA-damaging agents in inducing apoptosis193. As current research continues to reveal a more detailed understanding of how the translational circuitry is rewired to weather cellular stresses encountered during tumorigenesis, new therapeutic targets and approaches for selective blockade of these adaptive stress responses will undoubtedly be discovered.
Conclusion and future perspectives
In recent years, many studies have revealed crucial new insights into the regulatory circuitry governing translation of the cancer genome, including cis-regulatory elements present in the 5′ and 3′ UTRs of mRNAs and codon usage bias embedded within the mRNA-coding region. This regulatory logic provides the basis for the selective translation of key mRNA networks governing distinct tumour cell behaviours, such as proliferation, cell survival, metastasis and adaptation to stress conditions. Although considerable gains have been made in understanding the molecular basis underlying translational specificity in cancer cells, we still lack a complete map of this regulatory landscape. In particular, major gaps exist in our understanding of how structural and sequence-specific cis-regulatory elements in mRNAs, such as IRESs and PRTEs, interface in trans with specific RBPs and with components of the translational machinery. Exciting technologies have recently been developed (TABLE 1), such as individual-nucleotide-resolution UV CLIP (iCLIP) and icSHAPE, that will enable future studies to address these crucial questions and open new doors into our understanding of cancer aetiology.
Importantly, translation deregulation is emerging not only as a primary downstream output of oncogenic signalling but also as a central mediator of cancer resistance to several clinical therapies13,195. These findings, along with a growing body of work that has shifted the view of translation from an ‘undruggable’ housekeeping function to a regulated network that is co-opted to support tumour growth and function, are driving increasing interest in targeting translational control as a means of selectively killing cancer cells30. Indeed, therapeutic approaches to inhibit the translation initiation machinery26,196 and even ribosome biogenesis itself 197–199 are already being tested in clinical trials for their efficacy against various cancers. Ultimately, as research continues to decipher the regulatory language underlying translation of the cancer genome, this will pave the way for new strategies that target even more refined levels of translation, providing novel therapeutic approaches to disrupt the translational control networks that cancer cells tailor to support the transformed phenotype.
Acknowledgments
The authors thank M. Barna for critical discussion and reading of the manuscript. This work was supported by funding from the US National Institutes of Health (R01 CA140456, R01 CA184624, R01 DK098057, R01 CA154916, RO1 HL119439 and PO1 CA165997). The authors apologize to those whose work could not be mentioned owing to space limitations. D.R. is a Leukaemia and Lymphoma Society Scholar.
Glossary
- mTOR
A serine/threonine kinase that forms two distinct molecular complexes (mTORC1 and mTORC2) and acts as a master regulator of protein synthesis, largely through mTORC1-dependent phosphorylation of eukaryotic translation initiation factor 4E-binding proteins (4EBPs) and ribosomal protein S6 kinase
- tRNA
Non-coding RNA with a unique L-shaped tertiary structure that contains a ‘charged’ amino acid covalently linked to the tRNA by aminoacyl transferases at one end and the tRNA anticodon loop that recognizes distinct mRNA codons through base-pairing interactions at the other end
- Internal ribosome entry site (IRES)
A complex structural element first discovered and characterized in viruses that facilitates translation initiation by recruiting the 40S ribosome to the mRNA in a cap-independent manner frequently aided by specific RNA-binding proteins known as IRES trans-acting factors (ITAFs)
- Messenger ribonucleoprotein (mRNP) complex
A complex of mRNA and RNA-binding proteins that can vary throughout the life of the mRNA and act to either promote or inhibit mRNA splicing, stability and translation depending on the nature of the bound proteins
- Upstream open reading frame (uORF)
An ORF comprising a start codon upstream of the primary ORF with an in-frame stop codon that can lie either 5′ or 3′ of the primary start codon. uORFs typically block translation of downstream ORFs, as ribosomes generally initiate translation at the first start codon they encounter and then disassociate from the mRNA upon translation termination
- Alternative cleavage and polyadenylation (APA)
A mechanism for generating mRNAs with different lengths of 3′ untranslated region that relies on the recognition of alternative poly(A) signals in association with U- and GU-rich downstream sequence elements by the cleavage and polyadenylation specificity factor (CPSF) complex and the cleavage stimulating factor (CSTF) complex
- Programmed translational readthrough (PTR)
A phenomenon that enables translation to continue past the normal stop codon in favour of termination at a downstream stop codon, potentially generating an elongated protein with a novel 3′ extension
- 5′-terminal oligopyrimidine tract (5′TOP)
A sequence-specific element located at the +1 position of the 5′ untranslated region that consists of a 5′ cytosine residue followed by a stretch of 7–13 pyrimidine nucleotides
- Pyrimidine-rich translational element (PRTE)
A 5′ untranslated region (5′UTR) sequence-specific motif related to the 5′-terminal oligopyrimidine tract that is not restricted to the +1 position of the 5′UTR and contains an invariant uridine at position 6 flanked by pyrimidines
- Ribosomopathies
Inherited human cancer susceptibility disorders — such as cartilage–hair hypoplasia syndrome, Shwachman–Diamond syndrome, 5q deletion syndrome, dyskeratosis congenita and Diamond–Blackfan anaemia — that are characterized by mutations in distinct components of the translational machinery, including enzymes involved in the modification and processing of rRNA, ribosome assembly factors and ribosomal proteins
- Methionine initiator tRNA (tRNAiMet)
A distinct species of tRNA charged with methionine that interacts with the ribosome P site as part of the eIF2–GTP–Met-tRNAiMet ternary complex that initiates translation at start codons
- Wobble position
The first position in a tRNA anticodon loop that can enable non-Watson–Crick base interactions with the third nucleotide in the codon triplet and be a site for tRNA modifications
- Unfolded protein response (UPR)
A conserved pathway activated by the accumulation of unfolded proteins in the endoplasmic reticulum (ER) that signals through three downstream effector arms (PRKR-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6)) to either relieve protein misfolding stress in the ER through increased production of chaperone proteins, enhanced protein degradation and dampened protein synthesis or commit the cell to apoptosis if stress is unresolvable
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Miluzio A, et al. Impairment of cytoplasmic eIF6 activity restricts lymphomagenesis and tumor progression without affecting normal growth. Cancer Cell. 2011;19:765–775. doi: 10.1016/j.ccr.2011.04.018. This study demonstrated that mice happloinsufficient for eIF6, which regulates the formation of functional 80S ribosomes, show delayed in vivo tumorigenesis and reduced tumour growth, thus uncovering a rate-limiting role for translation initiation independent of the eIF4F complex. [DOI] [PubMed] [Google Scholar]
- 2.Barna M, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. This is the first study to genetically demonstrate that the ability of MYC to drive increased protein synthesis is a key determinant of oncogenicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furic L, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci USA. 2010;107:14134–14139. doi: 10.1073/pnas.1005320107. This paper describes the generation of a knock-in mouse that expresses a non-phosphorylatable form of eIF4E and demonstrates a crucial role for eIF4E phosphorylation during in vivo tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh AC, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP–eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faller WJ, et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517:497–500. doi: 10.1038/nature13896. This exciting study demonstrates that oncogenic activation of translation elongation through eEF2 can be rate limiting for tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfe AL, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70. doi: 10.1038/nature13485. In this study, ribosome profiling in cancer cells revealed genome-wide translational effects of eIF4A inhibiton and uncovered the G-quadruplex as a novel 5′UTR cis-regulatory element. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubio CA, et al. Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 2014;15:476. doi: 10.1186/s13059-014-0476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh AC, et al. Cell type-specific abundance of 4EBP1 primes prostate cancer sensitivity or resistance to PI3K pathway inhibitors. Sci Signal. 2015;8:ra116. doi: 10.1126/scisignal.aad5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pourdehnad M, et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci USA. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Truitt ML, et al. Differential requirements for eIF4E dose in normal development and cancer. Cell. 2015;162:59–71. doi: 10.1016/j.cell.2015.05.049. This paper describes the generation of the first genetic loss-of-function mouse for eIF4E and unexpectedly reveals that eIF4E dose is limiting for tumorigenesis but not for normal development. Translation profiling further uncovers an eIF4E-dependent oncogenic translation programme enriched for oxidative stress response genes and marked by a novel functional cis-regulatory element termed the CERT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leprivier G, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–1079. doi: 10.1016/j.cell.2013.04.055. This paper shows that cancer cells can reprogramme translation at the elongation step through AMPK–eEF2K signalling in order to adapt to metabolic stress conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci USA. 2012;109:18767–18772. doi: 10.1073/pnas.1203853109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boussemart L, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 14.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robichaud N, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34:2032–2042. doi: 10.1038/onc.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones RM, et al. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol Cell Biol. 1996;16:4754–4764. doi: 10.1128/mcb.16.9.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, Schmidt EV. Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2α in response to growth induction by c-myc. Proc Natl Acad Sci USA. 1993;90:6175–6178. doi: 10.1073/pnas.90.13.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–5709. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bah A, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 20.She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorrello NV, et al. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 22.Raught B, et al. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004;23:1761–1769. doi: 10.1038/sj.emboj.7600193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petroulakis E, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–446. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, et al. Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Mol Cell Biol. 2014;34:4088–4103. doi: 10.1128/MCB.01035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. This study, along with reference 117, uses novel ATP active-site inhibitors of mTOR and unbiased genome-wide profiling to define the mTOR-dependent translatome and identifies the 5′TOP and the PRTE as cis-regulatory elements that control translation of key mRNA subsets. It also describes a new role for mTOR–4EBP signalling in specializing translation of the cancer genome to direct tumour invasion and metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajasekhar VK, et al. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell. 2003;12:889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–1103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–266. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 30.Bhat M, et al. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261–278. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- 31.Blagden SP, Willis AE. The biological and therapeutic relevance of mRNA translation in cancer. Nat Rev Clin Oncol. 2011;8:280–291. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]
- 32.Kozak M. Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc Natl Acad Sci USA. 1986;83:2850–2854. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelletier J, Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5′ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985;40:515–526. doi: 10.1016/0092-8674(85)90200-4. This is one of the first studies to show a role for 5′UTR secondary structure in influencing mRNA translation. [DOI] [PubMed] [Google Scholar]
- 34.Koromilas AE, Lazaris-Karatzas A, Sonenberg N. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992;11:4153–4158. doi: 10.1002/j.1460-2075.1992.tb05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray BK, et al. ATP-dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J Biol Chem. 1985;260:7651–7658. [PubMed] [Google Scholar]
- 36.Svitkin YV, et al. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA. 2001;7:382–394. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozen F, et al. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol Cell Biol. 1990;10:1134–1144. doi: 10.1128/mcb.10.3.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feoktistova K, Tuvshintogs E, Do A, Fraser CS. Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proc Natl Acad Sci USA. 2013;110:13339–13344. doi: 10.1073/pnas.1303781110. This study describes a new function of eIF4E in stimulating eIF4A helicase activity that occurs independently of eIF4E binding to the 5′ cap. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parsyan A, et al. mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol. 2011;12:235–245. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- 40.Manzella JM, Blackshear PJ. Regulation of rat ornithine decarboxylase mRNA translation by its 5′-untranslated region. J Biol Chem. 1990;265:11817–11822. [PubMed] [Google Scholar]
- 41.Kevil C, Carter P, Hu B, DeBenedetti A. Translational enhancement of FGF-2 by eIF-4 factors, and alternate utilization of CUG and AUG codons for translation initiation. Oncogene. 1995;11:2339–2348. [PubMed] [Google Scholar]
- 42.Kevil CG, et al. Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int J Cancer. 1996;65:785–790. doi: 10.1002/(SICI)1097-0215(19960315)65:6<785::AID-IJC14>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Bordeleau ME, et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest. 2008;118:2651–2660. doi: 10.1172/JCI34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiegering A, et al. Targeting translation initiation bypasses signaling crosstalk mechanisms that maintain high MYC levels in colorectal cancer. Cancer Discov. 2015;5:768–781. doi: 10.1158/2159-8290.CD-14-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cencic R, et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS ONE. 2009;4:e5223. doi: 10.1371/journal.pone.0005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin X, Sarnow P. Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J Biol Chem. 2004;279:13721–13728. doi: 10.1074/jbc.M312854200. [DOI] [PubMed] [Google Scholar]
- 47.Gerlitz G, Jagus R, Elroy-Stein O. Phosphorylation of initiation factor-2α is required for activation of internal translation initiation during cell differentiation. Eur J Biochem. 2002;269:2810–2819. doi: 10.1046/j.1432-1033.2002.02974.x. [DOI] [PubMed] [Google Scholar]
- 48.Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat Cell Biol. 1999;1:190–192. doi: 10.1038/11109. [DOI] [PubMed] [Google Scholar]
- 49.Coldwell MJ, Mitchell SA, Stoneley M, MacFarlane M, Willis AE. Initiation of Apaf-1 translation by internal ribosome entry. Oncogene. 2000;19:899–905. doi: 10.1038/sj.onc.1203407. [DOI] [PubMed] [Google Scholar]
- 50.Sherrill KW, Byrd MP, Van Eden ME, Lloyd RE. BCL-2 translation is mediated via internal ribosome entry during cell stress. J Biol Chem. 2004;279:29066–29074. doi: 10.1074/jbc.M402727200. [DOI] [PubMed] [Google Scholar]
- 51.Nanbru C, et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J Biol Chem. 1997;272:32061–32066. doi: 10.1074/jbc.272.51.32061. [DOI] [PubMed] [Google Scholar]
- 52.Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002;16:3087–3099. doi: 10.1101/gad.248902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006;7:404–410. doi: 10.1038/sj.embor.7400623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene. 2006;25:4613–4619. doi: 10.1038/sj.onc.1209483. [DOI] [PubMed] [Google Scholar]
- 55.Graber TE, Holcik M. Cap-independent regulation of gene expression in apoptosis. Mol Biosyst. 2007;3:825–834. doi: 10.1039/b708867a. [DOI] [PubMed] [Google Scholar]
- 56.Wurth L, Gebauer F. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim Biophys Acta. 2015;1849:881–886. doi: 10.1016/j.bbagrm.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Ray D, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 2009;34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaudhury A, et al. TGF-β-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. 2010;12:286–293. doi: 10.1038/ncb2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hussey GS, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41:419–431. doi: 10.1016/j.molcel.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee AS, Kranzusch PJ, Cate JH. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522:111–114. doi: 10.1038/nature14267. Using transcriptome-wide PAR-CLIP analysis, this study identified a novel role for eIF3 in interacting non-canonically with stem–loops in the 5′UTR of select mRNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Somers J, Poyry T, Willis AE. A perspective on mammalian upstream open reading frame function. Int J Biochem Cell Biol. 2013;45:1690–1700. doi: 10.1016/j.biocel.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hopkins BD, et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science. 2013;341:399–402. doi: 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ingolia NT, et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–1379. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee S, et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci USA. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ye Y, et al. Analysis of human upstream open reading frames and impact on gene expression. Hum Genet. 2015;134:605–612. doi: 10.1007/s00439-015-1544-7. [DOI] [PubMed] [Google Scholar]
- 71.Jin X, Turcott E, Englehardt S, Mize GJ, Morris DR. The two upstream open reading frames of oncogene mdm2 have different translational regulatory properties. J Biol Chem. 2003;278:25716–25721. doi: 10.1074/jbc.M300316200. [DOI] [PubMed] [Google Scholar]
- 72.Brown CY, Mize GJ, Pineda M, George DL, Morris DR. Role of two upstream open reading frames in the translational control of oncogene mdm2. Oncogene. 1999;18:5631–5637. doi: 10.1038/sj.onc.1202949. [DOI] [PubMed] [Google Scholar]
- 73.Landers JE, Cassel SL, George DL. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res. 1997;57:3562–3568. [PubMed] [Google Scholar]
- 74.Mehta A, Trotta CR, Peltz SW. Derepression of the Her-2 uORF is mediated by a novel post-transcriptional control mechanism in cancer cells. Genes Dev. 2006;20:939–953. doi: 10.1101/gad.1388706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu L, et al. Mutation of the CDKN2A 5′ UTR creates an aberrant initiation codon and predisposes to melanoma. Nat Genet. 1999;21:128–132. doi: 10.1038/5082. [DOI] [PubMed] [Google Scholar]
- 76.Wethmar K, et al. C/EBPβΔORF mice — a genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010;24:15–20. doi: 10.1101/gad.557910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPβ at the core of the TGFβ cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 78.Begay V, et al. Deregulation of the endogenous C/EBPβ LIP isoform predisposes to tumorigenesis. J Mol Med (Berl ) 2015;93:39–49. doi: 10.1007/s00109-014-1215-5. [DOI] [PubMed] [Google Scholar]
- 79.Jia J, Yao P, Arif A, Fox PL. Regulation and dysregulation of 3′UTR-mediated translational control. Curr Opin Genet Dev. 2013;23:29–34. doi: 10.1016/j.gde.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 81.Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan J, Marr TG. Computational analysis of 3′-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Res. 2005;15:369–375. doi: 10.1101/gr.3109605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iwakawa HO, Tomari Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25:651–665. doi: 10.1016/j.tcb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 86.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 87.Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336:237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meijer HA, et al. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science. 2013;340:82–85. doi: 10.1126/science.1231197. [DOI] [PubMed] [Google Scholar]
- 90.Mathonnet G, et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007;317:1764–1767. doi: 10.1126/science.1146067. [DOI] [PubMed] [Google Scholar]
- 91.Eichhorn SW, et al. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 2014;56:104–115. doi: 10.1016/j.molcel.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bethune J, Artus-Revel CG, Filipowicz W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 2012;13:716–723. doi: 10.1038/embor.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508:66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fukao A, et al. MicroRNAs trigger dissociation of eIF4AI and eIF4AII from target mRNAs in humans. Mol Cell. 2014;56:79–89. doi: 10.1016/j.molcel.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 95.Fukaya T, Iwakawa HO, Tomari Y. MicroRNAs block assembly of eIF4F translation initiation complex in Drosophila. Mol Cell. 2014;56:67–78. doi: 10.1016/j.molcel.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 96.Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447:875–878. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- 97.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 98.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 99.Thomson JM, et al. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Novoa I, Gallego J, Ferreira PG, Mendez R. Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat Cell Biol. 2010;12:447–456. doi: 10.1038/ncb2046. [DOI] [PubMed] [Google Scholar]
- 101.Bava FA, et al. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature. 2013;495:121–125. doi: 10.1038/nature11901. This study identifies a novel function of CPEB1 in regulating 3′UTR shortening through APA in hundreds of mRNAs involved in cellular proliferation, cancer progression and pre-mRNA splicing. [DOI] [PubMed] [Google Scholar]
- 102.Masamha CP, et al. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510:412–416. doi: 10.1038/nature13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Izquierdo JM. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J Biol Chem. 2008;283:19077–19084. doi: 10.1074/jbc.M800017200. [DOI] [PubMed] [Google Scholar]
- 105.Dixon DA, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mazan-Mamczarz K, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA. 2003;100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eswarappa SM, et al. Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell. 2014;157:1605–1618. doi: 10.1016/j.cell.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014;15:R14. doi: 10.1186/gb-2014-15-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park SM, et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin Invest. 2015;125:1286–1298. doi: 10.1172/JCI78440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ito T, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466:765–768. doi: 10.1038/nature09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kharas MG, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16:903–908. doi: 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Castello A, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. This important study was the first to use quantitative MS in combination with UV crosslinking to identify the complete RBP–mRNA interactome, uncovering an unexpected number and diversity of RBPs within the cell. [DOI] [PubMed] [Google Scholar]
- 114.Kwon SC, et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 115.Baltz AG, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 116.Preitner N, et al. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell. 2014;158:368–382. doi: 10.1016/j.cell.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thoreen CC, et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. This paper, along with reference 26, identifies the 5′TOP and the PRTE as cis-regulatory elements that control translation of key mRNA subsets downstream of mTOR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xue S, et al. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature. 2015;517:33–38. doi: 10.1038/nature14010. This paper describes the generation of the first targeted knockout of a cellular IRES in mice and demonstrates a key functional role for IRES-driven translation in vivo. It also reveals that IRES-dependent translation is enabled by a new cis-regulatory element termed the TIE, which blocks cap-dependent translation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Levy S, Avni D, Hariharan N, Perry RP, Meyuhas O. Oligopyrimidine tract at the 5′ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc Natl Acad Sci USA. 1991;88:3319–3323. doi: 10.1073/pnas.88.8.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Avni D, Shama S, Loreni F, Meyuhas O. Vertebrate mRNAs with a 5′-terminal pyrimidine tract are candidates for translational repression in quiescent cells: characterization of the translational cis-regulatory element. Mol Cell Biol. 1994;14:3822–3833. doi: 10.1128/mcb.14.6.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jefferies HB, Reinhard C, Kozma SC, Thomas G. Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc Natl Acad Sci USA. 1994;91:4441–4445. doi: 10.1073/pnas.91.10.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pende M, et al. S6K1−/−/S6K2−/− mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ruvinsky I, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miloslavski R, et al. Oxygen sufficiency controls TOP mRNA translation via the TSC-Rheb-mTOR pathway in a 4E-BP-independent manner. J Mol Cell Biol. 2014;6:255–266. doi: 10.1093/jmcb/mju008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Patursky-Polischuk I, et al. The TSC-mTOR pathway mediates translational activation of TOP mRNAs by insulin largely in a raptor- or rictor-independent manner. Mol Cell Biol. 2009;29:640–649. doi: 10.1128/MCB.00980-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patursky-Polischuk I, et al. Reassessment of the role of TSC, mTORC1 and microRNAs in amino acids-meditated translational control of TOP mRNAs. PLoS ONE. 2014;9:e109410. doi: 10.1371/journal.pone.0109410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Feldman ME, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sheridan CM, et al. YB-1 and MTA1 protein levels and not DNA or mRNA alterations predict for prostate cancer recurrence. Oncotarget. 2015;6:7470–7480. doi: 10.18632/oncotarget.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tcherkezian J, et al. Proteomic analysis of cap-dependent translation identifies LARP1 as a key regulator of 5′TOP mRNA translation. Genes Dev. 2014;28:357–371. doi: 10.1101/gad.231407.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mura M, et al. LARP1 post-transcriptionally regulates mTOR and contributes to cancer progression. Oncogene. 2015;34:5025–5036. doi: 10.1038/onc.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fonseca BD, et al. La-related protein 1 (LARP1) represses terminal oligopyrimidine (TOP) mRNA translation downstream of mTOR complex 1 (mTORC1) J Biol Chem. 2015;290:15996–16020. doi: 10.1074/jbc.M114.621730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Meyer KD, et al. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhou J, et al. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 137.Carlile TM, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li X, et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol. 2015;11:592–597. doi: 10.1038/nchembio.1836. [DOI] [PubMed] [Google Scholar]
- 139.Lovejoy AF, Riordan DP, Brown PO. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE. 2014;9:e110799. doi: 10.1371/journal.pone.0110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schwartz S, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pavon-Eternod M, et al. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–7280. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Winter AG, et al. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc Natl Acad Sci USA. 2000;97:12619–12624. doi: 10.1073/pnas.230224097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhou Y, Goodenbour JM, Godley LA, Wickrema A, Pan T. High levels of tRNA abundance and alteration of tRNA charging by bortezomib in multiple myeloma. Biochem Biophys Res Commun. 2009;385:160–164. doi: 10.1016/j.bbrc.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pavon-Eternod M, Gomes S, Rosner MR, Pan T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA. 2013;19:461–466. doi: 10.1261/rna.037507.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- 146.Felton-Edkins ZA, et al. The mitogen-activated protein (MAP) kinase ERK induces tRNA synthesis by phosphorylating TFIIIB. EMBO J. 2003;22:2422–2432. doi: 10.1093/emboj/cdg240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wei Y, Tsang CK, Zheng XF. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009;28:2220–2230. doi: 10.1038/emboj.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci USA. 2010;107:11823–11828. doi: 10.1073/pnas.1005188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lampson BL, et al. Rare codons regulate KRas oncogenesis. Curr Biol. 2013;23:70–75. doi: 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci USA. 2004;101:12588–12591. doi: 10.1073/pnas.0404957101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gingold H, et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. This is a key study showing a link between tRNA expression patterns and codon usage bias in gene expression programmes that support cancer cell behaviours. [DOI] [PubMed] [Google Scholar]
- 153.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Spinola M, et al. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene. 2005;24:5502–5509. doi: 10.1038/sj.onc.1208687. [DOI] [PubMed] [Google Scholar]
- 155.Begley U, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007;28:860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chan CT, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Van den Born E, et al. ALKBH8-mediated formation of a novel diastereomeric pair of wobble nucleosides in mammalian tRNA. Nat Commun. 2011;2:172. doi: 10.1038/ncomms1173. [DOI] [PubMed] [Google Scholar]
- 158.Songe-Moller L, et al. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol. 2010;30:1814–1827. doi: 10.1128/MCB.01602-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fu D, et al. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol. 2010;30:2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kryukov GV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 161.Endres L, et al. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS ONE. 2015;10:e0131335. doi: 10.1371/journal.pone.0131335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cozen AE, et al. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods. 2015;12:879–884. doi: 10.1038/nmeth.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- 165.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 166.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leprivier G, Rotblat B, Khan D, Jan E, Sorensen PH. Stress-mediated translational control in cancer cells. Biochim Biophys Acta. 2015;1849:845–860. doi: 10.1016/j.bbagrm.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 168.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brugarolas J, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 172.Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–279. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 173.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 174.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2α kinase: relevance to anemias. Blood. 2007;109:2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Braunstein S, et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007;28:501–512. doi: 10.1016/j.molcel.2007.10.019. This study identifies a hypoxia-induced switch from cap-dependent to IRES-dependent translation that supports tumorigenesis by driving the expression of key pro-angiogenic and pro-survival mRNAs. [DOI] [PubMed] [Google Scholar]
- 176.Morfoisse F, et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep. 2014;6:155–167. doi: 10.1016/j.celrep.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 177.Gu L, et al. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell. 2009;15:363–375. doi: 10.1016/j.ccr.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen TM, et al. Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation. Nucleic Acids Res. 2014;42:2932–2944. doi: 10.1093/nar/gkt1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shi Y, et al. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene. 2015;35:1015–1024. doi: 10.1038/onc.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 180.Andreev DE, et al. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife. 2015;4:e03971. doi: 10.7554/eLife.03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Somers J, et al. A common polymorphism in the 5′ UTR of ERCC5 creates an upstream ORF that confers resistance to platinum-based chemotherapy. Genes Dev. 2015;29:1891–1896. doi: 10.1101/gad.261867.115. This article describes a common polymorphic variant that creates a new uORF in the 5′UTR of the DNA repair enzyme ERCC5 and promotes tumour resistance to platinum-based chemotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 184.Pola C, Formenti SC, Schneider RJ. Vitronectin-αvβ3 integrin engagement directs hypoxia-resistant mTOR activity and sustained protein synthesis linked to invasion by breast cancer cells. Cancer Res. 2013;73:4571–4578. doi: 10.1158/0008-5472.CAN-13-0218. [DOI] [PubMed] [Google Scholar]
- 185.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hart LS, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–4634. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cho PF, et al. A new paradigm for translational control: inhibition via 5′-3′ mRNA tethering by Bicoid and the eIF4E cognate 4EHP. Cell. 2005;121:411–423. doi: 10.1016/j.cell.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 188.Morita M, et al. A novel 4EHP-GIGYF2 translational repressor complex is essential for mammalian development. Mol Cell Biol. 2012;32:3585–3593. doi: 10.1128/MCB.00455-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.von Stechow L, et al. The E3 ubiquitin ligase ARIH1 protects against genotoxic stress by initiating a 4EHP-mediated mRNA translation arrest. Mol Cell Biol. 2015;35:1254–1268. doi: 10.1128/MCB.01152-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yi T, Papadopoulos E, Hagner PR, Wagner G. Hypoxia-inducible factor-1α (HIF-1α) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. J Biol Chem. 2013;288:18732–18742. doi: 10.1074/jbc.M113.471466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Uniacke J, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486:126–129. doi: 10.1038/nature11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Avdulov S, et al. eIF4E threshold levels differ in governing normal and neoplastic expansion of mammary stem and luminal progenitor cells. Cancer Res. 2015;75:687–697. doi: 10.1158/0008-5472.CAN-14-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Martinez A, et al. Phosphorylation of eIF4E confers resistance to cellular stress and DNA-damaging agents through an interaction with 4E-T: a rationale for novel therapeutic approaches. PLoS ONE. 2015;10:e0123352. doi: 10.1371/journal.pone.0123352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kenney JW, Moore CE, Wang X, Proud CG. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul. 2014;55:15–27. doi: 10.1016/j.jbior.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 195.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Graff JR, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Drygin D, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 198.Bywater MJ, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. This surprising study showed that therapeutic targeting of ribosome biogenesis through the inhibition of RNA polymerase I could selectively kill cancer cells in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.CX-5461 inhibits RNA Pol I in blood cancers. Cancer Discov. 2014;4:OF5. doi: 10.1158/2159-8290.CD-NB2014-154. No authors listed. [DOI] [PubMed] [Google Scholar]
- 200.Duncan R, Milburn SC, Hershey JW. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J Biol Chem. 1987;262:380–388. [PubMed] [Google Scholar]
- 201.Hiremath LS, Webb NR, Rhoads RE. Immunological detection of the messenger RNA cap-binding protein. J Biol Chem. 1985;260:7843–7849. [PubMed] [Google Scholar]
- 202.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. This manuscript provides the first functional evidence that eIF4E overexpression can be oncogenic and demonstrates that deregulated translation can act as a primary driver of tumorigenesis. [DOI] [PubMed] [Google Scholar]
- 203.Lazaris-Karatzas A, Sonenberg N. The mRNA 5′ cap-binding protein, eIF-4E, cooperates with v-myc or E1A in the transformation of primary rodent fibroblasts. Mol Cell Biol. 1992;12:1234–1238. doi: 10.1128/mcb.12.3.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ruggero D, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–486. doi: 10.1038/nm1042. This study is the first demonstration that eIF4E overexpression is sufficient to drive spontaneous tumorigenesis in vivo. [DOI] [PubMed] [Google Scholar]
- 205.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–173. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 206.Cheng EH, Gorelick FS, Czernik AJ, Bagaglio DM, Hait WN. Calmodulin-dependent protein kinases in rat glioblastoma. Cell Growth Differ. 1995;6:615–621. [PubMed] [Google Scholar]
- 207.Parmer TG, et al. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer. 1999;79:59–64. doi: 10.1038/sj.bjc.6690012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev. 2011;21:474–483. doi: 10.1016/j.gde.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Boon K, et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20:1383–1393. doi: 10.1093/emboj/20.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci USA. 2000;97:11198–11202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Grandori C, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 212.Arabi A, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 213.Cairns CA, White RJ. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 1998;17:3112–3123. doi: 10.1093/emboj/17.11.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.White RJ, Trouche D, Martin K, Jackson SP, Kouzarides T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature. 1996;382:88–90. doi: 10.1038/382088a0. [DOI] [PubMed] [Google Scholar]
- 215.Hannan KM, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862–8877. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004;18:423–434. doi: 10.1101/gad.285504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stefanovsky VY, et al. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol Cell. 2001;8:1063–1073. doi: 10.1016/s1097-2765(01)00384-7. [DOI] [PubMed] [Google Scholar]
- 218.Zhao J, Yuan X, Frodin M, Grummt I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell. 2003;11:405–413. doi: 10.1016/s1097-2765(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 219.Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kondrashov N, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Marcel V, et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013;24:318–330. doi: 10.1016/j.ccr.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ruggero D, et al. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- 223.Yoon A, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
- 224.Bellodi C, Kopmar N, Ruggero D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010;29:1865–1876. doi: 10.1038/emboj.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bellodi C, et al. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010;70:6026–6035. doi: 10.1158/0008-5472.CAN-09-4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bellodi C, et al. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep. 2013;3:1493–1502. doi: 10.1016/j.celrep.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gao L, et al. Genome-wide small nucleolar RNA expression analysis of lung cancer by next-generation deep sequencing. Int J Cancer. 2015;136:E623–E629. doi: 10.1002/ijc.29169. [DOI] [PubMed] [Google Scholar]
- 228.Jha P, et al. Genome-wide small noncoding RNA profiling of pediatric high-grade gliomas reveals deregulation of several miRNAs, identifies downregulation of snoRNA cluster HBII-52 and delineates H3F3A and TP53 mutant-specific miRNAs and snoRNAs. Int J Cancer. 2015;137:2343–2353. doi: 10.1002/ijc.29610. [DOI] [PubMed] [Google Scholar]
- 229.Martens-Uzunova ES, et al. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene. 2012;31:978–991. doi: 10.1038/onc.2011.304. [DOI] [PubMed] [Google Scholar]
- 230.Ravo M, et al. Small non-coding RNA deregulation in endometrial carcinogenesis. Oncotarget. 2015;6:4677–4691. doi: 10.18632/oncotarget.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ronchetti D, et al. Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Med Genom. 2013;6:27. doi: 10.1186/1755-8794-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wang X, et al. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc Natl Acad Sci USA. 1999;96:13118–13123. doi: 10.1073/pnas.96.23.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. This article provides the first description of ribosome profiling, a technology that has provided unparalleled insights into translational control of the cancer genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jan CH, Williams CC, Weissman JS. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. 2014;346:1257521. doi: 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Konig J, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chu C, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416. doi: 10.1016/j.cell.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.McHugh CA, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236. doi: 10.1038/nature14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc Natl Acad Sci USA. 2011;108:10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kertesz M, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Underwood JG, et al. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Spitale RC, et al. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lucks JB, et al. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci USA. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Spitale RC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–490. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sugimoto Y, et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 2015;519:491–494. doi: 10.1038/nature14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Khoddami V, Cairns BR. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol. 2013;31:458–464. doi: 10.1038/nbt.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hussain S, et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013;4:255–261. doi: 10.1016/j.celrep.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Squires JE, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chan CT, et al. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Basanta-Sanchez M, Temple S, Ansari SA, D’Amico A, Agris PF. Attomole quantification and global profile of RNA modifications: epitranscriptome of human neural stem cells. Nucleic Acids Res. 2015;44:e26. doi: 10.1093/nar/gkv971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zheng G, et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods. 2015;12:835–837. doi: 10.1038/nmeth.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]