

The SPAST gene, which produces two isoforms of the microtubule-severing protein spastin, is the chief gene mutated in hereditary spastic paraplegia. Truncated M1 spastin proteins are toxic and have the potential to accumulate in these patients.
Abstract
The SPAST gene, which produces two isoforms (M1 and M87) of the microtubule-severing protein spastin, is the chief gene mutated in hereditary spastic paraplegia. Haploinsufficiency is a popular explanation for the disease, in part because most of the >200 pathogenic mutations of the gene are truncating and expected to produce only vanishingly small amounts of shortened proteins. Here we studied two such mutations, N184X and S245X, and our results suggest another possibility. We found that the truncated M1 proteins can accumulate to notably higher levels than their truncated M87 or wild-type counterparts. Reminiscent of our earlier studies on a pathogenic mutation that generates full-length M1 and M87 proteins, truncated M1 was notably more detrimental to neurite outgrowth than truncated M87, and this was true for both N184X and S245X. The greater toxicity and tendency to accumulate suggest that, over time, truncated M1 could damage the corticospinal tracts of human patients. Curiously, the N184X mutation triggers the reinitiation of translation at a third start codon in SPAST, resulting in synthesis of a novel M187 spastin isoform that is able to sever microtubules. Thus microtubule severing may not be as reduced as previously assumed in the case of that mutation.
INTRODUCTION
More than 200 different mutations to the SPAST gene, also termed SPG4, account for the majority of cases of autosomal-dominant hereditary spastic paraplegia (HSP). Spasticity and weakness of lower limbs observed in HSP-SPG4 patients are caused by progressive degeneration of axons mainly within the corticospinal tracks (Hazan et al., 1999; Fonknechten et al., 2000; Shoukier et al., 2009). The SPAST open reading frame (ORF) has two initiation codons. A weak Kozak sequence surrounding the first initiation codon leads to leaky scanning of the first AUG and preferred translation from the second AUG. As a result, a 616–amino acid (68 kDa) isoform called M1 and a 530–amino acid (60 kDa) isoform called M87 are synthesized simultaneously (Claudiani et al., 2005). M87 is the predominant spastin isoform in all tissues at all stages of development, whereas M1 is only detectably present in adult spinal cord (Solowska et al., 2008, 2010). M87 is a microtubule-severing enzyme that breaks longer microtubules into shorter ones and thereby regulates the number and mobility of microtubules, as well as the distribution of their dynamic plus ends (Errico et al., 2002; Evans et al., 2005; Roll-Mecak and Vale, 2005; Baas et al., 2006). Because the overwhelming majority of pathogenic mutations destroy enzymatic activity, inadequate microtubule severing resulting from inactivation of one spastin allele (haploinsufficiency) has been proposed as the mechanism underlying the disease (Hazan et al., 1999; Bürger et al., 2000; Fonknechten et al., 2000; Lindsey et al., 2000). The role of M1 in the etiology of HSP-SPG4 is poorly understood. The 86–amino acid N-terminal domain present in only M1 spastin contains a hydrophobic region spanning amino acids 49–80 that forms a hairpin that can partially insert M1 into endoplasmic reticulum (ER) membrane to participate in ER shaping (Sanderson et al., 2006; Park et al., 2010; Blackstone et al., 2011, 2012). It is not clear, however, to what extent the M1 isoform participates in microtubule severing.
Because genetic analyses of HSP-SPG4 patients have not revealed any correlation between spastin levels and the severity of neurodegenerative symptoms (Yip et al., 2003; Shoukier et al., 2009), we proposed that SPAST mutations might also result in synthesis of neurotoxic spastin proteins (Solowska and Baas, 2015). We previously showed that full-length human spastin carrying a pathological missense mutation had detrimental effects on neurite outgrowth when expressed in cultured neurons and on motor function in Drosophila (Solowska et al., 2008, 2014). The majority of pathogenic SPAST mutations, however, result in synthesis of truncated proteins that are believed to be less stable than full-length proteins. Moreover, the presence of premature termination codons (PTCs) responsible for the synthesis of truncated proteins typically leads to nonsense-mediated decay (NMD) of the affected mRNA (Lykke-Andersen and Jensen, 2015; Popp and Maquat, 2016). Therefore it has been assumed that the levels of truncated spastin proteins would be too low to cause axonal degeneration observed in HSP-SPG4, and this has been a principal argument for haploinsufficiency as the basis of the disease as opposed to toxicity of mutant spastin proteins. To investigate this, we tested the accumulation and toxicity of spastin isoforms encoded by N184X and S245X, two SPAST-truncating mutations found in HSP patients (Lindsey et al., 2000; McDermott et al., 2006; Shoukier et al., 2009).
RESULTS
N184X nonsense mutation promotes reinitiation of translation at a third start codon present in human spastin
Most SPAST mutations, including splice-site mutations, insertions/deletions (with the exception of in-frame exon deletions), and missense point mutations, generate PTCs. Here we examined whether truncated spastin proteins resulting from the presence of PTCs participate in the etiology of HSP-SPG4, in light of the predominant view that they do not. We generated human spastin constructs SP N184X and SP S245X carrying PTCs found in HSP-SPG4 patients (Lindsey et al., 2000; McDermott et al., 2006; Shoukier et al., 2009). Truncated spastin proteins encoded by SP N184X and SP S245X would presumably lack microtubule-binding domain (MTBD) and the ATPase associated with various cellular activities (AAA) domain necessary for microtubule severing (Figure 1A). Indeed, microtubules in cells transfected with SP S245X construct were indistinguishable from the intact, long microtubules in nonexpressing cells. Surprisingly, however, microtubules in cells transfected with SP N184X construct were clearly severed and as short as microtubules in cells transfected with wild-type (WT) M87 spastin (Figure 1B). DNA sequencing confirmed the presence of N184X and S245X mutations in spastin cDNA (Figure 2A). Because of a leaky scanning of the first AUG with a weak Kozak consensus sequence tgaATGa, we predicted expression of two truncated spastins—M1 (∼20 kDa) and M87 (∼12 kDa) in cells transfected with SP N184X, and M1 (∼27 kDa) and M87 (∼17 kDa) in cells transfected with SP S245X (Figure 2B). To test this prediction, we transfected rat fibroblasts synthesizing the tetracycline-controlled transactivator Tet-On Advanced (RFL-6 TET cells) with SP N184X or SP S245X in pTRE-Tight vector and induced spastin synthesis with 1 µg/ml doxycycline (Dox). Subsequent immunoblotting with SP5 anti-spastin antibody confirmed the presence of truncated spastins in cell lysates (Figure 2, C and D). Of interest, however, unlike the WT M1 isoform, which, due to inefficient initiation of translation, is always expressed at a lower level than the WT M87 isoform, the truncated M1 spastins, despite a weak Kozak consensus sequence, were expressed at higher levels than their truncated M87 counterparts. Indeed, we were unable to detect M87 N184X protein by immunoblotting (Figure 2D). These results were the first indication that truncated M1 proteins might accumulate significantly more than corresponding M87 proteins.
FIGURE 1:
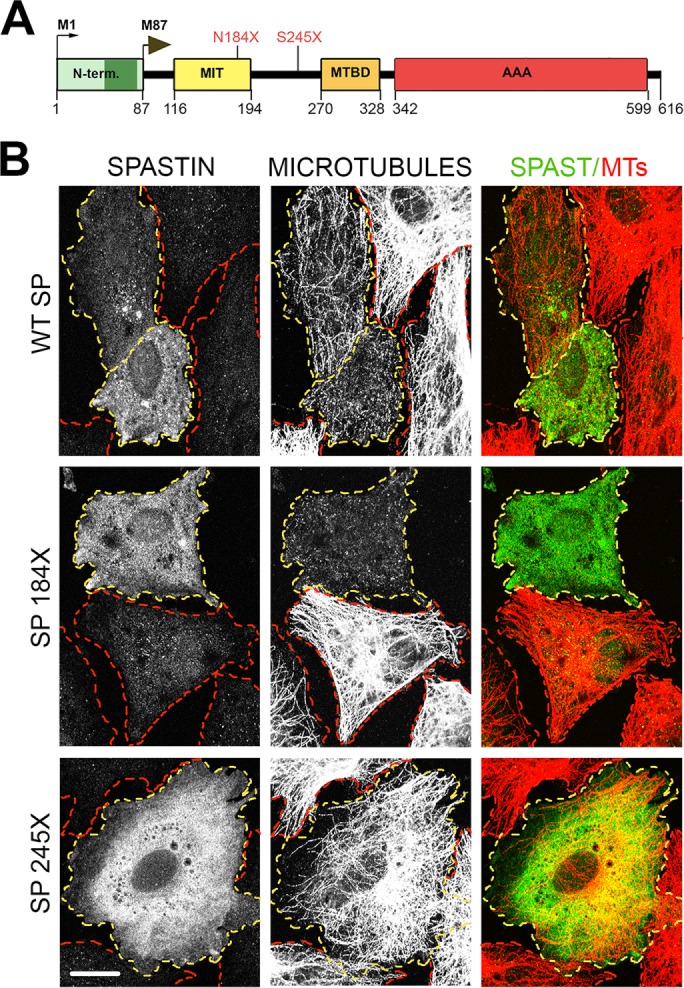
Effects of truncating mutations on spastin microtubule-severing activity. (A) Schematic representation of spastin functional domains and localization of two nonsense mutations N184X and S245X (red). AAA, ATPase associated with various cellular activities (amino acids 342–599); MIT, microtubule-interacting and trafficking domain (amino acids 116–194); MTBD, microtubule-binding domain (amino acids 270–328); N-term, N-terminal domain (amino acids 1–86), present only in the M1 spastin isoform, includes a hydrophobic region (dark green, amino acids 49–80). The premature termination codons N184X and S245X generated in WT spastin cDNA would result in synthesis of truncated spastins lacking MTBD and AAA domain required for microtubule severing. (B) Microtubules are severed in rat fibroblasts transfected with WT spastin cDNA and, unexpectedly, also in cells transfected with spastin cDNA carrying the N184X mutation, but not in cells transfected with spastin cDNA with the S245X mutation. The presence of WT and mutated spastins was detected with the anti–spastin SP5 antibody. Microtubules were stained with the anti-tubulin antibody. Cells expressing spastins are outlined in yellow and nonexpressing cells in red. Scale bar, 20 μm.
FIGURE 2:

N184X PTC promotes reinitiation of translation from the third start codon. (A) Sequencing confirms the presence of N184X and S245X mutations in spastin cDNAs. (B) Schematic representation of truncated spastin proteins encoded by cDNAs with N184X or S245X mutations. A poor Kozak consensus sequence at the M1 initiation codon leads to a preferred initiation of translation at the M87 initiation codon. As a result, both truncated M1 and M87 spastin isoforms should be expressed simultaneously. (C) Two antibodies against amino acids 90–283 (SP5) and 337–465 (SP/AAA) were generated to detect truncated and full-length spastin proteins. (D) Immunoblotting with anti–spastin SP5 antibody confirmed the presence of truncated spastins in lysates from cells transfected with SP N184X or SP S245X and treated with Dox. Despite a weak Kozak sequence at the M1 initiation codon, the M1-truncated spastins were expressed at higher levels than corresponding M87 isoforms. Indeed, we were unable to detect M87 N184X protein. (E) No bands corresponding to full-length spastin isoforms were detected by immunoblotting of lysate from cells transfected with SP N184X construct. Instead, a band of ∼48 kDa was identified by immunoblotting with anti–spastin SP/AAA antibody. (F) Kozak consensus sequence for optimal recognition of the AUG start codon includes purine A/G in position –3 and G in position +4 (bold letters; Kozak, 2002). Examination of human spastin mRNA shows that sequence surrounding M1 AUG deviates significantly from the consensus motif, with neither the purine in position –3 nor the guanine in position +4. Deviations from an optimal Kozak consensus sequence are shown in red. M87 AUG is in a better context, with a guanine in position +4. The third AUG start codon at spastin M187 is surrounded by a sequence most closely resembling the optimal Kozak consensus sequence. (G) Schematic representation of protein expressed in cells transfected with SP N187X. (H) Human and mouse spastin have the third initiation codon.
To investigate further the unexpected severing activity observed in cells transfected with SP N184X construct, we used SP/AAA anti-spastin antibody to test whether full-length spastin was expressed as a result of the readthrough of the 184 PTC. Immunoblot analysis of the lysates from cells transfected with SP N184X revealed the presence of a prominent ∼48-kDa band but did not detect any bands corresponding to full-length, 68-kDa M1 or 60-kDa M87 spastin isoforms, which were readily detectable in a lysate from WT spastin–transfected cells, despite a significantly lower protein load indicated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels (Figure 2E). Instead, examination of human spastin mRNA revealed the presence of an in-frame M187 initiation codon surrounded by Kozak consensus sequence better than that surrounding M1 or M87 (Figure 2F; Kozak, 2002). The reinitiation of translation from this third start codon explains the presence of ∼48-kDa M187 protein detected by immunoblotting. MTBD and AAA domain present in M187 spastin are sufficient for the microtubule severing observed in cells transfected with SP N184X construct (White et al., 2007). A schematic representation of proteins encoded by SP N184X is shown in Figure 2G (also see Table 1). The third initiation codon with Kozak sequence identical to human is also found in mouse spastin DNA (Figure 2H).
TABLE 1:
Characteristics of spastin isoforms encoded by spastin constructs.
| Construct | Spastin isoforms expressed simultaneouslya | Amino acids | Molecular weight (kDa) | Severing activityb |
|---|---|---|---|---|
| WT SP | WT M1 | 1–616 | 68 | + |
| WT M87 | 87–616 | 60 | ++++ | |
| SP N184X | M1 N184X | 1–184 | 20 | None |
| M87 N184X | 87–184 | 12c | None | |
| M187 | 187–616 | 48 | ++++ | |
| SP S245X | M1 S245 | 1–245 | 27 | None |
| M87 S245X | 87–245 | 17 | None |
aM1 and M87 isoforms are expressed simultaneously as a result of leaky scanning of the first initiation codon and preferred translation from the second initiation codon. M187 isoform is expressed as a result of reinitiation of translation from the third initiation codon.
bLow expression levels of WT M87 or M187 isoform resulted in a loss of ∼90% of microtubules, and significantly higher expression of WT M1 resulted in ∼50% microtubule loss.
cM87 N184X isoform was not detected.
Because the reinitiation of translation would result in synthesis of M187 in patients carrying the N184X mutation, we compared the microtubule-severing activity of M187, WT M87, and WT M1. We transfected RFL-6 TET cells with WT M87 or WT M1 construct expressing only WT M87 or WT M1 isoform, with M187 construct expressing only M187 isoform, or with SP N184X construct expressing simultaneously M187 and truncated M1 and M87 isoforms (see Materials and Methods). The synthesis of spastin proteins was induced with 1 µg/ml Dox. After 21 h, cells were fixed and stained with anti-spastin SP/AAA antibody, which detects only full-length M1, M87, or M187 proteins but not truncated spastin in SP N184X–transfected cells. Microtubules were stained with anti-tubulin antibody. For WT M87, M187, and SP N184X transfected cells, the fluorescence intensity of spastin staining and tubulin staining was measured in 55–60 randomly chosen cells with ∼1-µm-long microtubules, and the 30 lowest values were used to calculate the lowest levels of spastin required for such severing. An example of a cell with a low level of M187 and short microtubules is shown in Figure 3A. The WT M1 isoform expressed even at high levels did not sever microtubules into short fragments (Figure 3B). Quantitative analysis revealed that the expression levels of WT M87 and M187 sufficient to cause the loss of ∼90% of microtubules were not statistically different. We also found that truncated spastins in cells transfected with SP N184X cDNA did not affect the microtubule-severing activity of the coexpressed M187 spastin. Despite significantly higher expression levels, the microtubule loss in cells transfected with WT M1 was significantly lower than that in cells expressing WT M87 or M187 spastin (Figure 3C). Table 1 summarizes the properties of spastin isoforms encoded by WT SP, SP N184X, and SP S245X constructs.
FIGURE 3:

Microtubule-severing activity of spastin isoforms. (A) Low expression levels of M187 spastin results in microtubules shorter than 1 μm. (B) In contrast to WT M87 or M187, even high levels of WT M1 spastin do not produce short microtubules. (C) Quantitative analyses indicate that equal expression levels of WT M87 and M187 result in equal microtubule loss. Coexpression of truncated spastin isoforms in cells transfected with SP N184X construct did not affect the severing activity of M187. Despite significantly higher expression level, the microtubule loss in cells transfected with WT M1 was significantly lower than that in cells transfected with WT M87, M187, or SP N184X. All statistical analyses were performed using one-way analysis of variance (ANOVA) with Bonferroni post hoc test. Scale bar, 20 μm.
Truncated M1 spastins accumulate to higher levels than WT spastin
Typically, the presence of PTCs responsible for the synthesis of truncated proteins promotes mRNA destabilization by NMD. The outcome of NMD, however, can vary at both transcript and protein levels. During mRNA splicing, a multisubunit landmark of proteins termed exon junction complexes (EJCs) are deposited 20–24 nucleotides upstream of the exon–exon junctions and then removed from the mRNA by the elongating ribosome. PTCs located >50–55 nucleotides upstream from an exon–exon junction prevent the removal of downstream EJCs, and such leftover EJC(s) serve as anchoring points for the assembly of MND factors and subsequent degradation of mRNA (Schweingruber et al., 2013; Lykke-Andersen and Jensen, 2015; Popp and Maquat, 2016). The N184X mutation, located 35 nucleotides upstream from the exon 3/4 junction, represents an interesting case in which translation of truncated M1/M87 proteins removes the first three EJCs, and then reinitiation of translation at M187 removes the remaining EJCs, preventing NMD (Figure 4, A and B). In the case of the S245X mutation, located >55 nucleotides upstream from the exon 5/6 junction, only the first four EJCs will be removed, leaving mRNA vulnerable to NMD (Figure 4C). However, because the removal of EJCs requires a pioneer round of translation, the truncated protein would still be synthesized.
FIGURE 4:

Hypothetical effects of premature termination codons 184X and 245X on stability of spastin mRNA. (A) Localization of spastin functional domains within 17 exons in spastin mRNA. The 184X termination codon UAA (red) is localized 7 bases upstream from the AUG of M187 initiation codon (green) and 35 bases upstream from exon 3/4 junction AG/AG (bold black). The 245X is localized in exon 5. (B) Pioneer round of translation of truncated 184X spastin would remove EJCs (red stars) from exons 1–3, and reinitiation of translation at M187 AUG would remove remaining EJCs from exons 4–16. (C) Pioneer round of translation of truncated 245X spastin would remove EJCs from exons 1–4. Remaining EJCs might render mRNA unstable.
Truncated proteins are usually less stable than their full-length counterparts, and indeed that seems to be the case regarding truncated M87 spastins. However, the high expression levels of truncated M1 spastins observed (Figure 2, D and E) indicate that these proteins are stable. To test the tendency of spastin proteins to accumulate, we generated a panel of constructs expressing separately M1 and M87 isoforms (see Materials and Methods). To directly compare accumulation of truncated spastins to accumulation of mutated full-length spastins, we included full-length M1 and M87 carrying the pathological missense C448Y mutation (Hazan et al., 1999; Fonknechten et al., 2000; Solowska et al., 2014). To ensure equal efficiency of translation, we replaced imperfect Kozak sequences at M1 and M87 initiation codons by optimal consensus sequences and then cloned the cDNAs into pTRE-Tight vector carrying an inducible promoter (Figure 5A). Individual spastin isoforms were synthesized in the presence of 0.25–2 µg/ml Dox in rat fibroblasts RFL-6 TET. We measured the levels of spastin expression in transfected cells by immunoblot analysis with anti–spastin SP5 antibody, which recognizes full-length and truncated spastins. We found that, at any given Dox concentration, the expression of M1 spastin was higher than that of its M87 counterpart (Figure 5, B and C).
FIGURE 5:

Relative accumulation of individually expressed WT and mutated spastin isoforms. (A) Schematic representation of M1 and M87 constructs in pTRE-Tight vector carrying Dox-inducible promoter TRE/CMV. Weak Kozak sequences at M1 and M87 initiation codons were replaced by optimal Kozak sequences (green). (B) Immunoblot analysis of lysates from RFL-6 TET cells transfected with M1 and M87 spastin constructs and treated with 0.0, 0.25, 0.5, 1.0, or 2.0 μg/ml Dox for 7 h. Each band represents aliquots collected from four independent transfections and combined for SDS–PAGE. Spastin proteins were detected using anti–spastin SP5 antibody and secondary antibody conjugated with horseradish peroxidase. After reaction with chemiluminescent peroxidase substrate, all blots were imaged using a high-resolution CCD camera. (C) SDS–PAGE analysis of lysates collected from four independent transfections of RFL-6 with M1 and M87 spastin constructs after treatment with 0.25 μg/ml Dox for 7 h. Spastin proteins were detected as in B. (D) Coomassie staining was used to ensure equal total protein loads in samples used for immunoblotting. (E) The optical densities of bands representing M1 and M87 spastins were measured using Image Lab software for identically generated images. Regardless of Dox concentration, the intensity of bands representing M1 was higher than that of M87 bands. (F) Quantitative analysis shows that accumulation of M87 C448Y and M87 S245X was not significantly different from that of WT M87. Compared to WT M1, all M87 spastins accumulate significantly less (p < 0.001) and all mutated M1 spastins accumulate more than WT M1 (p < 0.001). Of interest, truncated M1 spastins accumulate to significantly higher level than full-length M1 C448Y spastin (p < 0.001). All statistical analyses were performed using one-way ANOVA with Dunnett’s post hoc test.
To quantify the accumulation of individual spastin isoforms at low expression levels, we performed four separate transfections and induced spastin synthesis with 0.25 µg/ml Dox for 7 h (Figure 5D). Immunoblot analysis showed again that the M1 spastins accumulate to significantly higher levels than M87 spastins (p < 0.001). Although the levels of truncated M87 S245X spastin were consistently lower and the levels of full-length M87 C448Y spastin consistently higher than the levels of WT M87 spastin, these differences were not statistically significant. Of interest, however, compared with WT M1, the levels of mutated M1 spastins were significantly higher (p < 0.001). Moreover, the accumulation of truncated M1 spastins was significantly higher than that of full-length M1 C448Y (p < 0.001) but the accumulation of M1 N184X spastin was not statistically different from that of M1 S245X spastin (Figure 5E). These results suggest that a lesser stability of the mRNAs with PTCs encoding truncated spastins might be at least partially compensated for by a greater metabolic stability of truncated M1.
Truncated M1 spastins have greater effect on neurite outgrowth than truncated M87 spastin
To examine the effect of truncated spastins on neuronal cells, we transfected rat primary cortical neurons with cDNAs encoding M1 N184X, M1 S245X, or M87 S245X spastin. Because the expression levels of M87 N184X were below detection, we were unable to test this isoform. Before neuronal cell transfection, we found that the optimal Kozak consensus sequence at the M1 N184X construct completely abolished reinitiation of translation at M187 (Figure 6A), and consequently no microtubule severing was observed in cells transfected with M1 N184X (Figure 6B). Therefore the effects of M1 N184X expression observed in neurons cannot be attributed to excessive microtubule severing. Because M1 S245X and M87 S245X spastins do not have MTBD and AAA domain, they also cannot sever microtubules.
FIGURE 6:
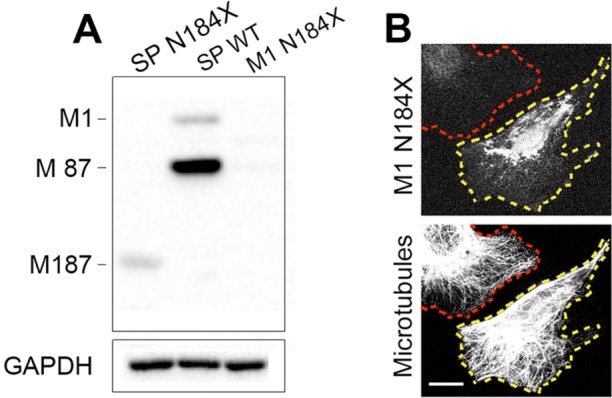
Optimal Kozak consensus sequence at M1 N184X prevents translation of M187 isoform. (A) Immunoblotting with SP/AAA anti-spastin antibody shows that M187 isoform present in cells transfected with SP N184X construct is not detected in cells transfected with WT spastin or with M1 N184X construct with an optimal Kozak sequence at M1 initiation codon. (B) The lack of microtubule severing in cells transfected with M1 N184X construct (outlined in yellow) confirms that efficient initiation of M1 translation prevents reinitiation of M187 translation downstream from N184X termination codon. Scale bar, 20 µm.
The neuronal cells transfected with constructs encoding individual truncated spastins were fixed 21 h later and double-stained with anti-spastin SP5 antibody and anti-tubulin antibody (Figure 7A). Neurons transfected with green fluorescent protein (GFP) were used to test the effect of a randomly expressed protein on neurite outgrowth (Figure 7B). Examination of transfected cells revealed considerable differences in toxicity between truncated spastins. Despite very low levels of expression, only ∼22% of neuronal cells (25 of 116 cells) synthesizing M1 N184X grew neurites. The percentage of cells with neurites was 39.4% (140 of 355 cells) for M1 S245X and 57.8% (288 of 498 cells) for M87 S245X. By comparison, ∼86% of control, mock-transfected cells or GFP-transfected cells grew neurites. To assess more thoroughly the effect of mutated spastins on neurite outgrowth, we measured the levels of spastins and the length of neurites from individual cells. The data, presented in Figure 7C, show that the level of M1 N184X expression is very low in all cells with neurites. The highest levels of expression were measured in M87 S245X-expressing cells with neurites. There was no overlap between spastin-expressing cells (Figure 7C, blue circles) and control, mock-transfected cells (Figure 7C, orange circles). Quantitative analysis confirmed these observations (Figure 7, D and E). Despite the highest levels of spastin (3472 ± 68.2 arbitrary fluorescence units [AFU]), the M87 S245X cells grew the longest neurites (36.1 ± 1.5 µm), but they were still significantly shorter than for control cells (89.8 ± 3.4 µm) or GFP-transfected cells (86.9 ± 3.3 µm). Even though the expression of M1 S245X was significantly lower (2978.4 ± 79.9 AFU) than that of M87 245X (3472 ± 68.2 AFU), the neurites of M1 S245X cells were significantly shorter (24.39 ± 1.6 vs. 36.1 ± 1.5 µm). Although the difference in neurite outgrowth between M1 S245X- and M1 N184X-expressing cells was not statistically significant (24.39 ± 1.6 and 18.1 ± 1.5 µm, respectively), the expression level of M1 N184X (1264.6 ± 33.8 AFU) was significantly lower than that of M1 S245X (2978.4 ± 79.9 AFU). The results of neuronal cell transfections are summarized in Table 2. Experiments described earlier showed that M1 N184X and M1 S245X can accumulate to significantly higher levels than M87 S245X (Figure 5, D and E). Therefore the lower levels of truncated M1 spastins than M87 S245X in neuronal cells with neurites most likely indicate their significantly higher toxicity. As a result, only cells expressing relatively low levels of truncated M1 spastins are able to grow neurites. Because the truncated spastins did not have MTBD and AAA domain necessary for spastin–spastin interaction, it is unlikely that detrimental effects of these proteins on neurite outgrowth were caused by dominant-negative inhibition of endogenous spastin-severing activity.
FIGURE 7:

Effect of truncated spastins on neurite outgrowth. (A) Rat cortical neurons were transfected with M1 N184X, M1 S245X, or M87 S245X construct and, 24 h after transfection, double labeled with SP5 anti-spastin antibody and anti-tubulin antibody. The blue arrowheads indicate spastin-expressing cells, and the orange arrowheads denote the nonexpressing cells. (B) Rat cortical neurons transfected with GFP. (C) Distribution of control, mock-transfected cells (n = 60; orange circles), and M1 N184X (n = 25; light blue circles; top), M1 S245X (n = 80; blue circles; middle), and M87 S245X (n = 96; dark blue circles; bottom) transfected neuronal cells according to neurite length (micrometers) and fluorescence intensity of spastins (AFU). (D) Quantitative analysis demonstrates that neuronal cells expressing truncated spastins grew processes significantly shorter than control cells or cells expressing GFP (p < 0.001). In addition, M1 S245X–expressing cells grew neurites significantly shorter than cells expressing M87 S245X spastin (p < 0.001). (E) Although there was no statistically significant difference in neurite length between the M1 N184X– and M1 S245X–expressing cells (see D), the levels of M1 S245X spastin in cells growing neurites were significantly higher than the levels of spastin in M1 N184X cells still able to grow neurites (p < 0.001). The M87 S245X cells that grew the longest neurites also expressed significantly higher levels of spastin than either M1 N184X or M1 S245X cells (p < 0.001). All statistical analyses were performed using one-way ANOVA with Dunnett’s post hoc test. Scale bar, 20 µm (A, B).
TABLE 2:
Effect of truncated spastins on neurite outgrowth in primary neuronal cells.
| Truncated spastin | Severing activity | Number of neurons | Expression level (AFU)a | Neurite length (μm)b | Relation of spastin expression level to neurite length |
|---|---|---|---|---|---|
| M87 S245X | None | 96 | 3472 ± 68.2 | 36.1 ± 1.5 | +++/+++ |
| M87 N184X | None | ND | Not detected | ND | ND |
| M1 S245X | None | 80 | 2978 ± 79.9 | 24.4 ± 1.6 | ++/++ |
| M1 N184X | None | 25 | 1264 ± 33.8 | 18.1 ± 1.5 | +/++ |
ND, not done.
aSpastin isoform expression in transfected neurons was significantly higher (p < 0.001) than background expression of endogenous spastin.
bNeurites of spastin transfected neurons were significantly shorter (p < 0.001) than neurites of nontransfected neurons or of neurons expressing GFP.
DISCUSSION
Our earlier studies showed that expression of full-length human spastin carrying the pathological missense mutation C448Y was detrimental to the outgrowth of neurites from cultured neurons, affected microtubule dynamics in fibroblasts, and had ill effects on motor function in Drosophila (Solowska et al., 2014). However, because the majority of pathogenic SPAST mutations are truncating, here we examined the hypothesis that truncated human spastins also display characteristics allowing them to contribute to HSP-SPG4. Most researchers have favored the view that due to potential instability of the mRNA and/or protein, such mutations would result in only vanishingly low levels of truncated spastin proteins, so low as to be inconsequential to the disease (for detailed discussion of the controversy, see Solowska and Baas, 2015). To investigate the potential merits of our alternative hypothesis, we introduced N184X and S245X pathogenic mutations (Lindsey et al., 2000; McDermott et al., 2006; Shoukier et al., 2009) into full-length human spastin cDNA and examined the accumulation and toxicity of resulting truncated spastin isoforms.
Significance of the third initiation codon present in human spastin ORF
The absence of the MTBD and AAA domain from truncated M1 and M87 spastin isoforms encoded by SP N184X or SP S245X constructs would eliminate their microtubule-severing activity. Unexpectedly, however, microtubules were severed in cells transfected with the SP N184X. Follow-up experiments revealed that N184X PTC activates a reinitiation of translation from a newly discovered third start codon located seven nucleotides downstream of 184X and results in a synthesis of an ∼48-kDa M187 spastin. This isoform includes the spastin regions encoding the MTBD and AAA domain sufficient for microtubule severing (White et al., 2007). The presence of N184X PTC seems to be necessary for initiation of M187 translation because this spastin isoform was never detected in cells transfected with WT spastin cDNA. Because the microtubule-severing activity of the M187 isoform is comparable to that of WT M87 spastin, patients carrying the N184X mutation would experience <50% loss of spastin activity. Moreover, the reinitiation of translation at M187 would prevent NMD of mutated spastin mRNA by removing EJCs from exons 4–16. However, the M187 isoform, unlike WT M87, lacks the MIT domain responsible for interaction with two ESCRT-III proteins, CHMP1B and IST1, and therefore cannot be recruited to endosomes or the midbody and involved in endosomal trafficking or cytokinesis (Connell et al., 2009; Guizetti et al., 2011; Allison et al., 2013). M187 spastin will also be unable to interact with centrosomal protein NA14, which targets WT M87 to the centrosome and midbody to facilitate microtubule severing in these locations (Errico et al., 2004; Goyal et al., 2014). Thus, with this particular mutation, microtubule severing would not be as compromised as expected before our findings, although it is also unlikely that the presence of M187 can fully compensate for the lost M87.
Significance of different accumulation and toxicity of truncated M1 and M87
The presence of PTCs resulting in decay of affected mRNAs would greatly decrease the expression of truncated and potentially toxic proteins (Rebbapragada and Lykke-Andersen, 2009; Lykke-Andersen and Jensen, 2015; Popp and Maquat, 2016). Some mRNAs, however, can escape degradation, and, indeed, truncated spastin transcripts with PTCs were detected in mouse models of HSP-SPG4 (Tarrade et al., 2006; Kasher et al., 2009). Because mRNAs in our experiments were transcribed from cDNAs, they were already spliced and did not carry EJCs. Therefore the effect of PTCs on the stability of such mRNAs could not be determined. Instead, we tested the accumulation of individually expressed WT and mutated spastins synthesized under control of identical regulatory elements. The transcription of spastin mRNA was controlled by the Dox-inducible TRE/CMV promoter, and equal efficiency of translation was ensured by replacing the endogenous imperfect Kozak sequences surrounding M1 and M87 initiation codons with optimal Kozak consensus sequences. When the levels of protein synthesis are equal, the greater accumulation would suggest that a given protein is degraded less effectively. The results of our experiments show that under conditions promoting equal levels of synthesis, M1 spastins always accumulate significantly more than M87 spastins, suggesting that M1 spastins may be significantly more stable. Relatively low accumulation of truncated M87 S245X, combined with unstable mRNA with S245X PTC and lack of detectable M87 N184X, might explain why truncated M87 spastins were not detected in brains of mouse models of HSP-SPG4 (Tarrade et al., 2006; Kasher et al., 2009; Fassier et al., 2013), in lymphoblastoid cell lines derived from human HSP-SPG4 patients (Riano et al., 2009), or in neuronal cells derived from pluripotent stem cells generated from fibroblasts of HSP-SPG4 patients (Denton et al., 2014; Havlicek et al. 2014). Such low metabolic stability of truncated proteins is typical and prevents their accumulation to potentially toxic levels. Contrary to this general rule, truncated M1 spastins accumulated to significantly higher levels not only compared with WT M1 and M87, but also compared with full-length M1 with missense mutation C448Y, which was found to be neurotoxic in our earlier studies (Solowska et al., 2014). The low level of M1 expression in vivo most likely results from down-regulation of M1 translation by GC-rich 5’ untranslated region, the upstream AUG sequence, and a weak Kozak consensus sequence at the first initiation codon. Because such translation-reducing measures are often used to prevent overproduction of toxic proteins (Kozak, 2002), one might assume that even WT M1 is harmful at higher levels. We speculate that increased ability of truncated M1 proteins to accumulate might, to at least some degree, compensate for the instability of corresponding mRNAs and lead to toxic levels of expression. Truncated M1 spastins were also significantly more detrimental to neurite outgrowth than truncated M87. Indeed, only neuronal cells expressing very low levels of M1 N184X were able to grow neurites, and, although the levels of M1 S245X in neurons with neurites were higher than that of M1 N184X, they were still significantly lower than that of M87 S245X. Despite the low levels of expression, the neurites grown by cells with M1 N184X or M1 S245X were significantly shorter, not only than the neurites grown by control cells or cells expressing GFP, but also than those in cells synthesizing truncated M87 S245X. The reduced neurite outgrowth caused by the presence of truncated spastins contrasts with the increase of neurite length and branching observed in neurons expressing moderately enhanced levels of WT spastin (Riano et al., 2009; Qiang et al., 2010). It is unlikely, however, that the observed detrimental effects on neurite outgrowth are caused by dominant-negative activity of truncated spastins because they lack the MTBD and AAA domain required for spastin–spastin interaction as well as microtubule binding (White et al., 2007). The higher toxicity of M1 than M87 is not limited to truncated spastins, as we also observed the same phenomenon in cultured neurons and Drosophila expressing full-length M1 and M87 spastins with the C448Y mutation (Solowska et al., 2014).
Together these results support the idea that pathogenic mutations of SPAST leading to HSP have different effects on M87 and M1 spastin isoforms. The reduction in the levels of functional WT M87 would presumably be responsible for any aspects of the disease that are caused by insufficient microtubule severing (if insufficient microtubule severing actually does contribute to the disease). The low stability of mRNA with PTCs combined with low stability of truncated M87 spastins makes the accumulation of this isoform to neurotoxic levels unlikely in vivo. In contrast, because WT M1 is expressed at very low levels and cuts microtubules significantly less effectively than WT M87, microtubule severing would not be affected greatly by M1 mutations. Instead, mutant/truncated forms of M1 would be responsible for gain-of-function neurotoxicity that is exacerbated over time by the potential accumulation of the toxic M1 in corticospinal axons.
Hypothetical mechanisms of truncated M1 toxicity
M87 is a soluble, cytoplasmic protein, and the hydrophobic amino acids 49–80 in the M1 N-terminal domain form a hairpin that partially inserts M1 into ER membrane. M1 hydrophobic hairpins can interact with hydrophobic hairpins of atlastin-1, a large, integral-membrane GTPase that mediates homotypic fusion of ER tubules, and REEP1, an ER morphogen that links ER membranes to microtubules (Evans et al., 2006; Sanderson et al., 2006; Park et al., 2010; Blackstone et al., 2011). Coordinated interaction of atlastin-1, M1 spastin, and REEP1 has been posited to be important for shaping of the ER tubules and for ER–microtubule interactions that build the tubular ER network (Park et al., 2010; Blackstone et al., 2011; Blackstone, 2012). It is not clear whether M1 inserted into ER membrane forms hexamers with M87 to sever microtubules. Because the truncated M1 spastins do have the N-terminal domain, they might still insert into ER membrane, but their interaction with atlastin-1 and REEP1 might be disrupted and affect the curvature of ER membranes. Lack of MTBD in truncated M1 might affect the interaction of atlastin-1, M1 spastin, and REEP1 complexes with microtubules. Of interest, mutations of atlastin-1 affecting interaction of this protein with spastin are associated with HSP-SPG3A, the second-most-common HSP (Zhao et al., 2001; Evans et al., 2006), and mutations in REEP1 that prevent interaction with microtubules were reported in patients with HSP-SPG31 (Beetz et al., 2008; Park et al., 2010). These findings support the view that mutations in proteins involved in ER shaping and microtubule interactions play a role in the pathogenesis of HSP.
The detrimental effects of M1 in neurons might be particularly severe because neurons compared with nonneuronal cells have comparatively limited ability to undergo the stress-induced up-regulation of autophagy responsible for degradation of membrane-associated proteins such as M1 (Martinez-Vicente and Cuervo, 2007; Maday and Holzbaur, 2016). Autophagosomes formed in the distal axon mature during retrograde transport toward the cell body for fusion with lysosomes (Maday and Holzbaur, 2014). Because the soma is the primary site for degradation of autophagic cargo, impaired retrograde transport would result in accumulation of undigested proteins and damaged organelles within axons. Such a decrease in retrograde transport was reported in HSP-SPG4 patient–derived neurons and was attributed to the loss of spastin microtubule-severing activity. As a result, axonal swellings filled with fragmented microtubules, and mitochondria were observed in affected axons (Denton et al., 2014; Havlicek et al., 2014). Alternatively, we proposed that mutant M1 might inhibit axonal transport through aberrant activation of protein kinases, which in turn inhibits relevant molecular motor proteins (Solowska et al., 2008; Leo et al., 2017). A limited ability of neurons to eliminate organelles affected by anomalous membrane-associated proteins combined with impaired retrograde transport might result in long-term exposure to truncated M1 spastins and lead to degeneration of the long axons of the corticospinal tract observed in HSP-SPG4.
MATERIALS AND METHODS
Reagents
Rabbit polyclonal antibody SP5 was generated against spastin amino acids 90–283, and SP/AAA antibody was generated against spastin amino acids 337–465 (Solowska et al., 2008). Anti-spastin antibodies were used for immunoblotting and immunostaining. Alexa Fluor 488–conjugated goat anti-rabbit antibody (Invitrogen) and Cy3-conjugated anti–β-tubulin mouse monoclonal antibody (Sigma-Aldrich, St. Louis, MO) were used for immunostaining. Mouse anti-GAPDH (ab9484; Abcam), and donkey anti-rabbit peroxidase-conjugated antibody (Jackson ImmunoResearch Laboratories) were used for immunoblotting.
SPAST constructs
The nonsense mutations N184X and S245X were generated in the full-length human spastin cDNA using the QuikChange ll XL site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The presence of the mutations was confirmed by DNA sequencing. The nomenclature of the mutations refers to the cDNA sequence (GenBank NM_014946), with the A of the M1 translation initiation codon as +1. To create constructs expressing only M1 spastin isoforms, the imperfect Kozak sequence tgaATGa surrounding M1 start codon was replaced by a good consensus Kozak sequence, accATGa, using site-directed mutagenesis. To prepare constructs expressing only M87 isoforms, the N-terminal part encoding the first 86 amino acids of spastin was deleted, and a good Kozak sequence accATGg, surrounding, the M87 start codon was created by site-directed mutagenesis. M187 encoding sequence was obtained by deleting the first exon from WT spastin cDNA. Mutated human spastin cDNAs were cloned into pTRE-Tight vector with Dox-inducible promoter (Clontech) for inducible expression of spastin proteins in cultured rat fibroblasts or pCMV vector (Stratagene) for expression in primary rat cortical neurons.
Cell transfections and immunostaining
Primary rat cortical neurons were cultured and transfected with mutated M1 or M87 spastin cDNA in pCMV vector (Stratagene) using the Amaxa Nucleofector and an approach identical to one we used previously for cultured rat hippocampal neurons (Yu et al., 2005). Rat fibroblasts stably transfected to synthesize the tetracycline-controlled transactivator Tet-On Advanced (RFL-6 TET cells) were cultured in Lab-Tek chambers (Nalgene) and transfected with spastin cDNAs in pTRE-Tight vector using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. Five hours after transfection, medium with 1 µg/ml Dox was added to induce spastin synthesis. Both neuronal cells and fibroblasts were fixed 24 h after transfection with 4% paraformaldehyde in phosphate-buffered saline (PBS) and then postextracted with 0.1% Triton X-100. SP5 anti-spastin antibody was used to detect WT and truncated spastins, and SP/AAA anti-spastin antibody was used to detect WT and M187 spastin. The cultures were double labeled by exposure first to anti-spastin antibody overnight at 4°C and then to Alexa Fluor 488–conjugated goat anti-rabbit (1:500; Invitrogen) and Cy3-conjugated anti–β-tubulin mouse monoclonal antibody (Sigma-Aldrich) for 1 h at 37°C. Double-labeled overlay images were obtained with a Zeiss Pascal LSM 5 confocal microscope. Spastin expression levels in transfected cells were measured using images obtained on an Axiovert 200 microscope (Carl Zeiss) equipped with a high-resolution charge-coupled device (CCD) camera (Orca ER, Hamamatsu). All images were generated using identical imaging criteria, such as gain and exposure time. Mean gray values (the sum of gray values of all of the pixels in a selected cell divided by the number of pixels) of raw images were obtained using AxioVision software and presented as AFUs. To detect neuronal cells expressing low levels of truncated spastins, fluorescence levels were first measured in control, mock-transfected cells immunostained with anti–spastin SP5 antibody, and then fluorescence was measured in identically stained cells transfected with spastin constructs. Expressing neuronal cells had fluorescence higher than control (mean ± SD AFU). After measurement of the intensity of fluorescence, brightness and contrast of all images were adjusted in an identical manner using Adobe Photoshop.
Cell transfections and immunoblotting
To compare accumulation levels of WT and mutated spastin isoforms, 3 × 105 RFL-6 TET cells were plated into one well of a 24-well plate and transfected using Lipofectamine 2000 (Invitrogen). Transfection for each individual construct was repeated four times. Spastin synthesis was induced 24 h after transfection with 0.25–2 µg/ml Dox added for 7 h. Transfection efficiency was evaluated in separate wells by counting the number of spastin-positive cells per 200 cells and found to be ∼30% for all constructs tested. Transfected cells were washed with PBS and collected in 200 µl of SDS–PAGE sample buffer (Sigma-Aldrich) with Halt protease inhibitor cocktail (Thermo Scientific). Aliquots containing equal total protein levels, as estimated by Coomassie staining, were used for SDS–PAGE electrophoresis. We used 10% gels for full-length spastins and 15% gels for truncated spastins. After electrophoresis, proteins were transferred to nitrocellulose. All membranes were collected and immunoblotted at the same time overnight at 4°C with the anti-–spastin SP5 antibody diluted 1:20,000. After washing, blots were incubated for 1 h with peroxidase-conjugated anti-rabbit antibody at 20°C, washed again, and incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) for 3 min. Each membrane was imaged using the ChemiDoc MP System (Bio-Rad) equipped with a high-resolution CCD camera. Intensity of spastin bands was measured using Image Lab software for 10-s exposures.
Acknowledgments
This work was supported by a grant to P.W.B. from the National Institute of Neurological Disorders and Stroke (R01 NS28785) and the Pennsylvania Department of Health CURE Program to Drexel University College of Medicine. A.N.R. is supported by a National Research Service Award (1F31NS093748-01A1) from the National Institute of Neurological Disorders and Stroke. P.W.B. is the 2017 recipient of the Advanced Scholarship for Research into Hereditary Spastic Paraplegia and Related Diseases from the Tom Wahlig Foundation.
Abbreviations used:
- HSP
hereditary spastic paraplegia
- NMD
nonsense-mediated decay
- PTC
premature termination codon.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0047) on May 11, 2017.
REFERENCES
- Allison R, Lumb JH, Fassier C, Connell JW, Ten Martin D, Seaman MN, Hazan J, Reid E. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J Cell Biol. 2013;202:527–543. doi: 10.1083/jcb.201211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Vidya Nadar C, Myers KA. Axonal transport of microtubules: the long and short of it. Traffic. 2006;7:490–498. doi: 10.1111/j.1600-0854.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- Beetz C, Schüle R, Deconinck T, Tran-Viet KN, Zhu H, Kremer BP, Frints SG, van Zelst-Stams WA, Byrne P, Otto S, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131:1078–1086. doi: 10.1093/brain/awn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C, O’Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger J, Fonknechten N, Hoeltzenbein M, Neumann L, Bratanoff E, Hazan J, Reis A. Hereditary spastic paraplegia caused by mutations in the SPG4 gene. Eur J Hum Genet. 2000;8:771–776. doi: 10.1038/sj.ejhg.5200528. [DOI] [PubMed] [Google Scholar]
- Claudiani P, Riano E, Errico A, Andolfi G, Rugarli EI. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp Cell Res. 2005;309:358–369. doi: 10.1016/j.yexcr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Connell JW, Lindon C, Luzio JP, Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10:42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells. 2014;32:414–423. doi: 10.1002/stem.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A, Ballabio A, Rugarli EI. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet. 2002;11:153–163. doi: 10.1093/hmg/11.2.153. [DOI] [PubMed] [Google Scholar]
- Errico A, Claudiani P, D’Addio M, Rugarli EI. Spastin interacts with the centrosomal protein NA14, and is enriched in the spindle pole, the midbody and the distal axon. Hum Mol Genet. 2004;13:2121–2132. doi: 10.1093/hmg/ddh223. [DOI] [PubMed] [Google Scholar]
- Evans K, Keller C, Pavur K, Glasgow K, Conn B, Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc Natl Acad Sci USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassier C, Tarrade A, Peris L, Courageot S, Mailly P, Dalard C, Delga S, Roblot N, Lefevre J, Job D, et al. Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knock-out mice. Dis Model Mech. 2013;6:72–83. doi: 10.1242/dmm.008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonknechten N, Mavel D, Byrne P, Davoine CS, Cruaud C, Bönsch D, Samson D, Coutinho P, Hutchinson M, McMonagle P, et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet. 2000;9:637–644. doi: 10.1093/hmg/9.4.637. [DOI] [PubMed] [Google Scholar]
- Goyal U, Renvoisé B, Chang J, Blackstone C. Spastin-interacting protein NA14/SSNA1 functions in cytokinesis and axon development. PLoS One. 2014;9:e112428. doi: 10.1371/journal.pone.0112428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizetti J, Schermelleh L, Mäntler J, Maar S, Poser I, Leonhardt H, Muller-Reichert T, Gerlich DW. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 2011;331:1616–1620. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- Havlicek S, Kohl Z, Mishra HK, Prots I, Eberhardt E, Denguir N, Wend H, Plötz S, Boyer L, Marchetto MC, et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum Mol Genet. 2014;23:2527–2541. doi: 10.1093/hmg/ddt644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Dürr A, Wincker P, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Gene. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CC, et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo L, Weissmann C, Burns M, Kang M, Song Y, Qiang L, Brady ST, Baas PW, Morfini G. Mutant spastin proteins promote deficits in axonal transport through an isoform-specific mechanism involving casein kinase 2 activation. Hum Mol Genet. 2017;26:2321–2334. doi: 10.1093/hmg/ddx125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JC, Lusher ME, McDermott CJ, White KD, Reid E, Rubinsztein DC, Bashir R, Hazan J, Shaw PJ, Bushby KM. Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet. 2000;37:759–765. doi: 10.1136/jmg.37.10.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016;36:5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- McDermott CJ, Burness CE, Kirby J, Cox LE, Rao DG, Hewamadduma C, Sharrack B, Hadjivassiliou M, Chinnery PF, Dalton A, et al. Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology. 2006;67:45–51. doi: 10.1212/01.wnl.0000223315.62404.00. [correction published in Neurology (2009) 72, 1534] [DOI] [PubMed] [Google Scholar]
- Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016;165:1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Yu W, Liu M, Solowska JM, Baas PW. Basic fibroblast growth factor elicits formation of interstitial axonal branches via enhanced severing of microtubules. Mol Biol Cell. 2010;21:334–344. doi: 10.1091/mbc.E09-09-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Riano E, Martignoni M, Mancuso G, Cartelli D, Crippa F, Toldo I, Siciliano G, Di Bella D, Taroni F, Bassi MT, et al. Pleiotropic effects of spastin on neurite growth, depending on expression levels. J Neurochem. 2009;108:1277–1288. doi: 10.1111/j.1471-4159.2009.05875.x. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD. The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr Biol. 2005;15:650–655. doi: 10.1016/j.cub.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Sanderson CM, Connell JW, Edwards TL, Bright NA, Duley S, Thompson A, Luzio JP, Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum Mol Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweingruber C1, Rufener SC, Zünd D, Yamashita A, Mühlemann O. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829:612–623. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV, Mannan AU. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet. 2009;17:187–194. doi: 10.1038/ejhg.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska J, Garbern J, Baas PW. Evaluation of loss-of-function as an explanation for SPG4-based hereditary spastic paraplegia. Hum Mol Genet. 2010;19:2767–2779. doi: 10.1093/hmg/ddq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, Baas PW. Hereditary spastic paraplegia SPG4: what is known and not known about the disease. Brain. 2015;138:2471–2484. doi: 10.1093/brain/awv178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, D’Rozario M, Jean DC, Davidson MW, Marenda DR, Baas PW. Pathogenic mutation of spastin has gain-of-function effects on microtubule dynamics. J Neurosci. 2014;34:1856–1867. doi: 10.1523/JNEUROSCI.3309-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, Baas P. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- White SR, Evans KJ, Lary J, Cole JL, Lauring B. Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J Cell Biol. 2007;176:995–1005. doi: 10.1083/jcb.200610072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip AG, Durr A, Marchuk DA, Ashley-Koch A, Hentati A, Rubinsztein DC, Reid E. Meta-analysis of age at onset in spastin-associated hereditary spastic paraplegia provides no evidence for a correlation with mutational class. J Med Genet. 2003;40:e106. doi: 10.1136/jmg.40.9.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Solowska JM, Qiang L, Karabay A, Baird D, Baas PW. Regulation of microtubule severing by katanin subunits during neuronal development. J Neurosci. 2005;25:5573–5583. doi: 10.1523/JNEUROSCI.0834-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]