

CK2 regulates NHE3 by phosphorylating a single C-terminal amino acid, which, when mutated, reduces basal NHE3 activity and its acute stimulation and inhibition. It also is necessary for binding of proteins throughout the C-terminus, which means that it determines the C-terminal structure.
Abstract
Casein kinase 2 (CK2) binds to the NHE3 C-terminus and constitutively phosphorylates a downstream site (S719) that accounts for 40% of basal NHE3 activity. The role of CK2 in regulation of NHE3 activity in polarized Caco-2/bbe cells was further examined by mutation of NHE3-S719 to A (not phosphorylated) or D (phosphomimetic). NHE3-S719A but not -S719D had multiple changes in NHE3 activity: 1) reduced basal NHE3 activity—specifically, inhibition of the PI3K/AKT-dependent component; 2) reduced acute stimulation of NHE3 activity by LPA/LPA5R stimulation; and 3) reduced acute inhibition of NHE3 activity—specifically, elevated Ca2+ related (carbachol/Ca2+ ionophore), but there was normal inhibition by forskolin and hyperosmolarity. The S719A mutant had reduced NHE3 complex size, reduced expression in lipid rafts, increased BB mobile fraction, and reduced binding to multiple proteins that bind throughout the NHE3 intracellular C-terminus, including calcineurin homologous protein, the NHERF family and SNX27 (related PDZ domains). These studies show that phosphorylation of the NHE3 at a single amino acid in the distal part of the C-terminus affects multiple aspects of NHE3 complex formation and changes the NHE3 lipid raft distribution, which cause changes in specific aspects of basal as well as acutely stimulated and inhibited Na+/H+ exchange activity.
INTRODUCTION
The intestinal brush border Na+/H+ exchanger 3 (NHE3) accounts for a major component of intestinal Na absorption both in the basal state and in the late postprandial state, during which it contributes to recovery from the early digestion-related fluid secretion that spreads digestive enzymes over the digestive/absorptive small intestinal surface (Zachos et al., 2005; Girardi et al., 2012). As part of its acute regulation, NHE3 traffics to and from the intestinal brush border, where 80–90% is resident under basal conditions, and it also moves through the early and apical recycling endosomes under basal conditions. Postprandial changes in NHE3 activity occur via changes in its rates of endocytosis and exocytosis. NHE3 inhibition occurs soon after eating, and this relates to increased rates of endocytosis of NHE3 exceeding changes in rates of its exocytosis; these changes are mimicked by inhibition of NHE3, an important contributor to intestinal fluid secretion in almost all diarrheal diseases (Moe et al., 1999; Zachos et al., 2005; Donowitz and Li, 2007; Girardi et al., 2012; Singh et al., 2014; Priyamvada et al., 2015).
NHE3 exists simultaneously in multiple pools both in the brush border (BB) and intracellularly (Donowitz and Li, 2007; Donowitz et al., 2009). These pools change in size and composition with the signal transduction that regulates NHE3 activity (Li et al., 2001, Donowitz and Li, 2007; Donowitz et al., 2009). For instance, BB NHE3 is present in both detergent-soluble and detergent-insoluble pools, defined via solubility in Triton X-100 (Akhter et al., 2002; Li et al., 2004). The detergent-insoluble pool can be further separated into lipid raft and non–lipid raft compartments. The former is defined as being affected by removal of cholesterol by methyl β-cyclodextrin (MβCD) exposure, and the latter is believed to represent NHE3 bound to the microvillar cytoskeleton.
The regulation of NHE3 requires its long, ∼377–amino acid intracellular C-terminus and involves two large multiprotein signaling complexes that form at two distinct sites on the C-terminus, as well as lipid-binding partners (Donowitz and Li, 2007; Donowitz et al., 2009; Mohan et al., 2010). Because of these complexes, the density gradient–based estimate of the size of NHE3 is as much as 1200–2000 kDa, unlike the 90–180 kDa predicted from its sequence and found on SDS gels (Donowitz et al., 2009). The proteins that have been identified in these complexes include multiple kinases, at least one phosphatase, multiple scaffolds, and other signaling molecules, although a full accounting of the proteins associated is lacking (Donowitz and Li, 2007; Donowitz et al., 2009). The changes in NHE3 activity are associated not only with changes in the amount of NHE3 present in the brush border but also with changes in the size and composition of the C-terminal signaling complexes and the distribution of NHE3 in the BB pools. For instance, NHE3 complexes are increased in size with carbachol inhibition and with lysophosphatidic acid (LPA) stimulation of NHE3 via LPA5 receptors and reduced in size with elevated Ca2+ inhibition of NHE3 (Li et al., 2004; Donowitz et al., 2009; Lin et al., 2011). In addition, NHE3 regulation depends on the state of phosphorylation of its C-terminus. NHE3 is phosphorylated under basal conditions, and most but not all rapid (minutes) and intermediate term (hours) regulation is associated with changes in phosphorylation of the NHE3 C-terminus, particularly the distal part of the C-terminus. Kinases shown to phosphorylate the NHE3 C-terminus and alter its activity include casein kinase 2 (CK2), CaMKII, PKA, cGKII, GSK-3, AKT, SGK1, and the MAP kinases ERK and RSK (Wiederkehr et al., 1999; Wang et al., 2005; Sarker et al., 2008; Zizak et al., 2012; Singh at al., 2014; Chen et al., 2015; No et al., 2015). Other kinases, including p38 MAP kinase and PDK1, affect NHE3 activity but have not been shown to phosphorylate it, whereas still others, such as PKC, phosphorylate NHE3, but the role of this phosphorylation in NHE3 regulation is not clear (Wiederkehr et al., 1999). Of these kinases, several phosphorylate/regulate NHE3 under basal conditions, whereas others affect NHE3 only in the postprandial state and diarrheal diseases. The former include CaMKII, ERK, and CK2. Three kinases require phosphorylation at three sites in the NHE3 distal C-terminus for regulation to occur (cGKII, CaMKII, and RSK in the starfish NHE3; Harada et al., 2010; Zizak et al., 2012; Chen et al., 2015).
We previously reported that the CK2 α subunit associates with the NHE3 C-terminus and phosphorylates it at a single downstream site, amino acid S719 (Sarker et al., 2008). This site, like most of the NHE3 C-terminal phosphorylation sites, is in the distal part of the C-terminus, which has been suggested to be in a highly intrinsically disordered area (Hendus-Altenburger et al., 2014). When expressed in NHE3-null fibroblasts, this phosphorylation accounts for ∼40% of basal NHE3 activity and regulates delivery of newly synthesized as well as basal NHE3 exocytosis but not endocytosis. Conclusions about the role of CK2 in regulating NHE3 activity have been based on mutations of the NHE3-S719 and a CK2 inhibitor, which had similar effects on basal NHE3 activity. In the present studies, we further investigated the mechanisms of how CK2 regulates the activity of NHE3 in an intestinal epithelial cell model. We find that whereas the NHE3-719D mutation behaves like wild type, the mutant that cannot be phosphorylated, NHE3-719A, reduces binding of proteins throughout the NHE3 C-terminus, reduces association of NHE3 with lipid rafts, and alters basal and regulated NHE3 activity. We speculate that phosphorylation of the single C-terminal amino acids is involved in organizing the tertiary structure of the NHE3 C-terminus to cause these multiple effects.
RESULTS
CK2 phosphorylation of NHE3-S719 is required for NHE3 basal activity and brush border membrane expression
Initial studies confirmed the role of CK2 phosphorylation in regulation of basal NHE3 activity (Sarker et al., 2008). The CK2-specific inhibitor 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazol (DMAT) reduced basal activity of NHE3-WT by 40% (Sarker et al., 2008). Similarly, the NHE3-S719A mutant had ∼50% lower activity than NHE3-WT, and DMAT failed to affect the activity of NHE3-S719A (Figure 1A). To begin exploring how CK2 regulates NHE3 activity, we examined cell surface expression of NHE3 using confocal microscopy of immunostained Caco-2/bbe cells. In this approach, brush border expression levels of WT and NHE3-S719A were determined by combining hemagglutinin (HA)-NHE3 intensities from the serial xy-sections that overlapped with wheat germ agglutinin (WGA)–Alexa 488, which was compared with the total HA-NHE3 signal. Quantitation was with MetaMorph software and is reported as percentage of total NHE3 in BB (Figure 1B). BB NHE3 was reduced from 52% in wild type (WT)-NHE3 to 26% in NHE3-S719A; this is a similar percentage reduction to the decrease in NHE3 transport activity. Thus CK2-dependent NHE3 basal activity relates to the amount of brush border expression of NHE3. This conclusion is similar to the one reached from studies of basal NHE3 activity in PS120 fibroblasts transfected with NHE3 (Sarker et al., 2008).
FIGURE 1:

NHE3-S719A mutant has reduced NHE3 activity due to reduced surface expression. Confluent Caco-2/bbe cells were grown for 12 d and then infected with Ad-HA-NHE3-WT or -S719A. (A) NHE3 activities were measured under basal conditions and with DMAT (30 µM, 30 min) treatment. NHE3-S719A mutant cells decreased basal NHE3 activity by ∼50% and lost the effect of DMAT, a specific inhibitor of CK2. DMAT also reduced the NHE3 activity of NHE3-WT cells by ∼40%. NHE3 activity was measured from the initial rates of Na+-dependent intracellular alkalinization. (B) Immunofluorescence detection of microvillus WGA and NHE3 colocalization indicates that the presence of NHE3-S719A is ∼50% less in microvillus than is NHE3-WT. Reconstructed xz-images and a single xy-image (0.5 μM) taken at the same distance from initially detected BB image for WT NHE3 and NHE3-S719A. n, number of separate experiments. p values are via paired t tests.
The phosphoinositide 3-kinase/AKT signaling pathway component of basal NHE3 activity requires CK2 phosphorylation of NHE3-S719
NHE3 basal activity depends on the phosphoinositide 3-kinase (PI3K)/AKT pathways (Li et al., 2004). Because the NHE3-719A mutant has reduced basal NHE3 activity, we questioned whether the NHE3 pool affected was that regulated by PI3K/AKT. To do so, we examined the effect of the PI3K inhibitor wortmannin on Na+/H+ exchange activity of NHE3-S719A and compared the effects with those on NHE3-WT. Wortmannin (100 nM) reduced NHE3-WT activity by 30–40% but did not affect the activity of NHE3-S719A (Figure 2A). This is consistent with the PI3K-dependent pool of NHE3 that is involved in basal NHE3 activity requiring CK2 phosphorylation of S719. Because PI3K and AKT are involved in the same pathway of NHE3 basal stimulation (Lee-Kwon et al., 2001; Li et al., 2004; Singh et al., 2014), we also examined the effect of an AKT inhibitor on NHE3-WT and NHE3-S719A. NHE3-S719A lost the previously demonstrated reduced NHE3 activity caused by the AKT inhibitor (AKTi; Singh et al., 2014; Figure 2B). Moreover, there was no additive effect when NHE3-WT cells were treated with both the CK2 inhibitor 4,5,6,7-tetrabromo-2-azabenzimidazole (4,5,6,7-tetrabromobenzotriazol [TBB]) and AKTi (Figure 2C). These results further support that the PI3K/AKT component of basal NHE3 activity requires basal CK2 phosphorylation of NHE3-S719.
FIGURE 2:

The Pl3K and AKT components of basal NHE3 activity require CK2 phosphorylation of NHE3-S719. (A) Treatment of NHE3-WT cells with 100 nM wortmannin for 30 min reduced NHE3 activity by ∼50%. This NHE3 activity was similar to that in NHE3-S719A, which was not further altered by wortmannin. (B) NHE3 activity was measured in cells treated with or without the AKT inhibitor AKTi-VIII at 10 µM for 1 h. In NHE3-WT cells, NHE3 activity was reduced by ∼30%, whereas there was no further effect of AKTi-VIII on NHE3-S719A. (C) Treatment of NHE3-WT cells with 30 µM TBB reduced NHE3 activity, but additional treatment with AKTi-VIII did not further inhibit NHE3 activity, suggesting that both inhibitions affect the same signaling pathway. n, number of experiments. p values are via unpaired t tests. (D) CK2 kinase assay with His-tagged NHE3 C-terminal fusion protein was performed in the presence and absence of TBB or AKTi-VIII. Samples were separated in 14% SDS–PAGE and stained with Pro-Q Diamond Phosphoprotein Assay Kit. Left, phosphorylated/nonphosphorylated NHE3 fusion proteins visualized by Typhoon imager; right, total protein after Coomassie blue stain. TBB blocked the CK2 phosphorylation of the NHE3 C-terminal fusion protein, but AKTi-VIII had no effect.
To confirm that the CK2 inhibitors were altering NHE3 phosphorylation, we determined the effect of TBB using an in vitro CK2 assay. This assay used recombinant histidine (His)-tagged NHE3 C-terminal fragment amino acids 668–647 with CK2 in the absence and presence of TBB. We performed a control experiment using an AKT inhibitor that we previously showed altered basal NHE3 activity (Akt inhibitor VIII [AKITi]). Phosphorylation of the NHE3 fusion protein was visualized by the Pro-Q Diamond Phosphostain (Figure 2D). CK2 phosphorylates the NHE3-S719–containing fusion protein, which was prevented by TBB but not by AKTi.
CK2 phosphorylation of NHE3 is necessary for acute stimulation of NHE3 by LPA5R/LPA
We next determined the role of CK2 phosphorylation of NHE3 in a known example of acute NHE3 stimulation—that by LPA in cells expressing LPA5R. LPA5 receptors are not endogenously expressed in Caco-2/bbe cells. Thus, to study the dependence on CK2 of LPA stimulation of NHE3, we transduced Caco-2/bbe cells by Ad-LPA5R (Figure 3). Whereas apical LPA failed to stimulate NHE3 in wild-type Caco-2/bbe/HA-NHE3 (unpublished data), LPA5R-expressing Caco-2/bbe cells responded to 1 μM apical LPA with acute stimulation of NHE3. However, LPA stimulation of NHE3 did not occur in Caco-2/bbe/NHE3-S719A cells (Figure 3B). In addition, the stimulatory effect of LPA on NHE3-WT was completely blocked by TBB (Figure 3C).
FIGURE 3:

LPA stimulates NHE3 activity in Caco-2/bbe cells expressing adenoviral-NHE3-WT and LPA5R but not in cells expressing NHE3-S719A and LPA5R. (A) Confluent monolayers of Caco-2 cells were infected with Ad-HA-LPA5R virus, and ∼2 d later, cell lysate was examined for LPA5R expression by Western analysis. Caco-2 cells without viral infection were used as control. Anti-LPA5R antibody detected a ∼37-kDa band, which was absent in control cells. (B) LPA (1 µM for 30 min) stimulated NHE3 activity in Caco-2/bbe/Ad-HA-NHE3-WT cells by ∼90% (p < 0.05) but had no effect in NHE3-S719A cells. (C) Stimulatory effect of LPA (1 μM) on NHE-WT in Caco-2 cells was completely inhibited by the CK2 inhibitor TBB (30µM). n, number of experiments. p values are comparison with basal NHE3 activity (paired t tests and ANOVA).
NHE3-S719A mutant is not inhibited by acutely elevated calcium but is inhibited similarly to wild-type NHE3 by forskolin and hyperosmolarity
Given what appeared to be differential regulatory roles for CK2 phosphorylation in basal NHE3 activity, we performed more-detailed studies of acute inhibition of NHE3. Acute NHE3 inhibition, which is an important aspect of normal digestive physiology, appears to be a differentially regulated process. In Caco-2/bbe cells, cAMP inhibition of NHE3 depends on either Na+/H+ exchange regulatory cofactor 1 (NHERF1) or NHERF2, whereas calcium inhibition of NHE3 is only NHERF2 dependent. In contrast, hyperosmolarity inhibits NHE3 by an NHERF-independent process. Forskolin inhibition of NHE3 was examined in Caco-2/bbe cells expressing NHE3-WT or NHE3-S719A. Forskolin treatment inhibited NHE3 activity in both the NHE3-WT– and NHE3-S719A mutant–expressing cells, with similar percentage inhibition (Figure 4A). Thus cAMP inhibition of NHE3 is not dependent on CK2 phosphorylation of NHE3.
FIGURE 4:

CK2 phosphorylation of NHE3-S719 is necessary for Ca2+ inhibition of NHE3 but not for inhibition of NHE3 by forskolin (FSK) and hyperosmolarity. (A) FSK (25 µM for 30 min) inhibits NHE3 activity in both NHE3-WT and S719A mutant cells by a similar percentage of basal activity. (B) Carbachol (CCh; 25 µM for 5 min) inhibits NHE3 activity in NHE3-WT cells by ∼40% but had no significant effect in NHE3-S719A cells. (C) NHE3-WT cells treated with calcium ionophore A23187 (1 µM for 10 min) reduced NHE3 activity by ∼30% (p < 0.05), but the effect of A23187 on NHE3-S719A cells was not significant. (D) Treatment of NHE3-WT cells with CCh or DMAT reduced NHE3 activity ∼40%, but there was no additional inhibition of NHE3 when DMAT-treated cells were further treated with CCh. (E) Under hyperosmolar conditions, NHE3 activity was reduced by ∼50% in both NHE3-WT and NHE3-S719A cells (p < 0.05). n, number of experiments. p values are by paired t tests and ANOVA.
We then examined the effect of elevated calcium on NHE3 activity in the same Caco-2/bbe cells. Carbachol treatment inhibited NHE3 in cells expressing NHE3-WT but not in cells expressing NHE3-S719A (Figure 4B). Similarly, we found CK2 phosphorylation dependence on NHE3-S719 when we treated cells with the calcium ionophore A23187 (Figure 4C). Additional evidence that elevated Ca2+ inhibition of NHE3 requires CK2 phosphorylation of NHE3 is provided in Figure 4D, which illustrates that there is a lack of additivity of carbachol and DMAT inhibition of NHE3, suggesting participation in a common pathway.
Hyperosmolarity inhibits NHE3 activity acutely by a process that does not involve NHE3 phosphorylation or trafficking of NHE3 in cell models (Nath et al., 1996). As shown in Figure 4E, hyperosmolar inhibition of NHE3 activity occurred similarly in NHE3-WT and NHE3-S719A. These results indicate that CK2 phosphorylation of NHE3-S719A is not necessary for the acute NHE3 inhibition caused by forskolin and hyperosmolarity but is necessary for that caused by acute calcium elevation.
NHE3-S719D behaves similarly to NHE3-WT in the PI3K-dependent component of basal activity and inhibition by forskolin and carbachol
Further studies evaluated several of the areas of NHE3 regulation that were abnormal with NHE3-S719A compared with wild type to test the hypothesis that any change in the phosphorylation of amino acid S719 would alter NHE3 activity and its regulation. As shown in Figure 5A, wortmannin (100 nM, 30 min) inhibited NHE3-S719D similarly to what occurred in NHE3-WT. Similarly, forskolin (25 μM, 30 min) and carbachol (25 μM, 5 min) inhibited NHE3-S719D similarly to the inhibition that occurred with NHE3-WT. These results show that not all changes in the amino acid at NHE3-S719 affect regulation of NHE3 and in particular that increasing the extent of phosphorylation of this amino acid does not further alter several aspects of regulation. In addition, we examined the effect of TBB on the basal activity of NHE3-S19D and found none. The latter supports that the effect of TBB on NHE3 activity is entirely by altering the phosphorylation of NHE3-S719 by CK2.
FIGURE 5:
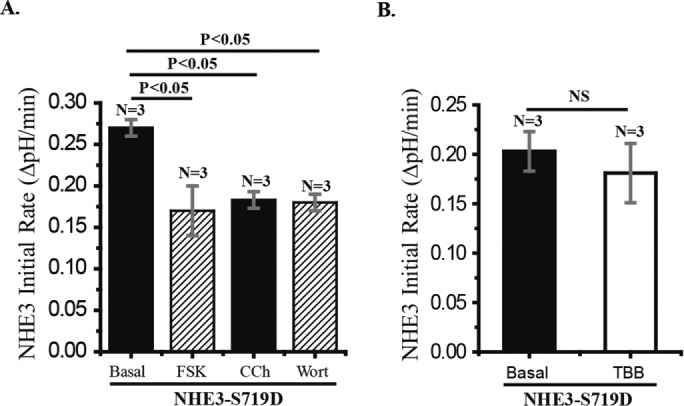
NHE3 activity of Caco-2 cells expressing NHE3-S719D is inhibited by forskolin, carbachol, and wortmannin, but TBB has no effect. (A) Caco-2/bbe cells were infected with Ad-HA-NHE3-S719D at 12 d postconfluency and activity measured ∼2 d later. Effects of wortmannin (100 nM, 30 min), forskolin (25 μM, 30 min), and carbachol (25 μM, 5 min) were determined. All three inhibited NHE3 activity similarly, and the magnitude of the inhibition was similar to that in NHE3-WT (Figures 2 and 4). (B) The effect of TBB (30 μM, 30 min) on NHE3 in Caco-2/bbe cells expressing NHE3-S719D was not significant. n, number of experiments. p values compared by paired t tests and ANOVA.
As summarized in Table 1, the transport changes that require phosphorylation of NHE3 at S719 depend on the PDZ domains of NHERF2 (Nath et al., 1996; Cha et al., 2005, 2014; Donowitz et al., 2009; Chen et al., 2010, 2015; Lin et al., 2011; Murtazina et al., 2011; Sarker et al., 2011). In addition, basal and stimulated trafficking of NHE3 from the early endosome to the plasma membrane (exocytosis) depends on sorting nexin 27 (SNX27; Singh et al., 2015). That these aspects of NHE3 regulation are related is suggested by high homology of the NHERF and SNX27 PDZ domains (Singh et al., 2015). Consequently, we examined whether NHE3-S719 affected SNX27 and NHERF binding.
TABLE 1:
Altered NHE3 transport activity of Caco-2/bbe cells expressing Ad-HA-NHE3- S719A in response to inhibitory or stimulatory agents compared with NHE3-WT activity.
| Na+/H+ exchange activity | ||
|---|---|---|
| Inhibitor/activator | NHE3-WT | NHE3-S719A |
| Basal | Normal | Reduced |
| DMAT | Inhibits | Lost effects |
| Wortmannin | Inhibits | Lost effects |
| AKTi VIII | Inhibits | Lost effects |
| LPA | Stimulates | Lost effect |
| Forskolin | Inhibits | Inhibits |
| Hyperosmolarity | Inhibits | Inhibits |
| Carbachol | Inhibits | Lost effects |
| A23187 | Inhibits | Lost effects |
NHE3-S719A mutant lost part of basal NHE3 activity, as well as calcium and LPA effects on NHE3 activity. Normal regulation: forskolin and hyperosmolarity. Altered regulation: basal, DMAT, wortmannin, AKTi-VIII, carbachol, A23187, LPA.
NHE3-S719A mutant fails to associate with SNX27
Glutathione S-transferase (GST)–SNX27 fusion protein was used to pull down NHE3 from Caco-2/bbe cell lysate expressing NHE3 WT or NHE3-S719A. GST-SNX27 pulled down NHE3-WT but not NHE3-S719A (Figure 6). Because binding to NHE3 by SNX27 is required for basal NHE3 activity, failure of SNX27 to bind NHE3-719A is at least one explanation for the lower plasma membrane expression and lower basal activity of NHE3. Thus two components that contribute to regulation of NHE3 activity and basal NHE3 exocytosis have been identified as abnormal in NHE3-S719A: 1) PI3-K/AKT stimulation and 2) SNX27 binding.
FIGURE 6:
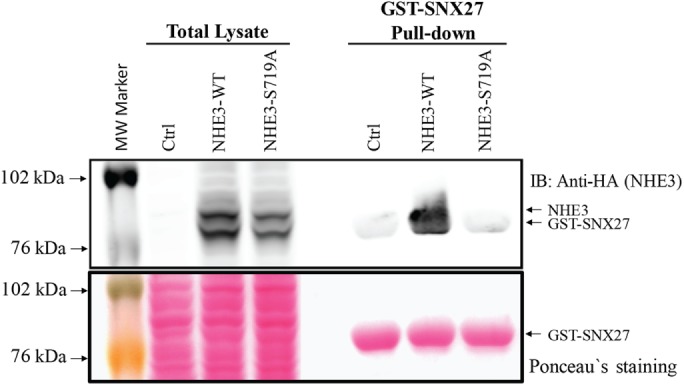
In vitro pull-down assay: NHE3-WT binds to SNX27, but NHE3-S719A binding to SNX27 is greatly reduced. GST-SNX27 pull-down assays were performed with cell lysates prepared from Caco-2/bbe cells grown in 10-cm Transwell plates infected with Ad-HA-NHE3-WT or NHE3-S719A mutant. Cells without adenoviral infection were used as control. The GST-SNX27 protein bands were visualized by Ponceau S staining (bottom), and NHE3-WT and S719A mutant proteins were visualized by Western blot with anti-HA rabbit polyclonal antibody (top). HA-NHE3 and GST-SNX27 protein bands overlapped partially. A large gel was used to separate NHE3 and GST-SNX27. NHE3-WT and SNX27 are visualized, but NHE3-S719A mutant binding to SNX27 was not identified. A single representative experiment, which was repeated twice with similar results, is shown.
NHE3-S719A mutant has decreased NHERF2 binding
NHERF2 is involved in multiple aspects of NHE3 regulation. Although preventing NHERF2 binding to NHE3 does not consistently alter basal NHE3 activity in intestinal cells, its binding is necessary for stimulation of NHE3 by d-glucose, LPA, and dexamethasone and acute inhibition by cGMP and elevated Ca2+ (Lee-Kwon et al., 2001; Kim et al., 2002; Yun, 2003; Cha et al., 2005, 2010, 2014; Zachos et al., 2009; Chen et al., 2010; Lin et al., 2010, 2011; Murtazina et al., 2011; Zhu et al., 2011). Because the NHE3-S719A mutant was not stimulated by LPA and not inhibited by elevated Ca2+, we speculated that interaction between NHE3-S719A and NHERF2 might be decreased. We examined this association by immunoprecipitation (IP) of FLAG-NHERF2 from Caco-2/bbe cells transduced with ad-FLAG-NHERF2 and also expressing NHE3-WT, -S719A, or -S719D. As shown in Figure 7, A and B, NHE3-S719A was coimmunoprecipitated significantly less than NHE3-WT by NHERF2. In contrast, NHE3-S719D was coimmunoprecipitated by FLAG-NHERF2 similarly to NHE3-WT (Figure 7, A and B). In addition, coIP of ezrin by NHERF2 was not altered, which could reflect that ezrin binding to NHERF2 does not depend on NHERF–NHE3 binding (Cha and Donowitz, 2008).
FIGURE 7:

NHERF2 and NHERF3 bind to NHE3-WT and NHE3-S719D normally but bind less to NHE3-S719A. The NHERF2 coIP of NHERF3 is similarly reduced in NHE3-S719A cells. (A) IP of FLAG-NHERF2 was performed using cell lysates prepared from Caco-2/bbe cells grown in 10-cm Transwell plates also infected with Ad-HA-NHE3-WT, -S719A, or- S719D and Ad-FLAG-NHERF2. IP FLAG–magnetic beads were washed, and bound proteins were eluted and separated by SDS–PAGE. Proteins were transferred to membrane and immunoblotted with anti-HA (monoclonal), anti-NHERF3 (polyclonal), anti-ezrin (polyclonal), and anti-FLAG (monoclonal) antibodies. Protein bands were visualized and quantitated with the Odyssey system and LI-COR software for the IR-Dye secondary antibodies. (B) NHE3, NHERF3, and NHERF2 protein bands from experiments as in A were quantitated with the Odyssey system and LI-COR software. Coimmunoprecipitated protein amount was normalized with immunoprecipitated protein amount (FLAG-NHERF2) and expressed as percentage of WT proteins. NHE3-S719A has greatly reduced binding to NHERF2 compared with WT and S719D. NHERF3 coIP by NHERF2 was also reduced in NHE3-S719A but normal in NHE3-S719D cells. (C, D) Caco-2/bbe/Lenti-NHERF3 knockdown cells were infected with Ad-HA-NHE3-WT, -S719A, or -S719D and Ad-FLAG-rat-NHERF3 (Lenti-NHERF3 short hairpin RNA has no effect on rat FLAG-NHERF3 expression). NHE3-WT and S719D showed similar binding with NHERF3, but NHE3-S719A had significantly less binding. n, number of experiments. p values are by unpaired t tests and ANOVA. (E) IPs of ezrin (mAb), FLAG (mAb), and immunoglobulin G (mAb) were performed using cell lysate prepared from Caco-2/bbe cells grown in 10-cm Transwells plates and infected with Ad-HA-NHE3-WT or -S719A and Ad-FLAG-NHERF2. Immunoblotting was with anti-HA, anti-ezrin, anti-NHERF3, and anti-FLAG antibodies. Binding of ezrin with NHE3-S719A was drastically reduced compared with NHE3-WT.
We previously reported that heterodimerization of NHERF3–NHERF2 is necessary for basal NHE3 activity as well as for elevated Ca2+ inhibition of NHE3 (Yang et al., 2014). We further used IP to determine whether the defect in Ca2+ inhibition of NHE3-S719A is related to changes in this heterodimerization of NHERF3-NHERF2 in cells expressing NHE3-WT or -S719A. Surprisingly, NHERF2 coIP of NHERF3 was also reduced in Caco-2/bbe cells expressing NHE3-S719A but not altered significantly in cells expressing NHE3-S719D (Figure 7, A and B). Because NHERF3 association with NHERF2 was reduced in NHE3-S719A-expressing cells (Figure 7, A and B), we further determined whether the NHE3 mutation affected binding to NHERF3. NHERF3-NHE3 coIP was examined in Caco-2/bbe/FLAG-NHERF3 cells. To examine NHERF3 coIP, we first knocked down endogenous NHERF3 and replaced it with FLAG-epitope–tagged NHERF3 (rat). Because NHERF3 is expressed endogenously in Caco-2/bbe cells in significant amounts, this was done to reduce the level of NHERF3 overexpression. The IP of NHERF3 coimmunoprecipitates NHE3, as previously reported, but the amount of coIP was reduced in Caco-2/bbe cells expressing NHE3-S719A but similar to that for NHE3-WT in cells expressing NHE3-S719D (Figure 7, C and D). Reverse coIP of NHERF2 by NHERF3 failed to detect endogenous NHERF2 (unpublished data). In addition, ezrin coIP of NHE3 was less with NHE3-S719A than with WT NHE3, whereas ezrin coIP of NHERF2 did not differ (Figure 7E). The latter findings support the view that it is the NHERF2–NHE3 binding that is affected by the S719A mutation.
NHE3-719A mutant has increased BB mobile fraction
Under basal conditions, NHE3-WT bound to NHERF2 is restricted in its BB mobility (Cha et al., 2004). In addition, because NHE3-S719A has reduced NHERF2 binding and all previous conditions in which the NHE3–NHERF2 association was reduced have been associated with increased NHE3 mobility, we predicted that NHE3-S719A would have increased apical membrane mobility. This prediction was supported, as shown in Figure 8. NHE3-S719A has increased mobility compared with NHE3-WT. Moreover, d-glucose stimulation of NHE3 is associated with increased NHE3 mobile fraction (Mf) and reduced binding to NHERF2. The d-glucose failed to increase NHE3 Mf in NHE3-S719A–expressing cells but increased Mf in NHE3-WT–expressing cells.
FIGURE 8:

Mobile fraction of NHE3-S719A is increased under basal conditions but is not increased by d-glucose. Caco-2/bbe cells were infected with Ad-HA-NHE3-WT or S719A cells. NHE3 mobility was measured under basal conditions (A, C) and 30 min after d-glucose treatment (B, D). (E) Mean ± SEM of multiple experiments as in A–D. n, number of experiments. p values by paired t tests.
NHE3-S719A mutant is present predominantly in smaller macromolecular complexes compared with NHE3-WT and NHE3-S719D
Multiple aspects of NHE3 association with signaling molecules that bind to its C-terminus and affect its regulation depend on NHERF in intestinal cells, with NHERF2 and NHERF3 being the primary NHERFs involved. NHERF2 and NHERF3 are involved in regulation of NHE3 at least in part by forming NHE3 macromolecular signaling complexes (Yang et al., 2014). Given reduced NHERF2 and NHERF3 interaction with the NHE3-S719A mutant, we hypothesized that the mutant would have altered associating protein lysate complexes and altered macrocomplex size. To test this hypothesis, we prepared total proteins from Caco-2/bbe cells expressing FLAG-NHERF2 and HA-NHE3-WT or -S719A or -S719D mutants. One milligram of total lysate was overlaid at the top of a 10–60% sucrose gradient and centrifuged at 40,000 rpm for 18 h at 4°C. NHE3-S719A was predominantly present in smaller macromolecular complexes than was NHE3-WT, whereas NHE3-S719D was present in complexes of similar size to NHE3-WT (Figure 9).
FIGURE 9:

NHE3-S719A predominantly associates with smaller protein complexes. Total cell lysate was prepared from Caco-2/bbe cells grown on Transwell filters and infected with Ad-HA-NHE3-WT, NHE3-S719A, or NHE3-S719D. The ∼2-mg total cell lysates were subjected to complex size analysis using sucrose gradient centrifugation as described in Materials and Methods. Because samples were collected from the bottom, starting complex sizes are larger. NHE3 was probed with HA mouse mAb. Distribution of NHE3-WT and S719D by size in protein complexes is similar, but S719A is predominantly in smaller protein complexes. A representative experiment, which was repeated three times with similar results, is shown.
NHE3-S719A mutant has reduced lipid raft distribution compared with NHE3 WT
Previous studies showed that membrane lipid rafts play an important role in NHE3 regulation and complex formation, including affecting the NHE3 pool involved in basal NHE3 activity, which depends on PI3K and AKT, NHE3 acutely stimulated by LPA, and NHE3 acutely inhibited by elevated calcium but not forskolin (Li et al., 2001; Murtazina et al., 2006). Consequently, we examined the lipid raft distributions of NHE3-WT and NHE3-S719A mutant by flotation studies of total Caco-2/bbe membrane using flotillin as a lipid raft marker. As shown in Figure 10, A and B, NHE3-S719A was distributed less in the lipid raft fractions than was NHE3-WT.
FIGURE 10:
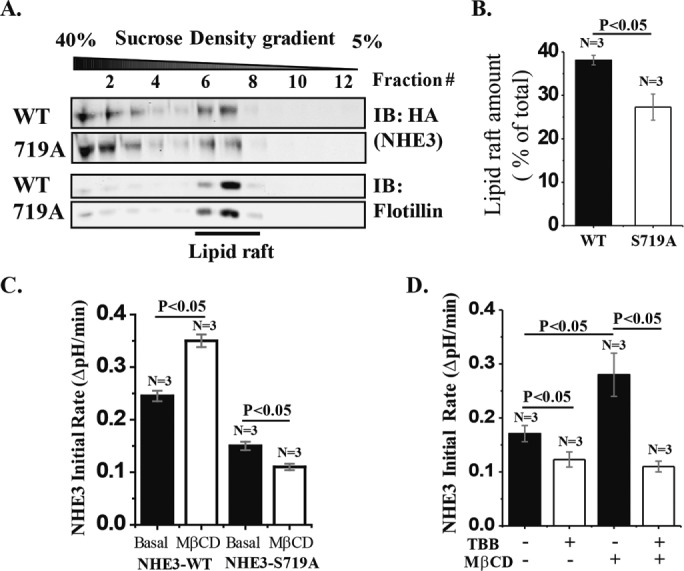
The lipid raft distribution of NHE3-S719A is reduced compared with WT-NHE3. (A) Total membrane preparations from Caco-2/bbe cells infected with Ad-HA-NHE3-WT or -S719A were subjected to lipid raft flotation analysis by sucrose gradient centrifugation. NHE3 was probed with HA mAb, and flotillin antibody was used as a lipid raft marker. NHE3 present at the low-density area overlapping with flotillin was considered as present in lipid rafts. (B) Percentage of NHE3 in lipid rafts calculated as total NHE3 intensity of the lipid raft component of NHE3 divided by total NHE3 intensity (lipid raft and nonlipid raft). Compared to NHE3-WT, S719A mutant was distributed ∼30% less in lipid rafts. (C) MβCD treatment (10 mM for 30 min) stimulated NHE3-WT activity (p < 0.05) but inhibited NHE3-S719A (p < 0.05). (D) MβCD stimulation of NHE3 activity in NHE3-WT cells was completely prevented by prior TBB treatment (p < 0.05). n, number of experiments. p values by paired t tests and ANOVA.
Having shown that the lipid raft distribution of NHE3-S719A was reduced compared with NHE3-WT, we examined the functional significance of this differential distribution by comparing the effect of MβCD on activities of NHE3-WT and NHE3-S719A. MβCD (10 mM for 30 min) stimulated NHE3 activity in cells expressing NHE3-WT, whereas in cells expressing NHE3-S719A, it significantly reduced NHE3 activity (Figure 10C). These results suggest that pools of NHE3-WT and NHE3-S719A or their regulatory proteins are distributed differently in lipid rafts under basal conditions, and at least a portion of the lipid raft pool of WT-NHE3 is inhibited by lipid raft contents, whereas a portion of the lipid raft pool of NHE3-719A is stimulated by lipid raft contents. In separate experiments, NHE3-WT–containing cells treated with the CK2 inhibitor TBB prevented MβCD stimulation of NHE3 (Figure 10D). This indicates that NHE3-S719 phosphorylation by CK2 is required for the lipid raft component that inhibits basal NHE3 activity.
Liquid chromatography–tandem mass spectrometry analysis of NHE3-WT and NHE3-S719A samples further reveals differential binding of proteins under basal conditions
Because the foregoing results showed that NHE3-S719A had changes in NHERF2, NHERF3, and SNX-27 binding (all contain closely related PDZ domains; Singh et al., 2015) and resulted in smaller multiprotein complexes, we questioned how this mutation altered the “NHE3 Interactome.” To address this question, we performed shotgun proteomic analysis on immunoprecipitated NHE3-WT and -S719A. As shown in Table 2, by this method, NHE3-S719A had reduced association not only with NHERF3 but also with multiple other proteins that bind throughout the NHE3 C-terminus or affected processes known to regulate NHE3 activity. These include proteins that bind to the most upstream part of the C-terminus (amino acids 475–589), including calcineurin homologous protein (CHP) and ezrin, and other proteins that bind to the NHE3 domain just downstream (amino acids 590–667), including CaM kinase IIδ. In addition, as shown in Figure 6, the NHE3-S719A mutant binds minimally to SNX27, which binds to the most C-terminal domain of NHE3 (amino acids 748–832).
TABLE 2:
Manually extracted list of NHE3 interacting proteins, most of which were previously shown to either associate with NHE3 or affect its regulation or expression.
| Normalized exclusive spectrum count | |||||
|---|---|---|---|---|---|
| Interacting protein | Accession no. | Molecular weight (kDa) | NHE3-WT | NHE3-S719A | S719A/WT |
| NHE3-complex related | |||||
| NHERF3 | gi: 21361142 | 57 | 4 | 1 | 0.25 |
| Calmodulin kinase IIδ | gi: 212549753 | 56 | 9 | 1 | 0.11 |
| CHP1 | gi: 6005731 | 22 | 21 | 10 | 0.48 |
| Ezrin | gi: 161702986 | 69 | 8 | 1 | 0.13 |
| RACK1 | gi|5174447 | 35 | 14 | 19 | 1.36 |
| Serine/threonine-protein phosphatase 2A | gi|21361399 | 65 | 8 | 11 | 1.38 |
| NHE3 signaling (sphingolipid metabolism) | |||||
| Sphingosine-1-phosphate lyase 1 | gi: 31982936 | 64 | 10 | 1 | 0.10 |
| Trafficking | |||||
| Golgi-to-endoplasmic reticulum trafficking protein 4 homologue | gi: 38570062 | 37 | 6 | 1 | 0.17 |
| Rho kinases (Rock) | gi: 41872583 | 161 | 8 | 1 | 0.13 |
| Rab 5C | gi: 354721184 | 27 | 6 | 1 | 0.17 |
| Heat shock 70-kDa protein 6 | gi:34419635 | 71 | 27 | 1 | 0.04 |
| Cytoskeleton association/tight junctions | |||||
| Myosin 9 | gi: 12667788 | 227 | 13 | 1 | 0.08 |
| Spectrin | gi: 154759259 | 285 | 48 | 18 | 0.38 |
| Tubulin α-4A chain isoform 2 | gi: 514052659 | 48 | 4 | 1 | 0.25 |
| Transporters | |||||
| Multidrug resistance protein 1 | gi: 42741659 | 141 | 7 | 1 | 0.14 |
| Glut1 | gi: 166795299 | 54 | 12 | 1 | 0.08 |
| Breakdown | |||||
| Ubiquitin-associated domain–containing protein 2 | gi: 221316645 | 39 | 10 | 1 | 0.10 |
| Proteosome | gi: 23110935 | 28 | 8 | 1 | 0.13 |
| FAS-associated factor2 | gi: 24797106 | 53 | 16 | 1 | 0.06 |
| Sequestosome | gi: 214830438 | 39 | 39 | 1 | 0.03 |
| 14-3-3 β/α | gi: 21328448 | 28 | 17 | 1 | 0.06 |
| 26S protease regulatory subunit 4 | gi: 24430151 | 47 | 23 | 1 | 0.04 |
| Calnexin precursor | gi:10716563 | 68 | 34 | 10 | 0.29 |
MS analysis of immunoprecipitated NHE3 and NHE3-S719A. A confluent monolayer of Caco-2/bbe cells grown on 10-cm Transwell plates was infected with Ad-FLAG-NHERF2 and Ad-HA-NHE3-WT or -S719A mutant. NHE3 IPs were performed using HA-conjugated beads, and proteins bound to HA-beads were eluted with 0.1 M glycine-HCl buffer (pH 3.0) and adjusted to pH 7. Eluted samples were used to identify proteins in NHE3 complex by liquid chromatography–tandem mass spectrometry. Identified proteins were normalized according to the ratio of NHE3-WT and NHE3-S719A total unique spectral counts (spectra on peptide sequences that are unique to a protein). Interacting proteins are listed with protein accession number, molecular weight, and normalized spectral counts.
Thus NHE3-S719A has changes in binding partners across the entire C-terminus. The NHE3-S719A mutant had reduced binding to additional proteins shown to be involved in NHE3 regulation (sphingosine-1-phosphate lyase1); proteins known to be involved in trafficking, including of NHE3 (Rho/ROCK, Rab 5C); and association with the chaperone HSP70 (Table 1). The full list of NHE3-interacting proteins identified by the proteomic analysis is given in Supplemental Table S1, which also indicates differences in binding to WT and NHE3-S719A.
DISCUSSION
The present studies reveal a major regulatory role for the single NHE3 amino acid, S719, that CK2 phosphorylates in the NHE3 C-terminus. The extent of changes caused by mutation of Ser to Ala was unexpected and includes the following: 1) Reduced size of the NHE3-containing complexes and association with multiple proteins that bind throughout the NHE3 C-terminus and not just in the area of the phosphorylation site. 2) Altered specific aspects of basal and acutely stimulated as well as acutely inhibited NHE3 activity while leaving other aspects intact. 3) Reduced percentage of total NHE3 present in lipid rafts. That WT-NHE3 and NHE3-S719D had similar basal activity and PI3K dependence, similar inhibition by forskolin and carbachol, and similar multiprotein complex size suggests that CK2 phosphorylation of S719 under basal conditions exerts its maximal effect on NHE3.
We suggest some possible unifying mechanisms that might account for such a major effect of this single phosphorylation, based on examining the specific changes in NHE3 activity that occurred, including 1) reduced basal NHE3 activity by effects on the component, which is PI3K/AKT dependent, 2) reduced acute stimulation of NHE3 by LPA acting via the LPA5 receptor, and 3) reduced acute inhibition by elevated Ca2+ but not the inhibition caused by forskolin or hyperosmolarity (Table 2). The common theme that comes from these changes in NHE3 activity relates to which aspects of NHE3 regulation are lipid raft dependent based on inhibition by MβCD, as previously reported (Murtazina et al., 2006; Zachos et al., 2014). These aspects include the PI3K- and AKT-dependent aspects of basal NHE3 activity; the acute stimulation caused by LPA via LPA5 receptors; and the acute inhibition caused by elevated Ca2+ but not that caused by forskolin and hyperosmolarity. Thus one conclusion suggested by these results is that the major effect of phosphorylation of S719 is to determine lipid raft distribution of NHE3. What is not clear from these results is whether the alteration in the NHE3 C-terminal–binding proteins led to the change in lipid raft distribution of NHE3 or resulted from this change in lipid raft distribution.
In addition to the specific transport processes involving NHE3 that are CK2 and lipid raft dependent, the overall effect of lipid rafts on NHE3 activity is inhibitory because removing this contribution with MβCD stimulates NHE3 activity. The present studies duplicate results in Caco-2/bbe cells previously published by our laboratory (Zachos et al., 2014), although we also reported that MβCD inhibited basal NHE3 activity in rabbit small intestine and OK cells (Murtazina et al., 2006), indicating cell-type variability. That this effect on basal NHE3 activity requires CK2 phosphorylation of S719 is supported because MβCD inhibited rather than stimulated NHE3 activity in the NHE3-S719A mutant. The specific lipid raft–related proteins that inhibit basal NHE3 activity were not established by these studies. However, a common theme of NHE3 regulation for which S719 is necessary is the involvement of the multi-PDZ domain–containing scaffold protein NHERF2. Of the four members of the NHERF family, NHERF2 is the member with the largest representation in lipid rafts, although because of technical issues in defining lipid raft involvement, there is some disagreement in published studies (Li at al., 2001; Sultan et al., 2013; Yang et al., 2013). Thus one explanation for at least some of the functional effects of CK2 phosphorylation of NHE3 at S719 is that this is necessary for the presence of NHE3 in lipid rafts, where it associates with NHERF2.
The phosphorylation of NHE3 at S719 affects NHE3 association with too many proteins to attribute the effects on basal and regulated function to just one, and studying NHE3 regulation in cells that lack NHERF2, such as PS120 fibroblasts, does not reproduce all of the findings described here. However, the reduced binding to NHERF2 potentially contributes to many of the findings with the S719 mutant, including that the presence of NHE3 in lipid rafts is at least partially dependent on its binding to NHERF2, as the NHE3 pool in lipid rafts is reduced in the small intestine of NHERF2-null mice (Sultan et al., 2013). The lack of binding of NHERF2 to NHE3 demonstrated for the NHE3-S719A mutant is consistent with NHE3 being in smaller complexes, having increased BB mobility, and failing to respond to LPA with acute stimulation and to elevated Ca2+ (carbachol and calcium ionophore) with inhibited activity, all consequences seen in NHE3 regulatory studies in which NHERF2 was not present via genetic manipulation. Of note, the reduced basal NHE3 activity with the S719A mutation, which is not duplicated by lack of NHERF2 binding, does occur with lack of NHE3 binding to SNX27 (Singh et al., 2015). This involves the SNX27-PDZ domain, which is highly homologous with the PDZ domains of the NHERF family. Thus the effects of the NHE3-S719A mutation on NHE3 activity could relate to lack of binding of related PDZ domains.
Also unexpected was the observation that the mutant NHE3-S719A but not S719D had a dramatic reduction in NHE3-associated complex size and that this reduced size was associated with reduced binding of NHE3 to multiple proteins, which bound throughout the NHE3 C-terminus and not just near the mutated amino acid. The proteins with reduced binding include those binding to 1) the very beginning of the C-terminus (CHP), which binds to NHE3 amino acids 464–484 by homology with the solved structure of NHE1 and is necessary for basal NHE3 activity (Li and Donowitz, unpublished data); 2) the proximal C-terminal stimulatory complex (amino acids 509–528; this includes AKT and GSK-3, which interact with the putative NHE3 α-helix that directly binds ezrin; Singh et al., 2014); 3) the large complex involved in both acute stimulation and inhibition (amino acids 586–605; this includes binding to CaMKIIδ and NHERFs, although CK2 binding to this area was not affected); and 4) the most distal C-terminus (SNX27 (Cha and Donowitz, unpublished data). Ezrin binding to NHE3 was also reduced, although it is not clear whether this is the ezrin that directly binds NHE3 or that which binds via NHERF binding. In addition, our NHE3-coprecipitation proteomics approach identified NHE3 proteins not previously known to associate with NHE3 or its complexes, which also had their association with NHE3 reduced by the S719A mutation. Some of these proteins would be expected to take part in the regulation of NHE3 trafficking (see Table 1; the list includes sphingosine-1-p lyase, Rock, Rab5C, and HSP70).
Lacking structural information concerning the NHE3 C-terminus and its complexes, we can only speculate why the single S719A mutation causes such major changes in the NHE3 complexes. It is not that any change in amino acids at this site would cause similar changes, because S719D behaved like wild type in those conditions that were compared. Based on homology modeling with NHE1, NHE3-S719 and the other, more C-terminal NHE3 amino acids have been suggested as being in a disordered part of the C-terminus (Norholm et al., 2011; Hendus-Altenburger et al., 2014). This is in contrast to the more upstream C-terminus, which contains multiple putative α-helixes and β-sheets, of which the most upstream α-helix has been structurally demonstrated to exist in complex with CHP in NHE1 (Ammar et al., 2006). We speculate that although theoretically disordered, at least partially because of the phosphorylation of NHE3-S719, the NHE3 C-terminus is rather highly structured and the distal C-terminus physically interacts with other parts of the C-terminus to influence NHE3 binding partners throughout the NHE3 regulatory domain. NHE3 exists in pools in the early endosomes and recycling endosomes as well as the brush border. Complexes are believed to form at both the brush border and intracellularly (Donowitz et al., 2009). Which site is primarily affected by the S719 mutation to alter the complex size is not known.
Although we suggested several possible wide-reaching mechanistic explanations for the changes in NHE3 activity with NHE3-S719A, the changes in lipid raft association and association with several specific binding partners that are altered were previously shown to affect NHE3 activity (Donowitz et al., 2009). For instance, binding of the NHE3 C-terminus to CHP and SNX27 was shown to be necessary for normal basal NHE3 activity, and binding to SNX27 is also necessary for the stimulated exocytosis involved in acute increases in NHE3 activity (Pang et al., 2001; Zaun et al., 2008; Singh et al., 2015). One of the implications of the present study is that changes in NHE3 lipid raft association via changes in its C-terminus may be a more general mechanism for NHE3 regulation than previously suspected and could explain previous studies that concentrated on effects of single binding partners of NHE3.
We previously demonstrated that acute NHE3 regulation involves a heterodimer of NHERF3 and NHERF2, with the NHERF3 binding to NHERF2 via its C-terminal four–amino acid PDZ domain recognition motif. This heterodimer is involved in setting basal NHE3 activity and is necessary for Ca2+ inhibition of NHE3 (Yang et al., 2014). The reduced NHERF2-NHERF3 coIP demonstrated in cells expressing NHE3-S719A is likely to contribute to the lack of Ca2+ inhibition of NHE3 and reduced basal NHE3 activity. In addition, the reduced NHERF2-NHERF3 coIP in the NHE3-S719A mutant indicates that much of this NHERF heterodimerization depends on the presence of NHE3 and further supports that the heterodimer is involved in NHE3 regulation. Because NHERF3 does not have a significant lipid raft distribution whereas NHERF2 has significant representation in lipid rafts allows the speculation that one function of the heterodimer is to serve as a switch that determines whether NHE3 regulation is lipid raft dependent or independent.
The multiple aspects of NHE3 regulation and complex formation that we showed were dependent on CK2 phosphorylation of a single C-terminal amino acid strongly support that the NHE3 C-terminus exists as a complex structure in terms of associating with multiple proteins and likely having interactions between the large complex-forming areas and other parts of the C-terminus. The relationship of these changes in NHE3 regulation with the lipid raft distribution of NHE3 and its C-terminal complex formation illustrates the need for more structural information concerning the NHE3 C-terminus. In addition, the alteration of NHE3 activity by changes in just one amino acid suggest that the NHE3 C-terminus might be a relevant drug target to alter NHE3 activity, which could be useful for treatment of multiple gastrointestinal conditions, including diarrhea and constipation.
MATERIALS AND METHODS
Chemicals, reagents, and antibodies
Reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. HOE-694 was a kind gift from Jurgen Peunter of Sonafi-Aventis (Frankfurt, Germany). TBB and DMAT were from Calbiochem/EMD-Millipore (Billerica, MA). Ca2+ ionophore 4-Br-A23187 was from Biomol (Plymouth Meeting, PA). EZ-Link Sulfo-NHS-SS-biotin and streptavidin-agarose were from Thermo-Pierce Chemical (Rockford, IL). Protein-G agarose beads were from Millipore. Glutathione Sepharose 4B beads were from GE Healthcare (Chicago, Il). 2,7-Bis(2-carboxyethyl)-5-carboxyfluorescein acetoxymethyl ester (BCECF-AM) and Alexa Fluor 488 and 568–conjugated goat anti-mouse and anti-rabbit secondary antibodies were from Invitrogen (Carlsbad, CA). Mouse monoclonal antibodies to the HA epitope were from Covance Research Products (Princeton, NJ). Rabbit polyclonal antibodies to HA were from Santa Cruz Biotechnology (Dallas, TX). Mouse monoclonal FLAG and ezrin antibodies were from Sigma-Aldrich and rabbit polyclonal ezrin antibodies were from Abcam (Cambridge, United Kingdom). Rabbit anti-NHERF2 was a gift from Chris Yun (Emory University School of Medicine, Atlanta, GA). Glyceraldehyde-3-phosphate dehydrogenase mouse monoclonal antibodies were from U.S. Biological (Swampscott, MA). Mouse Anti–flotillin-1 was from BD Biosciences (Franklin Lakes, NJ). DNA primers were from Operon Biotechnologies (Huntsville, AL). Restriction endonucleases were from New England Bio Labs (Ipswich, MA).
Cell culture
The Caco-2/bbe cell line originally derived from a human colon adenocarcinoma was obtained from M. Mooseker (Yale University) and J. Turner (University of Chicago) and grown on membranes (Transwell or “filterslips”) as described previously (Janecki et al., 1998, 2000) in DMEM containing 25 mM NaHCO3 supplemented with 0.1 mM nonessential amino acids, 10% fetal bovine serum (heat inactivated, 55°C for 30 min), 4 mM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin, pH 7.4, in 5% CO2/air at 37°C.
Adenoviral HA-NHE3, FLAG-NHERF2, FLAG-NHERF3 (rat), and HA-LPA5R preparation, purification, and expression
Triple HA-tagged rabbit NHE3-WT, -S719A, -S719D, and 3X-FLAG-NHERF2 (human) and 3X-FLAG-NHERF3 (rat) were cloned into the adenoviral shuttle vector ADLOX.HTM under the control of a cytomegalovirus (CMV) promoter. Double HA-tagged LPA5R was also cloned in ADLOX.HTM shuttle vector under the CMV promoter. Virus was generated and purified as described previously (Sarker et al., 2008). To express the adenovirus, Caco-2/bbe cells on Transwell inserts were treated with 6 mM ethylene glycol tetraacetic acid in serum-free Caco-2 medium for 2–3 h at 37°C before viral infection to allow maximum virus exposure to both the apical and basolateral surfaces. Viral particles were diluted in serum-free Caco-2 medium, and the cells were infected by incubation at 37°C for 6–7 h, after which the growth medium was replaced. For transport assays, immunostaining, or Western blot analyses, cells were used ∼44 h after infection and serum starved for at least 4 h.
Stable Caco-2/bbe cells with lentivirus short hairpin RNA NHERF3 knockdown
Several stable cell lines of Caco-2/bbe with NHERF3 knocked down were prepared using three separate lentivirus short hairpin RNA constructs (pLKO.1 puromycin), as previously described (Sarker et al., 2011). Caco-2/bbe cells with the most NHERF3 knockdown (>70% knockdown) were used.
NHE3 activity measurement
Monolayers of polarized Caco-2/bbe cells were grown on polycarbonate membranes (0.4-μm pore size) attached to plastic coverslips (called filterslips) for 12 d and infected with adeno-HA-NHE3-WT or -S719A/D mutants. Approximately 44 h after infection, cells were serum starved for at least 4 h, and Na+/H+ exchange activity (NHE3) was determined with the intracellular pH (pHi)-sensitive dye BCECF-AM (60-min loading) as described previously (Levine et al., 1995; Sarker et al., 2011) in the presence of HOE-694 (50 μM) to inhibit the contributions of NHE1 and NHE2. Exposure to DMAT (30 μM) or TBB (20 μM), forskolin (25 μM), wortmannin (0.1 µM), LPA (1 µM), and MβCD (10 mM) was during the final 30 min of dye loading/NH4Cl prepulse, but Akt inhibitor Akti-VIII (10 µM) was incubated for 1 h. In some experiments, 25 μM carbachol or 1 µM A23187 was added to the TMA perfusion after dye loading/NH4Cl prepulse with cells exposed to carbachol or A23187 only during the 4- to 5-min tetramethyl ammonium (TMA) exposure. Hyperosmolarity was generated by adding 50 mM mannitol to Na+ medium. For transport assays, filterslips were mounted in a cuvette, placed in a fluorometer (Photon Technology International), and perfused at both monolayer surfaces with TMA medium. The Na+/H+ exchange was started by replacing the TMA medium with Na+ medium at the apical monolayer surface while TMA perfusion continued basolaterally. Changes in pHi were monitored by recording the emission signal at 530 nm after excitation alternating between 440 and 490 nm. The fluorescence ratio was calibrated to pHi with the high potassium/nigericin method as described previously (Sarker et al., 2008, 2011). Rates of Na+-dependent intracellular alkalization (efflux of H+ in μM/s) were calculated for a given pHi within the linear phase (∼1 min) to quantitate the initial rate of intracellular alkalization and reported as ΔpH/min.
Immunoprecipitation and immunoblotting
Cells were grown in 10-cm Transwell plates for 12 d. Two days after adenoviral infection, Caco-2/bbe cells were serum starved for at least 4 h and washed three times with cold phosphate-buffered saline (PBS). Cells were scraped in PBS and collected in 1.5-ml Eppendorf tubes. Cells were mixed with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) lysis buffer containing protease inhibitors (20 mM HEPES, pH 7.4, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4) and homogenized by passing them through a 1-ml syringe/26-gauge needle (25×) and rotated for ∼1 h at 4°C to maximize solubilization. After removal of insoluble cell debris by centrifugation (10,000 rpm × 10 min), the protein concentration was measured with the Bio-Rad protein assay, Bradford dye–binding method. A total of ∼1.5 mg protein was used for IP using anti-FLAG magnetic beads or anti-HA resin or with specific antibody. In case of unconjugated antibody, Protein-G beads were used to precipitate antibody–protein complexes. After washing the beads four times, proteins were eluted with 2× Laemmli sample buffer. Proteins were then separated by SDS–PAGE (10%), transferred onto nitrocellulose membranes, and immunoblotted. Fluorescently labeled IR-Dyes 800 and 680 conjugated with rabbit polyclonal or mouse monoclonal antibodies (mAbs) were used as secondary antibodies (1:10,000). Protein bands were visualized and quantitated with the Odyssey system and LI-COR software for the IR-Dye secondary antibodies as described previously (Sarker et al., 2011).
Immunofluorescence
Caco-2/bbe cells were seeded on 0.4-μm Transwell polycarbonate semipermeable inserts. On day 12 after confluence, cells were infected with Ad-HA-NHE3-WT or -S719A mutant as described. Two days after infection, cells were serum starved for 4 h, kept at 4°C for 30 min, and then fixed with 3% paraformaldehyde (PFA) in cold PBS for 30 min. The fixed cells were washed with PBS and treated with 20 mM l-glycine for 10 min. Cells were incubated with WGA-488 (1:500 dilution; Thermo Fisher) for 30 min, washed three times with PBS, blocked, and permeabilized in PBS containing 1% bovine serum albumin (BSA) and 10% fetal bovine serum plus 0.075% saponin for 45 min. Mouse monoclonal HA antibodies (1:100) were incubated for 1 h at room temperature in blocking solution. Cells were then washed three times with PBS for 10 min for each wash. Cells were incubated with anti-mouse Alexa Fluor 568 (1:100 dilution) secondary antibody (Invitrogen) and Hoechst 33342 (1:500 dilution; Invitrogen) for 40 min at room temperature. Cells were washed three times with PBS, mounted with Fluorogel mounting buffer (Electron Microscopy Sciences, Hatfield, PA), and imaged with a Zeiss LSM510 confocal fluorescence microscope with AIM acquisition software and 8-bit file formats, using a water immersion objective (C-Apochromat/63× 1.2 W correction). Serial xy-sections were obtained at 0.5-μm steps, and xz-images were reconstructed with MetaMorph image analysis software (Molecular Devices). MetaMorph software was used to quantify the NHE3 total and surface amount of cells. The fluorescence intensity of each 0.5-μm xy-section containing NHE3 from top to bottom (18–20 sections) was determined separately, and total intensity was considered to represent total NHE3. The xy-sections that contained overlapping NHE3 and WGA signal were considered BB NHE3 (normally the upper two to six sections), and this summed NHE3 signal was considered BB NHE3.
Reduced glutathione resin pull down
Reduced glutathione (GSH)-resin pull-down assays were performed by incubating 5 µg of purified recombinant GST-tagged SNX27 with 1.5 mg of Caco-2/bbe/Ad-HA-NHE3/Ad-FLAG-NHERF2 cell lysate and 15 µl of GSH resin (Glutathione Sepharose 4B resin). The volume of the final mixture was adjusted to 750 μl with lysis buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1% Triton X-100, and protease inhibitors). After overnight rotation at 4°C, GSH resin was separated and washed four times with lysis buffer. Protein bound to GST was eluted with 75 μl of 2× Laemmli sample buffer by incubation at 80°C for 7 min. The input and eluted samples were analyzed by 10% SDS–PAGE and Western blotting using HA-polyclonal antibody.
Fluorescence recovery after photobleaching analysis
Fluorescence recovery after photobleaching (FRAP) was performed on a stage heated to 37°C of a Zeiss LSM 510 confocal microscope equipped with a C-Apochromat 63×/1.2 Korr water-immersion objective as described previously (Lin et al., 2011) with minor revision. Caco-2/SGLT1 cells 12 d postconfluency were infected with adeno-HA-NHE3-WT or -S719A mutant. For the FRAP study, cells were washed with DMEM/F-12 without phenol red twice and incubated for 1 h with blocking buffer (1% BSA in PBS). Cells were immunostained with monoclonal anti-HA antibody 1–2 h at 4°C and with Alexa Fluor 488 as secondary antibody for 1 h at 4°C in 1% BSA in PBS. Cells were then treated with d-glucose (25 mM) or d-mannose (25 mM) for 10 min. Transwell filter was cut out, placed on the glass slides with the apical surface outward, covered by a drop of d-glucose or d-mannose medium, and finally sealed with a coverslip with nail polish. FRAP was performed within 1 h (Lin et al., 2011). All data are shown as mean ± SE of the number of cells analyzed, which were obtained from at least three identical experiments unless stated otherwise. Statistical comparison was performed by unpaired Student’s t test.
Sucrose gradient ultracentrifugation for complex size analysis
Experiments were performed as described previously with slight modification (Li and Donowitz, 2014). Caco-2/bbe cells were grown on 10-cm Transwell plates for 12 d and infected with adeno-HA-NHE3-WT or -S719A/D mutants along with adeno-FLAG-NHERF2. At ∼44 h postinfection, Caco-2/bbe cells were serum starved for at least 4 h and washed three times with cold PBS. Harvested cells were homogenized by passing them through a 1-ml syringe/26-gauge needle and then solubilized with 5% sucrose in HEPES lysis buffer plus 0.5% Triton X-100 and protease inhibitors. Step gradients of 11 ml were prepared by overlaying 1 ml each of the following sucrose gradients: 60, 50, 40, 30, 25, 22.5, 20, 17.5, 15, 12.5, and 10%. Each gradient was prepared with the same HEPES buffer plus 0.1% Triton X-100. Samples (1 ml) were overlaid at the top of the gradient and centrifuged in a Beckman SW41Ti swing rotor at 40,000 rpm at 4°C for 16 h. Twenty-four fractions (0.5 ml of each) were collected. An 80-μl amount of each fraction was analyzed with SDS–PAGE and Western blotting.
Sucrose gradient ultracentrifugation for lipid rafts
Methods were slightly modified from that previously reported (Li et al., 2001; Akhter et al., 2002). Caco-2/bbe cells were grown on 10-cm Transwell plates for 12 d and infected with adeno-HA-NHE3-WT or -S719A mutant. Cells were washed three times with cold PBS. Harvested cells were homogenized by passing them through a 1-ml syringe/26-gauge needle and sonicated in TNE buffer (Tris–NaCl–EDTA) containing 25 mM Tris, pH 7.4, 150 mM NaCl, 50 mM NaF, 5 mM EDTA, 1 mM Na3VO4, and protease inhibitors. Nuclei and debris were removed by centrifugation at 3000 × g for 15 min at 4°C. The total membranes were pelleted by ultracentrifugation at 150,000 × g for 30 min at 4°C. Total membranes were resuspended by 1-ml syringe/26-gauge needle and then solubilized with 0.5 ml of cold TNE buffer supplemented with 0.5% Triton X-100 at 4°C for 30 min on a rotary shaker. Equal amount of samples were adjusted to 40% sucrose (final volume, 1 ml) before being overlaid with step gradients, 2 ml of 40%, 3 ml of 35%, 3 ml of 15%, and 3 ml of 5% sucrose. Each gradient was prepared with TNE buffer and adjusted to 0.5% Triton X-100. Samples were centrifuged in a Beckman SW41Ti rotor at 40,000 rpm at 4°C for 18 h. Twelve fractions (1 ml each) were collected from the bottom of the tubes. An 80-µl amount of each fraction was analyzed with SDS–PAGE and Western blotting, including for flotillin as a lipid raft marker. For MβCD treatment, serum-starved cells were washed once with Hanks balanced salt solution (HBSS) and incubated without or with 10 mM MβCD in HBSS at 37°C for 30 min.
Proteomic protein identification/mass spectroscopy analysis
Caco-2/bbe cells grown on 10-cm Transwell plates were infected with Ad-FLAG-NHERF2 and Ad-HA-NHE3-WT or -S719A mutant. NHE3 IPs were performed using HA-conjugated beads according to the procedure mentioned earlier, except for the elution step. Proteins bound to HA-beads were eluted with 0.1 M glycine-HCl buffer (pH 3.0). Eluted samples were immediately adjusted to ∼pH 7 using 1 M HEPES buffer. Proteins were reduced in 50 μl of 2.5 mM dl-dithiothreitol (DTT) at 60°C for 1 h, alkylated in 50 μl of 5 mM iodoacetamide for 45 min at room temperature in the dark, and digested with trypsin (V5111; Promega) in 250 mM TEAB according to the filter-aided sample preparation method (Wisniewski et al., 2009). Peptides from protein digests were desalted using Oasis HLB uElution solid-phase extraction plates (Waters) and analyzed by reverse-phase liquid chromatography coupled to tandem mass spectrometry using an EasyLC nanoLC 1000 (Thermo Scientific) interfaced with a Q-Exactive Plus (QE Plus; Thermo Scientific) mass spectrometer. Desalted peptides were resuspended in 10 μl of 2% acetonitrile in 0.1% formic acid and separated on a 75 µm × 150 mm ProntoSIL-120-5-C18 H column (5 µm, 120 Å; www.bischoff-chrom.com) using a 2–90% acetonitrile gradient at 300 nl/min over 90 min. Eluting peptides were sprayed through a 1-µm emitter tip (New Objective; www.newobjective.com) at 2.0 kV directly into the QE Plus. Survey scans (full mass spectroscopy [MS]) were acquired from 350 to 1800 m/z with data-dependent monitoring of up to 15 peptide masses (precursor ions), each individually isolated in a 2.0-Da window and fragmented using HCD activation collision energy 25 with a 30-s dynamic exclusion. Precursor and the fragment ions were analyzed at resolutions 70,000 and 35,000, respectively, with automatic gain control target values at 3e6 with 60-ms maximum injection time (IT) and 2e5 with 250-ms maximum IT, respectively. Isotopically resolved masses in precursor (MS) and fragmentation (MS2) spectra were extracted from raw MS data without deconvolution and with deconvolution using Xtract or MS2 Processor in Proteome Discoverer (PD) software (version 1.4; Thermo Scientific). All extracted data were searched using Mascot (2.5.1; www.matrixscience.com) against the RefSeq2015 protein database. The following criteria were set for all database searches: sample’s species; trypsin as the enzyme, allowing one missed cleavage; cysteine carbamidomethylation as fixed modification; and methionine oxidation and asparagine or glutamine deamidation as variable modifications. Mascot results were imported into Scaffold (version 4.4.1.1; Proteomic Software). Protein and peptide identifications were filtered in Scaffold at 1% false discovery rate confidence threshold, based on a concatenated decoy database search in Mascot. Protein amounts were based on the number of Exclusive Unique Spectrum Counts after normalizing counts to the same number of Exclusive Unique Spectrum Counts for immunoprecipitated NHE3. The Database for Annotation, Visualization and Integrated Discover bioinformatics tool (National Institute of Allergy and Infectious Diseases) was used to categorize the list of significantly enriched Gene Ontology.
In vitro CK2 phosphorylation assay
In vitro phosphorylation of NHE3-S719 was carried out by recombinant CK2 with purified His-tagged NHE3-C terminal fragment amino acids 668–775, with phosphorylation detected using the Pro-Q Diamond Phosphoprotein Gel Stain kit (Invitrogen). The kinase reaction was done by incubating 2 µg of NHE3 fusion protein, 0.3 mM ATP, kinase assay buffer (50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, and 0.01% Brij 35, pH 7.5, at 25°C), 200 U of CK2 (recombinant α, β subunits; New England BioLabs) with or without 30 μM TBB or 10 μM AKTi-VIII for 30 min at 30°C (final volume, 30 µl). Reaction was terminated by adding 30 µl of 2× Laemmli sample buffer and heated at 80°C for 10 min. Samples were separated by a 14% SDS–PAGE minigel. The gel was then fixed and stained with Pro-Q Diamond phosphoprotein gel stain kit according to the manufacturer’s protocol. After destaining and washing, the gel was scanned with light-based scanners (Typhoon 9400; GE Healthcare) and then further stained with Coomassie blue.
Statistical analysis
Results are expressed as mean ± SE. Statistical evaluation was performed by analysis of variance (ANOVA) when more than three conditions were compared in a single experiment or by Student’s t tests (paired and unpaired).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK26523, R01DK61765, P01DK072084, and P30DK089502. We acknowledge use of the Kudsi Imaging Core Facility of the Hopkins Digestive Disease Basic and Translational Research Core Center.
Abbreviations used:
- AKTi
AKT inhibitor VIII
- DMAT
2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazol
- LPA
lysophosphatidic acid
- MβCD
methyl β-cyclodextrin
- NHE3
Na+/H+ exchanger 3
- NHERF
Na+/H+ exchange regulatory cofactor
- SNX27
sorting nexin 27
- TBB
4,5,6,7-tetrabromo-2-azabenzimidazole, 4,5,6,7-tetrabromobenzotriazol
- Wort
wortmannin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-12-0862) on May 11, 2017.
REFERENCES
- Akhter S, Kovbasnjuk O, Li X, Cavet M, Noel J, Arpin M, Hubbard AL, Donowitz M Na(+)/H(+) exchanger 3 is in large complexes in the center of the apical surface of proximal tubule-derived OK cells. Am J Physiol Cell Physiol. 2002;283:C927–C940. doi: 10.1152/ajpcell.00613.2001. [DOI] [PubMed] [Google Scholar]
- Ammar YB, Takeda S, Hisamitsu T, Mori H, Wakabayashi S. Crystal structure of CHP2 complexed with NHE1-cytosolic region and an implication for pH regulation. EMBO J. 2006;25:2315–2325. doi: 10.1038/sj.emboj.7601145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Chen T, Sarker R, Yang J, Raben D, Tse CM, Kovbasnjuk O, Donowitz M. Lysophosphatidic acid stimulation of NHE3 exocytosis in polarized epithelial cells occurs with release from NHERF2 via ERK-PLC-PKCδ signaling. Am J Physiol Cell Physiol. 2014;307:C55–C65. doi: 10.1152/ajpcell.00045.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Donowitz M. The epithelial brush border Na+/H+ exchanger NHE3 associates with the actin cytoskeleton by binding to ezrin directly and via PDZ domain-containing Na+/H+ exchanger regulatory factor (NHERF) proteins. Clin Exp Pharmacol Physiol. 2008;35:863–871. doi: 10.1111/j.1440-1681.2008.04931.x. [DOI] [PubMed] [Google Scholar]
- Cha B, Kenworthy A, Murtazina R, Donowitz M. The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J Cell Sci. 2004;117:3353–3365. doi: 10.1242/jcs.01180. [DOI] [PubMed] [Google Scholar]
- Cha B, Kim JH, Hut H, Hogema BM, Nadarja J, Zizak M, Cavet M, Lee-Kwon W, Lohmann SM, Smolenski A, et al. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem. 2005;280:16642–16650. doi: 10.1074/jbc.M500505200. [DOI] [PubMed] [Google Scholar]
- Chen M, Sultan A, Cinar A, Yeruva S, Riederer B, Singh AK, Li J, Bonhagen J, Chen G, Yun C, et al. Loss of PDZ-adaptor protein NHERF2 affects membrane localization and cGMP- and [Ca2+]- but not cAMP-dependent regulation of Na+/H+ exchanger 3 in murine intestine. J Physiol. 2010;588:5049–5063. doi: 10.1113/jphysiol.2010.198721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Kocinsky HS, Cha B, Murtazina R, Yang J, Tse CM, Singh V, Cole R, Aronson PS, de Jonge H, et al. Cyclic GMP kinase II (cGKII) inhibits NHE3 by altering its trafficking and phosphorylating NHE3 at three required sites: identification of a multifunctional phosphorylation site. J Biol Chem. 2015;290:1952–1965. doi: 10.1074/jbc.M114.590174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;3:825–872. doi: 10.1152/physrev.00030.2006. [DOI] [PubMed] [Google Scholar]
- Donowitz M, Mohan S, Zhu CX, Chen TE, Lin R, Cha B, Zachos NC, Murtazina R, Sarker R, Li X. NHE3 regulatory complexes. J Exp Biol. 2009;212:1638–1646. doi: 10.1242/jeb.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi AC, Sole, Di F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am J Physiol Cell Physiol. 2012;302:C1569–C1587. doi: 10.1152/ajpcell.00017.2012. [DOI] [PubMed] [Google Scholar]
- Harada K, Fukuda E, Hirohashi N, Chiba K. Regulation of intracellular pH by p90Rsk-dependent activation of an Na(+)/H(+) exchanger in starfish oocytes. J Biol Chem. 2010;285:24044–24054. doi: 10.1074/jbc.M109.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendus-Altenburger R, Kragelund BB, Pedersen SF. Structural dynamics and regulation of the mammalian SLC9A family of Na+/H+ exchangers. Curr Top Membr. 2014;73:69–148. doi: 10.1016/B978-0-12-800223-0.00002-5. [DOI] [PubMed] [Google Scholar]
- Janecki A J, Montrose MH, Zimniak P, Zweibaum A, Tse C M, Khurana S, Donowitz M. Subcellular redistribution is involved in acute regulation of the brush border Na/H exchanger isoform 3 in human colon adenocarcinoma cell line Caco-2. Protein kinase C-mediated inhibition of the exchanger. J Biol Chem. 1998;273:8790–8798. doi: 10.1074/jbc.273.15.8790. [DOI] [PubMed] [Google Scholar]
- Janecki AJ, Janecki M, Akhter S, Donowitz M. Basic fibroblast growth factor stimulates surface expression and activity of Na(+)/H(+) exchanger NHE3 via mechanism involving phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:8133–8142. doi: 10.1074/jbc.275.11.8133. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee-Kwon W, Park JB, Ryu SH, Yun CH, Donowitz M. Ca2+-dependent inhibition of Na+/H+ exchanger 3 (NHE3) requires an NHE3-E3KARP-alpha-actinin-4 complex for oligomerization and endocytosis. J Biol Chem. 2002;277:23714–23724. doi: 10.1074/jbc.M200835200. [DOI] [PubMed] [Google Scholar]
- Lee-Kwon W, Johns DC, Cha B, Cavet M, Park J, Tsichlis P, Donowitz M. Constitutively active phosphatidylinositol 3-kinase and AKT are sufficient to stimulate the epithelial Na+/H+ exchanger 3. J Biol Chem. 2001;276:31296–31304. doi: 10.1074/jbc.M103900200. [DOI] [PubMed] [Google Scholar]
- Levine SA, Nath SK, Yun CH, Yip JW, Montrose M, Donowitz M, Tse CM. Separate C-terminal domains of the epithelial specific brush border Na+/H+ exchanger isoform NHE3 are involved in stimulation and inhibition by protein kinases/growth factors. J Biol Chem. 1995;270:13716–13725. doi: 10.1074/jbc.270.23.13716. [DOI] [PubMed] [Google Scholar]
- Li X, Donowitz M. Fractionation of subcellular membrane vesicles of epithelial and non-epithelial cells by OptiPrep™ density gradient ultracentrifugation. Methods Mol Biol. 2014;174:85–99. doi: 10.1007/978-1-4939-0944-5_6. [DOI] [PubMed] [Google Scholar]
- Li X, Galli T, Leu S, Wade J B, Weinman E J, Leung G, Cheong A, Louvard D, Donowitz M. Na+/H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J Physiol. 2001;537:537–552. doi: 10.1111/j.1469-7793.2001.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Leu S, Cheong A, Zhang H, Baibakov B, Shih C, Birnbaum MJ, Donowitz M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: role in absorption and differentiation. Gastroenterology. 2004;126:122–135. doi: 10.1053/j.gastro.2003.10.061. [DOI] [PubMed] [Google Scholar]
- Lin R, Murtazina R, Cha B, Chakraborty M, Sarker R, Chen TE, Lin Z, Hogema BM, de Jonge HR, Seidler U, et al. D-glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2-dependent process. Gastroenterology. 2011;140:560–571. doi: 10.1053/j.gastro.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Yeruva S, He P, Singh AK, Zhang H, Chen M, Lamprecht G, de Jonge HR, Tse M, Donowitz M, et al. Lysophosphatidic acid stimulates the intestinal brush border Na(+)/H(+) exchanger 3 and fluid absorption via LPA(5) and NHERF2. Gastroenterology. 2010;138:649–658. doi: 10.1053/j.gastro.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe OW. Acute regulation of proximal tubule apical membrane Na/H exchanger NHE-3, role of phosphorylation, protein trafficking, and regulatory factors. J Am Soc Nephrol. 1999;10:2412–2425. doi: 10.1681/ASN.V10112412. [DOI] [PubMed] [Google Scholar]
- Mohan S, Tse CM, Gabelli SB, Sarker R, Cha B, Fahie K, Nadella M, Zachos NC, Tu-Sekine B, Raben D, et al. NHE3 activity is dependent on direct phosphoinositide binding at the N terminus of its intracellular cytosolic region. J Biol Chem. 2010;285:34566–34578. doi: 10.1074/jbc.M110.165712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtazina R, Kovbasnjuk O, Chen TE, Zachos NC, Chen Y, Kocinsky HS, Hogema BM, Seidler U, de Jonge HR, Donowitz M. NHERF2 is necessary for basal activity, second messenger inhibition, and LPA stimulation of NHE3in mouse distal ileum. Am J Physiol Cell Physiol. 2011;301:C126–C136. doi: 10.1152/ajpcell.00311.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtazina R, Kovbasnjuk O, Donowit M, Li X. Na+/H+ exchanger NHE3 activity and trafficking are lipid raft-dependent. J Biol Chem. 2006;281:17845–17855. doi: 10.1074/jbc.M601740200. [DOI] [PubMed] [Google Scholar]
- Nath SK, Hang CY, Levine SA, Yun CH, Montrose MH, Donowitz M, Tse CM. Hyperosmolarity inhibits the Na+/H+ exchanger isoforms NHE2 and NHE3: an effect opposite to that on NHE1. Am J Physiol. 1996;270:G431–G441. doi: 10.1152/ajpgi.1996.270.3.G431. [DOI] [PubMed] [Google Scholar]
- No YR, He P, Yoo BK, Yun CC. Regulation of NHE3 by lysophosphatidic acid is mediated by phosphorylation of NHE3 by RSK2. Am J Physiol Cell Physiol. 2015;309:C14–C21. doi: 10.1152/ajpcell.00067.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørholm AB, Hendus-Altenburger R, Bjerre G, Kjaergaard M, Pedersen SF, Kragelund BB. The intracellular distal tail of the Na+/H+ exchanger NHE1 is intrinsically disordered: implications for NHE1 trafficking. Biochemistry. 2011;50:3469–3480. doi: 10.1021/bi1019989. [DOI] [PubMed] [Google Scholar]
- Pang T, Su X, Wakabayashi S, Shigekawa M. Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J Biol Chem. 2001;276:17367–17372. doi: 10.1074/jbc.M100296200. [DOI] [PubMed] [Google Scholar]
- Priyamvada S, Gomes R, Gill RK, Saksena S, Alrefai WA, Dudeja PK. Mechanisms underlying dysregulation of electrolyte absorption in inflammatory bowel disease-associated diarrhea. Inflamm Bowel Dis. 2015;21:2926–2935. doi: 10.1097/MIB.0000000000000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker R, Grønborg M, Cha B, Mohan S, Chen Y, Pandey A, Litchfield D, Donowitz M, Li X. Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol Biol Cell. 2008;19:3859–3870. doi: 10.1091/mbc.E08-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker R, Valkhoff VE, Zachos NC, Lin R, Cha B, Chen TE, Guggino S, Zizak M, de Jonge H, Hogema B, Donowitz M. NHERF1 and NHERF2 are necessary for multiple but usually separate aspects of basal and acute regulation of NHE3 activity. Am J Physiol Cell Physiol. 2011;300:C771–C782. doi: 10.1152/ajpcell.00119.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Lin R, Yang J, Cha B, Sarker R, Tse CM, Donowitz M. AKT and GSK-3 are necessary for direct ezrin binding to NHE3 as part of a C-terminal stimulatory complex: role of a novel Ser-rich NHE3 C-terminal motif in NHE3 activity and trafficking. J Biol Chem. 2014;289:5449–5461. doi: 10.1074/jbc.M113.521336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Yang J, Cha B, Chen T, Sarker R, Yin J, Avula LR, Tse M, Donowitz M. Sorting nexin 27 regulates basal and stimulated brush border trafficking of NHE3. Mol Biol Cell. 2015;26:2030–2043. doi: 10.1091/mbc.E14-12-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan A, Luo M, Yu Q, Riederer B, Xia W, Chen M, Lissner S, Gessner JE, Donowitz M, Yun CC, et al. Differential association of the Na+/H+ exchanger regulatory factor (NHERF) family of adaptor proteins with the raft- and the non-raft brush border membrane fractions of NHE3. Cell Physiol Biochem. 2013;32:1386–1402. doi: 10.1159/000356577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Sun H, Lang F, Yun CC. Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am J Physiol Cell Physiol. 2005;289:C802–C810. doi: 10.1152/ajpcell.00597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederkehr MR, Zhao H, Moe OW. Acute regulation of Na/H exchanger NHE3 activity by protein kinase C: role of NHE3 phosphorylation. Am J Physiol. 1999;276:C1205–C1217. doi: 10.1152/ajpcell.1999.276.5.C1205. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Yang J, Singh V, Cha B, Chen TE, Sarker R, Murtazina R, Jin S, Zachos NC, Patterson GH, Tse CM, et al. NHERF2 protein mobility rate is determined by a unique C-terminal domain that is also necessary for its regulation of NHE3 protein in OK cells. J Biol Chem. 2013;288:16960–16974. doi: 10.1074/jbc.M113.470799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Singh V, Chen TE, Sarker R, Xiong L, Cha B, Jin S, Li X, Tse CM, Zachos NC, Donowitz M. NHERF2/NHERF3 protein heterodimerization and macrocomplex formation are required for the inhibition of NHE3 activity by carbachol. J Biol Chem. 2014;289:20039–20053. doi: 10.1074/jbc.M114.562413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CC. Concerted roles of SGK1 and the Na+/H+ exchanger regulatory factor 2 (NHERF2) in regulation of NHE3. Cell Physiol Biochem. 2003;13:29–40. doi: 10.1159/000070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Alamelumangpuram B, Lee LJ, Wang P, Kovbasnjuk O. Carbachol-mediated endocytosis of NHE3 involves a clathrin-independent mechanism requiring lipid rafts and Cdc42. Cell Physiol Biochem. 2014;33:869–881. doi: 10.1159/000358659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Kovbasnjuk O, Donowitz M. Regulation of intestinal electroneutral sodium absorption and the brush border Na+/H+ exchanger by intracellular calcium. Ann NY Acad Sci. 2009;1165:240–248. doi: 10.1111/j.1749-6632.2009.04055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- Zaun HC, Shrier A, Orlowski A. Calcineurin B homologous protein 3 promotes the biosynthetic maturation, cell surface stability, and optimal transport of the Na+/H+ exchanger NHE1 isoform. J Biol Chem. 2008;218:12456–12467. doi: 10.1074/jbc.M800267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Cha B, Zachos NC, Sarker R, Chakraborty M, Chen TE, Kovbasnjuk O, Donowitz M. Elevated calcium acutely regulates dynamic interactions of NHERF2 and NHE3 proteins in opossum kidney (OK) cell microvilli. J Biol Chem. 2011;286:34486–34496. doi: 10.1074/jbc.M111.230219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizak M, Chen T, Bartonicek D, Sarker R, Zachos NC, Cha B, Kovbasnjuk O, Korac J, Mohan S, Cole R, et al. Calmodulin kinase II constitutively binds, phosphorylates, and inhibits brush border Na+/H+ exchanger 3 (NHE3) by a NHERF2 protein-dependent process. J Biol Chem. 2012;287:13442–13456. doi: 10.1074/jbc.M111.307256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.