

The small GTPase RhoG and its exchange factor, Trio, regulate the formation and size of circular dorsal ruffles and associated functions, including macropinocytosis and cell migration.
Abstract
Circular dorsal ruffles (CDRs) are actin-rich structures that form on the dorsal surface of many mammalian cells in response to growth factor stimulation. CDRs represent a unique type of structure that forms transiently and only once upon stimulation. The formation of CDRs involves a drastic rearrangement of the cytoskeleton, which is regulated by the Rho family of GTPases. So far, only Rac1 has been consistently associated with CDR formation, whereas the role of other GTPases in this process is either lacking or inconclusive. Here we show that RhoG and its exchange factor, Trio, play a role in the regulation of CDR dynamics, particularly by modulating their size. RhoG is activated by Trio downstream of PDGF in a PI3K- and Src-dependent manner. Silencing RhoG expression decreases the number of cells that form CDRs, as well as the area of the CDRs. The regulation of CDR area by RhoG is independent of Rac1 function. In addition, our results show the RhoG plays a role in the cellular functions associated with CDR formation, including macropinocytosis, receptor internalization, and cell migration. Taken together, our results reveal a novel role for RhoG in the regulation of CDRs and the cellular processes associated with their formation.
INTRODUCTION
In many cells types, such as epithelial cells, fibroblasts, and smooth muscle cells, stimulation by growth factors promotes the formation of a unique type of structure called the circular dorsal ruffle (CDR; Buccione et al., 2004). CDRs (also called waves or ring ruffles) are F-actin– and cortactin-rich membrane protrusions that form transiently at the dorsal surface of the cell in response to growth factor stimulation (Mellstrom et al., 1988). CDRs form very rapidly and only once after growth factor treatment (Krueger et al., 2003), and one of their most notable features is their circularity (Mellstrom et al., 1988). CDRs appear at the dorsal surface, where they expand and then constrict toward their center in a coordinated manner to maintain a circular appearance, until they finally close, typically 5–30 min after stimulation (Krueger et al., 2003). They are different from the widely characterized ruffles forming at the cell edge, sometimes referred to as peripheral ruffles (PRs). PRs are usually linear structures that persist and undergo cycles of assembly and disassembly (Buccione et al., 2004). They also extend dorsally but are located at the periphery of the cell, typically at the leading edge (Abercrombie et al., 1970). Despite their differences, CDRs share many structural components with PRs, including actin, dynamin2, cortactin, N-WASP, and Arp2/3, among others (Krueger et al., 2003).
The function of CDRs has not been conclusively established. Previous work has shown that CDRs may be involved in cell migration, when cells switch from a static to a migratory phenotype (Krueger et al., 2003; Sero et al., 2011). Other reports associate the formation of CDRs with macropinocytosis and internalization of membrane receptors such as receptor tyrosine kinases (RTKs) and integrins (Dowrick et al., 1993; Dharmawardhane et al., 2000; Orth et al., 2006; Gu et al., 2011).
The signaling events downstream of the platelet-derived growth factor (PDGF) receptor that drive CDR formation require the function of phosphoinositide 3-kinase (PI3K; Wymann and Arcaro, 1994; Hooshmand-Rad et al., 1997) and the small GTPase Rac (Hooshmand-Rad et al., 1997; Goicoechea et al., 2006; Vidali et al., 2006). Rac1-/- cells are unable to form CDRs, suggesting that Rac1 is essential for their formation (Vidali et al., 2006). However, overexpression of constitutively active Rac1 induced PRs but no CDRs (Lanzetti et al., 2004), suggesting other signaling pathways working in parallel with Rac1 are required for CDR formation.
Although many of the components of the CDRs have been identified, the molecular mechanisms controlling their formation and disappearance have not been fully characterized. We hypothesized that RhoG, a Rac1-related small GTPase, was involved in CDR formation because RhoG has been associated with the formation of actin-rich ruffles at the dorsal surface of the cell (Gauthier-Rouviere et al., 1998; Katoh et al., 2000; Ellerbroek et al., 2004; van Buul et al., 2007; Yamaki et al., 2007). These RhoG-mediated dorsal ruffles typically form as intersecting, reticulated linear ruffles and are phenotypically different than CDRs (Blangy et al., 2000; Ellerbroek et al., 2004). However, they share many molecular components with CDRs. In addition, RhoG activity has been shown to associate with processes such as macropinocytosis, phagocytosis, bacterial internalization, and leukocyte transmigration, all of which share similarities with CDR formation (Wennerberg et al., 2002; deBakker et al., 2004; Ellerbroek et al., 2004; Patel and Galan, 2006; van Buul et al., 2007). RhoG has also been shown to play roles during cell migration, neurite outgrowth, cell survival, and changes in gene expression, suggesting that its cellular functions are complex (Katoh et al., 2000, 2006; Estrach et al., 2002; May et al., 2002; Murga et al., 2002; Vigorito et al., 2003; Yamaki et al., 2007; Meller et al., 2008).
Here we describe the role of RhoG and its guanine nucleotide exchange factor (GEF), Trio, in the regulation of CDR formation. We show that, in vascular smooth muscle cells, RhoG and Trio control the size of CDRs in a Rac1-independent manner and that perturbing their expression levels and/or activity also affects micropinocytosis, receptor internalization, and cell migration.
RESULTS
RhoG expression modulates the formation of CDRs
In smooth muscle cells, PDGF treatment induces the formation of CDRs (Goicoechea et al., 2006; Huynh et al., 2013). Because RhoG was previously associated with peripheral dorsal ruffles and other actin-rich dorsal structures, we hypothesized that RhoG may play a role during CDR formation. To test our hypothesis, we examined CDR formation in rat aortic smooth muscle cells (A7r5) transfected with either control or RhoG-specific small interfering RNA (siRNA; RhoG knockdown [KD]). Initially, we looked at the morphology of the CDRs in a time course after PDGF treatment. Our results show that in both control and RhoG KD cells, CDRs form with similar kinetics, appearing as early as 1 min, peaking at ∼2.5 min, and starting to decrease at 5 min (Figure 1, A and C, and Supplemental Movies S1 and S2). By 15 min, most CDRs had disappeared, and no CDRs were detected at later time points. Even though RhoG knockdown was very efficient (Figure 1B), silencing RhoG expression did not inhibit the rate of CDR formation, as we had hypothesized. However, at both 2.5 and 5 min after PDGF addition, the percentage of cells displaying CDRs was significantly lower in the absence of RhoG (Figure 1C).
FIGURE 1:

PDGF-induced CDR formation is affected by RhoG silencing. (A) A7r5 cells were transfected with siRNA against RhoG (RhoG KD) or with a nontargeting siRNA (CTRL). After 72 h, cells were serum starved for 2 h and stimulated with PDGF-BB (20 ng/ml) for 1, 2.5, and 5 min. Cells were fixed and processed for immunofluorescence using cortactin (red) as marker of dorsal ruffles, Alexa 488–phalloidin (green) to stain F-actin, and Hoechst (blue) to visualize nuclei. Representative images of the 2.5-min time point. Scale bar, 20 μm. (B) KD efficiency was tested for each experiment by immunoblot in cell lysates 72 h after transfection. (C) Number of cells with CDRs expressed as percentage of cells that stained positive for at least one CDR. (D) Average number of CDRs per cell (in cells positive for CDRs). (E) Average CDR area. Black bars, CTRL cells; gray bars, RhoG KD. Results are shown as mean ± SEM from three independent experiments (≥100 cells per condition/experiment).
Typically, most PDGF-treated cells form between one and five CDRs, with an average of approximately two per cell. In RhoG KD cells, the average number of CDRs per cell was not significantly different than in CTRL cells (Figure 1D). The most striking effect of silencing RhoG expression related to the area of the CDRs (Figure 1, A and E). In the absence of RhoG, the average area of the CDRs decreased almost twofold. At 2.5 min after PDGF treatment, the average CDR area was 951.22 ± 90.49 μm2 in CTRL cells compared with 502.27 ± 30.50 μm2 in RhoG KD cells (Figure 1E). This decrease is observed at every time point tested, ruling out the possibility that the CDRs form early or are in the process of disassembling at the time of the measurement (Figure 1E). To determine whether these results were specific for smooth muscle cells, we silenced RhoG in a human fibroblast cell line, MRC5, and tested their ability to form CDRs in response to PDGF (Supplemental Figure S1). Our results show that silencing RhoG (Supplemental Figure S1A) has a similar effect in fibroblasts as in smooth muscle cells, with significantly fewer cells forming CDRs (Supplemental Figure S1C), which were smaller in area (Supplemental Figure S1, B and D). Overall our results suggest that RhoG plays a role in both the formation of CDRs and the regulation of their size.
Dynamics of CDR formation in live cells
Time-lapse microscopy analysis corroborated the results from fixed cells, with the maximum area of CTRL CDRs being significantly larger than that of CDRs in Rho KD cells (Figure 2, A and B). Quantitative analysis shows that, in most cases, CDRs appear at or close to their maximum size and start closing right away (Figure 2A and Supplemental Movies S1 and S2). Occasionally, a small increase in area is observed before closing starts, but with our live-imaging setup, we rarely observe CDR expansion. Our results show that in RhoG KD cells, the lifetime of the CDRs is significantly shorter (Figure 2C), which means that they close significantly faster than CTRL CDRs, although with similar kinetics (Figure 2D). These results suggest that the speed at which they close may be comparable in terms of area/time. Using the area for each ruffle at each time point, we calculated the rate of closing in CTRL and RhoG KD cells and found that the closing rates are almost identical in both conditions, suggesting that the faster closing is the result of a smaller starting area, which closes at the same rate (Figure 2E). This size reduction was consistent throughout the lifetime of the CDRs, ruling out the possibility that the time course of CDR formation is affected in RhoG KD cells (e.g., CDRs formed earlier/later or disassembled faster). Taken together, these results suggest that RhoG controls the maximum CDR size without affecting their closing speed.
FIGURE 2:

Dynamics of CDR formation in live cells. A7r5 cells transfected with siRNA targeting RhoG (RhoG KD) or with a nontargeting siRNA (CTRL) were analyzed by time-lapse microscopy immediately after the addition of 20 ng/ml PDFG-BB. Pictures were taken at intervals of 35 s, and CDR properties were evaluated using ImageJ. (A) A representative series for CTRL and RhoG KD after formation and disassembly of CDRs. Scale bar, 20 μm. (B) Average maximal area that each CDR reached during the total time recorded. (C) Average CDR lifetime. (D) Maximum area of each CDR was standardized to 1, and CDR disassembly was then plotted vs. time. Complete disassembly is achieved significantly faster in average CTRL cells than with RhoG KD (*p < 0.0001) but with similar kinetics. Results for A–C are expressed as mean ± SEM from two independent experiments (a combined total of 72 cells were analyzed in CTRL, and 82 cells were analyzed in RhoG KD). (E) For each CDR, the disassembly rate was calculated from the slope of a linear regression calculated for each CDR disassembly event. The difference between these two sets of data is not significant.
PDGF induces RhoG activation
The activation of the small GTPases RhoA, Rac1, and Cdc42 in response to PDGF was described by Gabunia et al. (2011) (RhoA), Buchanan et al. (2000) and Ryu et al. (2002) (Rac1), and Jimenez et al. (2000) (Cdc42). In contrast, the activation of RhoG in response to PDGF has not been tested. However, RhoG has been shown to respond to other growth factors such as epidermal growth factor (Samson et al., 2010). Our results show that RhoG is rapidly activated after PDGF stimulation in A7r5 cells. Activation is detected as early as 1 min and returns to basal level after 5 min (Figure 3, A and B). We obtained similar results in human fibroblasts (MRC5; Supplemental Figure S1E). Of interest, CDR formation and RhoG activity follow a similar pattern in response to PDGF (Figures 1C and 3, A and B), with RhoG activity peaking slightly earlier than the CDRs. This suggests that RhoG activity may be implicated during CDR formation. To corroborate that RhoG activation depends on the kinase activity of PDGF receptor (PDGFR), we pretreated the cells with the PDGFR inhibitor AG1295. Our results show that AG1295 impairs PDGF-mediated RhoG activation, suggesting that it depends on PDGFR kinase activity (Figure 3, C and D).
FIGURE 3:
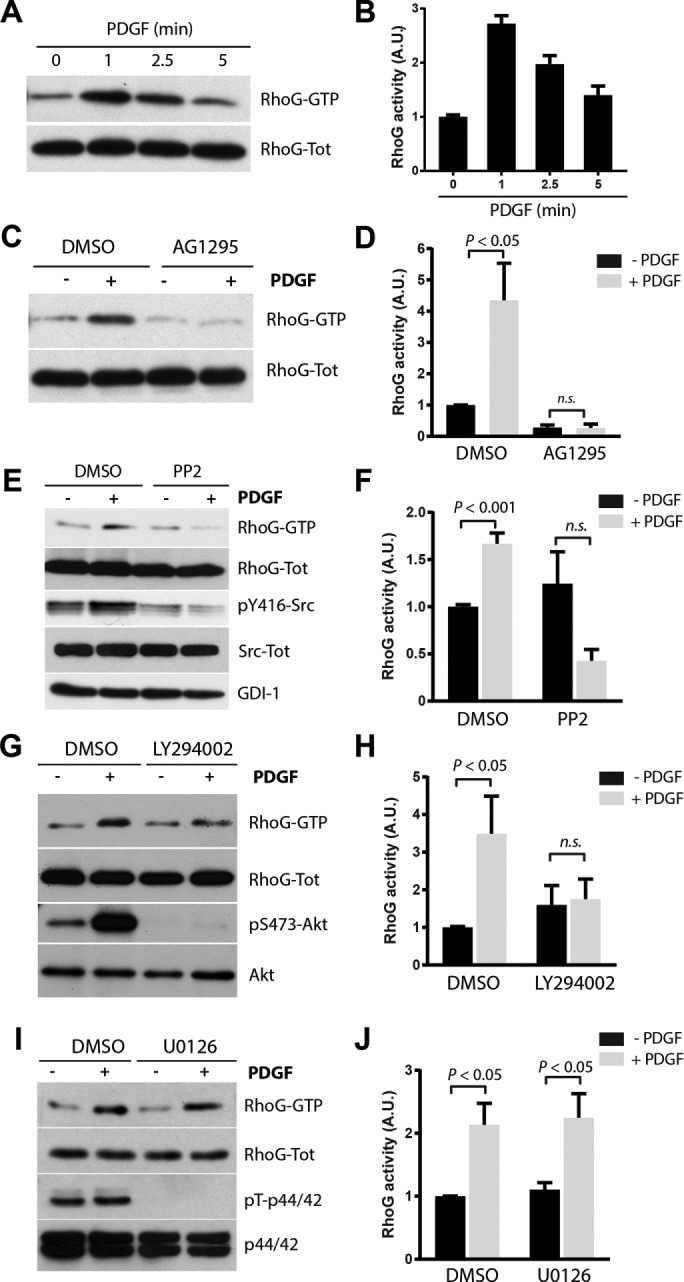
PDGF induces RhoG activation. (A) A7r5 cells were serum starved for 2 h and then stimulated with PDGF-BB (20 ng/ml) for 1, 2.5, and 5 min. Active RhoG (RhoG-GTP) was then precipitated from total lysates using GST-ELMO and immunoblotted with RhoG antibodies. (B) Quantification of three independent assays. (C) Cells pretreated with 10 μM AG1295 to inhibit PDGFR before PDGF-BB stimulation. (D) Quantification of three independent assays. Serum-starved cells were pretreated with 10 μM PP2 (E), 25 μM LY294002 (G), or 10 μM U0126 (I) for 30 min and then stimulated with PDGF for 1 min. Active RhoG (RhoG-GTP) was then measured as in A. Phospho–Tyr-416 Src, phospho–Ser-473 Akt, and phospho–Thr-202/Tyr-204 p44/42 antibodies were used as control to determine the efficiency of the inhibitors. Total Src, Akt, p44/42 and RhoGDI were used as loading controls (F, H, and J). Quantification of at least three independent assays for PP2, LY294002, and U0126, respectively. For all experiments, RhoG activity is calculated as the ratio of active (RhoG-GTP)/total RhoG signal (RhoG-Tot) and expressed in arbitrary units (A.U.). All results are shown as mean ± SEM of at least three independent experiments.
Src and PI3K control RhoG activation downstream from PDGFR
RhoG activation by PDGF has not been described previously, and so we sought to characterize the signaling pathways involved in its activation. To this end, we tested whether inhibition of known PDGF-activated signaling pathways affected the rapid activation of RhoG. We measured the RhoG activation in response to PDGF levels in the presence or absence of the following inhibitors: Src (PP2 and SU6656), PI3K (LY294002), and p44/42 mitogen-activated protein kinase (MAPK; U0126). Our results show that inhibiting both Src and PI3K abolished the PDGF-mediated activation of RhoG, whereas inhibition of p44/42 MAPK had no effect (Figure 3, E–J, and Supplemental Figure S2). These results suggest that RhoG activation downstream of PDGF depends on the activity of Src and PI3K but not on p44/42 MAPK.
Because both Src and PI3K appear to function upstream of RhoG after PDGF stimulation, we also analyzed their role in CDR formation. Both Src and PI3K play key roles during formation of CDRs in different cells lines (Wymann and Arcaro, 1994; Scaife et al., 2003; Veracini et al., 2006). Our results confirm these reports and show that when either Src or PI3K is inhibited, formation of CDRs is dramatically reduced (Figure 4, A and B). In the case of Src inhibition, formation of CDRs was completely abolished, whereas on PI3K inhibition, some CDRs were still observed. The few CDRs that formed were significantly smaller than those in CTRL cells (Figure 4C).
FIGURE 4:

Effect of Src and PI3K inhibition on CDR formation. (A) A7r5 cells were serum starved for 2 h and then treated with 10 μM PP2 or 25 μM LY294002 for 30 min, followed by a 2.5-min treatment with PDGF-BB (20 ng/ml). Cells were then fixed and processed for immunofluorescence using cortactin (green) as marker of dorsal ruffles, Alexa 594–phalloidin (red) and Hoechst (blue). Scale bar, 20 μm. (B) Number of cells with CDRs expressed as percentage of cells that stained positive for at least one CDR. Results are shown as mean ± SEM from three independent experiments (≥200 cells per condition/experiment). (C) Average CDR area (shown only for CTRL and LY294002 treatment because PP2-treated cells did not form CDRs). Results are shown as mean ± SEM from three independent experiments (≥200 CDRs).
Role of Rac1 and Cdc42 in CDR formation
Rac1 activity is required for CDR formation (Lanzetti et al., 2004; Vidali et al., 2006). There is also indirect evidence that Cdc42 plays a role in CDR formation (Machuy et al., 2007; Cortesio et al., 2010; King et al., 2011). Because RhoG can act upstream of both Rac1 and Cdc42 (Gauthier-Rouviere et al., 1998; Katoh and Negishi, 2003; Franke et al., 2012), we investigated whether RhoG was acting through these GTPases during CDR formation. If RhoG’s function during CDR formation depends on the downstream activation of Rac and/or Cdc42, then silencing or overexpressing Rac1 or Cdc42 should recapitulate the effects of RhoG KD or overexpression, respectively.
We compared the effects of silencing or overexpressing Rac1 or Cdc42 on CDR formation to those observed for RhoG KD or overexpression (Figure 5 and Supplemental Figure S3). As expected based on previous reports (Lanzetti et al., 2004; Vidali et al., 2006), our results show that Rac1 KD completely abolished CDR formation (Figure 5, A and C). In contrast, the effects of Cdc42 KD were similar to those of RhoG, showing partial inhibition of CDR formation (Figure 5, A and C). Similar to what we observed with RhoG KD, silencing Cdc42 significantly decreased the area of CDRs (Figure 5D) without affecting the number of CDRs per cell (Figure 5E). This decrease in area was comparable to that observed in RhoG KD cells (Figure 5D). In Rac1 KD cells, only a small subset of cells formed ruffles (only 31 CDRs formed in >2000 cells analyzed). Thus, even though the area of CDRs is smaller in Rac1 siRNA cells, the results may represent cells with incomplete KD. Of interest, simultaneous KD of RhoG and Cdc42 had an additive effect, with almost complete inhibition of CDR formation (Figure 5C). However, the effect of the double KD on CDR area was not additive because the CDRs that were still able to form in the double KD (Cdc42/RhoG KD) cells were similar in size to those in the single KDs (RhoG KD or Cdc42 KD; Figure 5D).
FIGURE 5:

Role of Rac1 and Cdc42 in CDR formation. (A) A7r5 cells were transfected with siRNA targeting RhoG (RhoG KD), Cdc42 (Cdc42 KD), Rac1 (Rac KD), or Cdc42 and RhoG combined (Cdc42/RhoG KD). Control cells were transfected with a nontargeting siRNA (siCTRL). (B) Cells were infected with a myc-tagged adenovirus control (CTRL) or with adenovirus encoding wild-type mycRhoG, mycCdc42, or mycRac or their constitutively active forms (mycRhoG Q61L, mycCdc42 Q61L, or mycRac Q61L). After 72 h, the cells were serum starved for 2 h and stimulated with PDGF-BB (20 ng/ml) for 2.5 min. In A, cells were then fixed and processed for immunofluorescence using cortactin (green) as marker of dorsal ruffles, Alexa 594–phalloidin (red), and Hoechst (blue), and in B, with cortactin (red), myc (green), and Hoechst (blue). Scale bar, 20 μM. (C–E) The experiments shown in A and B were quantified as follows: (C) percentage of cells with CDRs, (D) average CDR area, and (E) average number of CDR per cell (in cells positive for CDRs). Results are mean ± SEM from three independent experiments (≥100 cells per condition/experiment).
With the exception of RhoG, the effect of overexpressing the Rho GTPases was not as pronounced as that of silencing them (Figure 5, C–E). Supporting the results from the KD experiments, RhoG overexpression promoted a significant increase in the area of CDRs (Figure 5D). Overexpression of a constitutively active form of RhoG (Q61L) showed an even greater increase in CDR area. RhoG overexpression also promoted a decrease in the percentage of cells showing CDRs (Figure 5C), again more pronounced when the RhoG Q61L was expressed. These results suggest that too much or too little RhoG can affect CDR formation and that its activity needs to be tightly regulated. In contrast, overexpression of Cdc42, Rac1, or their constitutively active forms (Rac1 Q61L and Cdc42 Q61L) had no effect on the percentage of cells showing CDRs or the area of CDRs (Figure 5, C and D). The combined expression of RhoG and Cdc42 showed an increase in CDR size similar in magnitude to that observed when RhoG alone was overexpressed, and in contrast to the double KD, no additive effect was observed. Finally, overexpression of RhoG, Rac1, or Cdc42 (wild type or Q61L) or RhoG/Cdc42 combined had no effect on number of CDRs per cell (Figure 5E).
Taken together, these results suggest that RhoG functions independently of Rac and in parallel with Cdc42 to regulate pathways that function to control the formation of CDRs, as well as of their size.
Effects of RhoG KD on PDGF-induced Rac1 and Cdc42 activation
It was previously shown that Rac can be activated downstream of RhoG through the RhoG effector ELMO, which forms a complex and activates the Rac-GEF Dock180 (Katoh and Negishi, 2003). In A7r5 cells, Rac1 is robustly activated after PDGF treatment as early as 1 min after treatment (Figure 6A), as previously described in other cell lines (Monypenny et al., 2009). To test whether RhoG functions upstream or independently of Rac1, as suggested by the results from Figure 5, we analyzed the effect of silencing RhoG on Rac1 activation in response to PDGF. In the absence of RhoG, Rac1 was efficiently activated by PDGF, suggesting that they are activated independently (Figure 6, B and C). Even though the quantification shows that the absolute amount of active Rac1 is lower in RhoG KD cells (both with and without PDGF treatment), the relative increase of Rac1 activity between CTRL and PDGF-treated cells (the fold increase in nontreated vs. PDGF) was not significantly affected by the absence of RhoG (Figure 6, B and C). These differences can be attributed to the decrease in Rac1 basal activity observed in nontreated cells after RhoG silencing (Figure 6, B and C, lane 1 vs. lane 3). This was previously shown by others and is consistent with the fact that a fraction of Rac1 is activated by RhoG through ELMO/Dock180 (Katoh and Negishi, 2003; Monypenny et al., 2009; Samson et al., 2010). Our results suggest that in these cells, a small fraction of the basal activity of Rac1 depends on RhoG. However, Rac1 activation in response to PDGF is independent of RhoG. We observed a similar response when we analyzed Cdc42 activity. Cdc42 is activated robustly after PDGF treatment in control cells (Supplemental Figure S4A), confirming previous results in other cell lines (Jimenez et al., 2000), and it responds efficiently to PDGF when RhoG is knocked down, suggesting that it also functions independently of RhoG (Supplemental Figure S4B).
FIGURE 6:
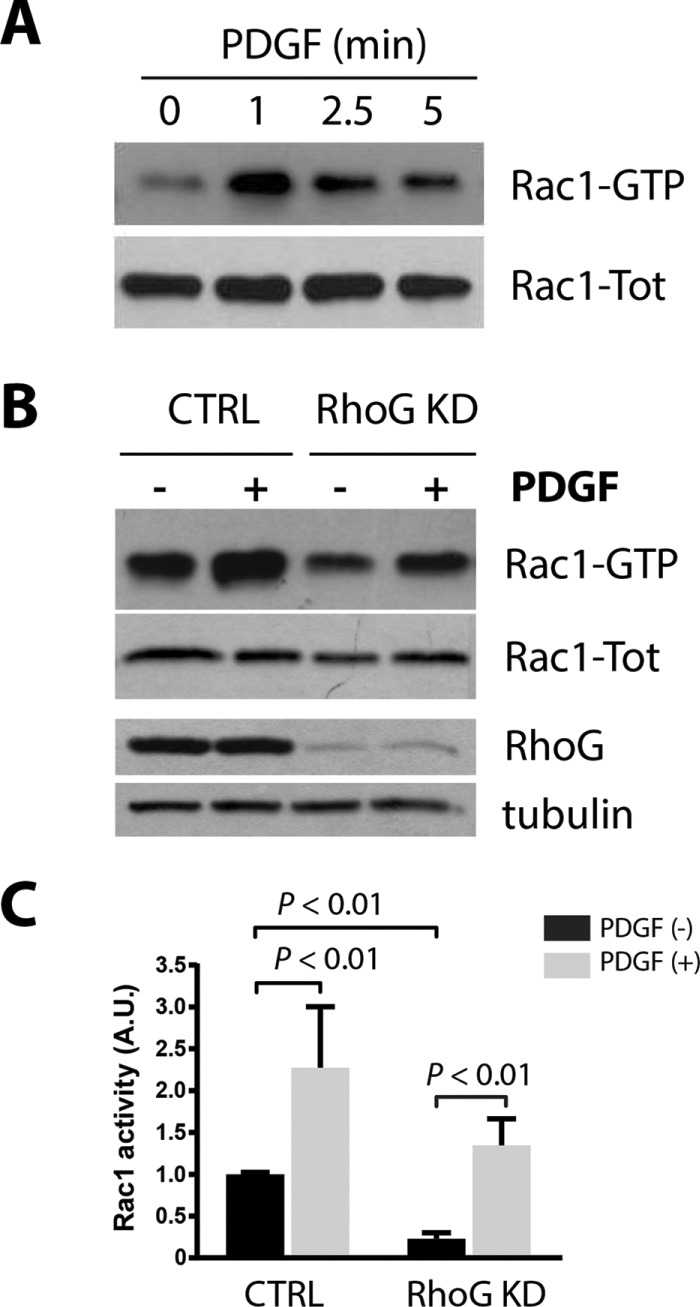
RhoG silencing effect on Rac1 activation. (A) A7r5 cells were serum starved for 2 h and then stimulated with PDGF-BB (20 ng/ml) for 1, 2.5, and 5 min. Active Rac1 (Rac1-GTP) was then precipitated from total lysates using GST-PBD and immunoblotted with Rac1 antibodies. (B) A7r5 cells were transfected with siRNA targeting RhoG (RhoG KD) or with a nontargeting siRNA (CTRL). After 72 h, cells were serum starved for 2 h and then treated with PDGF-BB (20 ng/ml) for 1 min. Active Rac1 (Rac1-GTP) was precipitated from total lysates using GST-PBD and immunoblotted with the indicated antibodies. Tubulin was used as loading control. (C) Quantification of three independent assays. Rac-GTP is calculated as a ratio of active (Rac1-GTP)/total Rac1 (Rac1-Tot) signal and expressed in arbitrary units (A.U.).
Trio regulates PDGF-mediated RhoG activation
Trio is a RhoGEF that possesses two separate GEF domains, GEFD1 and GEFD2, which control the activity of Rac/RhoG and RhoA, respectively (Debant et al., 1996; Blangy et al., 2000). It was previously shown that Trio regulates RhoG activity in the formation of actin-rich dorsal structures (Blangy et al., 2000; van Rijssel et al., 2012b). Trio also colocalizes with the actin- and ICAM-rich rings that form during leukocyte transendothelial migration (van Rijssel et al., 2012b). To determine whether Trio is also involved in the regulation of RhoG during CDR formation, we first analyzed its localization in PDGF-treated A7r5 cells by immunofluorescence microscopy. Our results show that Trio colocalizes with F-actin at CDRs, suggesting that it may play a role in regulating RhoG during CDR formation (Figure 7A). Silencing Trio expression using siRNA (Figure 7, B and C) or inhibiting Trio using the chemical inhibitor ITX3 (Bouquier et al., 2009; Figure 7, D and E) significantly decreased the activation of RhoG in response to PDGF. Both the basal levels of RhoG activation and its response to PDGF were reduced by ITX3. Even though Trio can act as both RhoG and Rac exchange factor, the reduction in Rac1 activity was not as pronounced as with RhoG, both at the basal level and in its response to PDGF (Figure 7, F and G). Inhibition of Trio with ITX3 also caused a significant decrease in both the number of cells that form CDRs and the average CDR area without significantly affecting the number of CDRs per cell (Figure 7, H–J). These results are almost identical to those observed in RhoG KD cells (Figure 1) and suggest that activation of RhoG in response to PDGF is mediated by Trio during PDGF-mediated CDR formation, whereas activation of Rac1 may be mediated by a different GEF.
FIGURE 7:

Trio regulates RhoG activation and CDR formation. (A) A7r5 cells were serum starved for 2 h and stimulated with PDGF-BB (20 ng/ml) for 2.5 min. Cells were then fixed and processed for immunofluorescence using Trio antibody (red), Alexa 488–phalloidin (green), and Hoechst (blue) to stain the nucleus. Scale bar, 20 μm. (B) A7r5 cells transfected with a siRNA targeting Trio (Trio KD) or nontargeting siRNA (CTRL) were serum starved for 2 h and then stimulated with PDGF-BB (20 ng/ml) for 1 min. Active RhoG (RhoG-GTP) was precipitated from total lysates using GST-ELMO and immunoblotted for RhoG. (C) Quantification of at least three independent assays. RhoG-GTP is calculated as the ratio of active/total RhoG signal and expressed in arbitrary units (A.U.). (D) Serum-starved A7r5 cells were preincubated with the Trio inhibitor ITX3 (100 μM) or with DMSO (CTRL) and then stimulated with PDGF-BB (20 ng/ml) for 1 min. Active RhoG (RhoG-GTP) was precipitated from total lysates using GST-ELMO and immunoblotted with the indicated antibodies. (E) Quantification of at least three independent assays. RhoG-GTP is calculated as the ratio of active/total RhoG signal and expressed in arbitrary units (A.U.). (F) Serum-starved A7r5 cells were preincubated with the Trio inhibitor ITX3 (100 μM) or with DMSO (CTRL) and then stimulated with PDGF-BB (20 ng/ml) for 1 min. Active Rac1 (Rac-GTP) was precipitated from total lysates using GST-PBD and immunoblotted with the indicated antibodies. (G) Quantification of at least three independent assays. Active Rac1 (Rac1-GTP) is calculated as the ratio of active/total Rac1 signal and expressed in arbitrary units (A.U.). (H) A7r5 cells were preincubated with ITX3 (100 μM) or DMSO (CTRL) and stimulated with PDGF-BB (20 ng/ml) for 2.5 min. Cells were fixed and processed for immunofluorescence using cortactin (green) as marker of dorsal ruffles, Alexa 594–phalloidin (red), and Hoechst (blue). Scale bar, 20 μm. Quantification of (I) percentage of cells with CDRs, (J) average area of each CDR, and (K) number of CDRs per cell (in cells positive for CDRs). Black bars, CTRL; gray bars, ITX3. Results are mean ± SEM from three independent experiments (≥100 cells per condition/experiment).
Effects of RhoG and Trio silencing during macropinocytosis and receptor internalization
What is the functional consequence of having smaller CDRs? One possibility is that smaller CDRs could be associated with dysfunctional endocytic process. Hasegawa et al. (2012) showed that silencing ARAP1, which reduces CDR area, inhibits dextran uptake through macropinocytosis. To determine whether RhoG plays a role during macropinocytosis, we analyzed the uptake of fluorescent-labeled dextran in A7r5 cells transfected with siRNA targeting RhoG. Our results show that PDGF treatment for 30 min stimulated dextran uptake (Figure 8, A and B). However, when RhoG expression was silenced, PDGF-mediated stimulation of dextran uptake was reduced to levels comparable to that in nontreated cells. Reexpression of mycRhoG (siRNA resistant) in RhoG KD cells restored levels of dextran uptake to control levels (Figure 8, A and B). A similar reduction in dextran uptake was observed when Trio expression was silenced (Figure 8, C and D). We were able to rescue the dextran uptake by reexpressing Trio-D1 green fluorescent protein (GFP; encoding the catalytic domain that activates Rac/RhoG; van Rijssel et al., 2012a).
FIGURE 8:

RhoG regulates macropinocytosis. A7r5 cells were transfected with the indicated siRNAs. RhoG or Trio expression was then rescued with adenovirus infection of mycRhoG or TrioD1-GFP, respectively. After 2 h of serum starvation, cells were incubated with 250 μg/ml Alexa Fluor 594–conjugated dextran (10,000 molecular weight) with or without 20 ng/ml PDGF-BB for 60 min. Cells were then lysed, and dextran internalization was measured at 590/617 nm in a fluorometer. Dextran concentration in each sample was extrapolated by a dextran standard curve, normalized by the amount of protein in each condition, and expressed as nanograms of dextran/microgram of protein. Results are shown as mean ± SEM from three independent experiments. (A) Effects of RhoG KD and rescue on dextran internalization. (B) The efficiency of RhoG KD and rescue was analyzed by immunoblotting with the indicated antibodies. (C) Effects of Trio KD and rescue on dextran internalization. (D) The efficiency of Trio KD and rescue (Trio-D1-GFP) was analyzed by immunoblotting with the indicated antibodies. (E) Internalization of PDGFR was assessed by protection to trypsin digestion assay as described in Materials and Methods. Cells transfected with siRNA targeting RhoG (RhoG KD) or a nontargeting control (CTRL) were serum starved for 2 h and stimulated with 20 ng/ml PDGF-BB for the indicated times. After trypsin digestion, cell lysates were analyzed by immunoblot for the indicated antibodies. PDGFR observed in this assay corresponds to the internalized fraction of receptor (protected from trypsin). RhoG blot shows the efficiency of knockdown. RhoGDI was used as a loading control. (F) To rule out changes in total levels of PDGFR between CTRL and RhoG KD, cells were treated as in E, incubated for 30 min with PDGF, and processed for immunoblotting without trypsin treatment.
It was previously shown that PDGFR, as well as other tyrosine kinase receptors and integrins, can be internalized by macropinocytosis to modulate proliferation and cell migration (Yamazaki et al., 2002; Krueger et al., 2003; Orth et al., 2006; Kawada et al., 2009; Gu et al., 2011; Schmees et al., 2012; Zhu et al., 2014). To determine whether PDGFR internalization is affected after RhoG silencing, we performed a trypsin-protection digestion assay (Ceresa et al., 1998). In this assay, cells are treated with trypsin to digest the fraction of the proteins present at the plasma membrane. The cells are then lysed and immunoblotted to detect the fraction that was “protected” from the trypsin digestion (internalized fraction). Our results show that in control cells, the pool of PDGFR protected from trypsin digestion increased soon after PDGF stimulation (30 min), suggesting that it gets rapidly internalized in response to PDGF. A significant fraction remains in the “protected” fraction even after 6 h of stimulation (Figure 8E, CTRL). In contrast, in RhoG KD cells, the fraction of PDGFR internalized was drastically decreased, suggesting that, in the absence of RhoG, most of the PDGFR remains on the cell surface after PDGF stimulation (Figure 8E, RhoG KD). This was not due to differences in the total PDGFR levels between CTRL and RhoG KD, because we did not observe any changes before or after PDGF treatment (Figure 8F).
Role of RhoG and Trio in vascular smooth muscle cell migration
Previous work showed that CDRs might be involved in cell migration, when cells switch from a static to a migratory phenotype (Krueger et al., 2003; Sero et al., 2011). To test the role of RhoG and Trio during PDGF-mediated migration, we used the xCELLigence real-time cell analyzer (RTCA), which uses noninvasive electrical impedance to monitor cell migration in real time. As previously demonstrated for many cell types (Andrae et al., 2008), PDGF treatment also stimulates migration in A7r5 cells (Figure 9, A and B, red vs. green line). However, when RhoG expression is silenced (Figure 9, A and E, magenta line) or Trio is inhibited (Figure 9B, magenta line), migration in the presence of PDGF is significantly impaired. In the absence of PDGF, however, we do not observe significant inhibition in migration when RhoG is silenced or Trio is inhibited (Figure 9, A and B, red and blue lines). In supporting of these results, RhoG silencing inhibited wound healing in response to PDGF, suggesting that migration is impaired in the absence of RhoG activity (Figure 9, C–E).
FIGURE 9:

RhoG and Trio regulate chemotaxis toward PDGF. (A) Real-time migration was performed with an xCELLigence RTCA DP System using CIM-plate 16. RhoG KD (shRhoG) or control (CTRL) cells were seeded on the upper part of the chamber and allowed to migrate toward a lower chamber containing 20 ng/ml PDFG-BB. (B) Alternatively, cells were allowed to migrate as described in the presence of 100 μM ITX3 or the equivalent volume of DMSO. Impedance measure every 5 min during the first 6 h of migration expressed as delta cell index, with each point corresponding to the average of three or four wells (±SD). Each graph is representative of three independent experiments. (C) Analysis of wound closure in a confluent monolayer of control or RhoG siRNA–transfected A7r5 cells (RhoG KD). Cells were imaged at different time points to monitor wound closure. Representative images for control (CTRL) and RhoG KD cells (RhoG KD) at 0 and 16 h after wounding. (D) Wound area at 0 and 16 h (error bars represent SEM; n = 3). (E) KD efficiency for A (left, shRNA-mediated KD) and C and D (right, siRNA-mediated KD) was analyzed by SDS–PAGE and Western blotting. (F) Working model. RhoG functions both upstream of Rac1 and contributes to regulate the formation of CDRs and independently of Rac1, where it functions downstream of Trio to regulate the size of the CDRs formed. Cdc42 also controls CDR formation and size, probably downstream of the Cdc42 GEF Tuba.
In summary, our results suggest that PDGF promotes the activation of RhoG. Activation of RhoG downstream of PDGF is regulated by the exchange factor Trio and plays a role in the formation of PDGF-mediated CDRs and the functions associated with CDR formation, including macropinocytosis, receptor internalization, and cell migration.
DISCUSSION
In this study, we demonstrate a role for the small GTPase RhoG and its exchange factor, Trio, in the regulation of CDRs downstream of PDGF. Our results show that Trio and RhoG influence the number of cells that form CDRs, as well as their size. Our working model proposes that a pool of RhoG functions upstream of Rac1, which in turn modulates formation of CDRs, whereas a second pool of RhoG functions downstream of Trio but independently of Rac1 to regulate the size of the CDRs formed (Figure 9F). We also found that Trio and RhoG modulate cellular processes associated with CDR formation, including micropinocytosis, receptor internalization, and cell migration.
Our results show that in rat vascular smooth muscle cells (A7r5) and human fibroblasts (MRC5), PDGF treatment stimulates a rapid and transient activation of RhoG, a process that requires the PDGFR kinase activity. In HeLa cells, RhoG can be activated in response to epidermal growth factor (EGF) but not PDGF or serum, suggesting that this response may be cell-type specific (Samson et al., 2010). The activation of RhoG downstream of PDGF requires both Src and PI3K. Again, this is in contrast to results in HeLa cells, in which inhibiting Src or PI3K had no effect on EGF-mediated RhoG activation (Samson et al., 2010). Taken together, these results suggest a different mechanism for RhoG activation downstream of PDGFR than with EGFR.
Silencing RhoG expression affects CDRs in two different ways. First, it reduces the number of cells that form CDRs in response to PDGF, and second, it induces a dramatic decrease in the average size of the CDRs that form. We believe that the decrease in CDR number may be related to the influence of RhoG on Rac1 activity. RhoG functions are frequently mediated by the downstream activation of Rac1 because the RhoG effector ELMO forms a complex with Dock180, which functions as a GEF for Rac (Gumienny et al., 2001; Brugnera et al., 2002; Katoh and Negishi, 2003). Rac1 plays a key role during CDR formation. Rac-null fibroblasts do not have CDRs (Vidali et al., 2006), whereas dominant-negative Rac abolishes both CDRs and peripheral ruffles (Suetsugu et al., 2003; Lanzetti et al., 2004). Our results agree with these findings and show that silencing Rac1 virtually eliminates CDR formation in response to PDGF. In A7r5 cells, Rac1 is efficiently activated in response to PDGF, even after RhoG silencing, suggesting that its ability to respond to PDGF is independent of RhoG. Similar results were reported in MEFs treated with PDGF (Monypenny et al., 2009) and in HeLa cells in response to EGF (Samson et al., 2010). There was a slight reduction, however, in the basal level of Rac1 in RhoG KD cells and the total levels of active Rac1 after PDGF treatment, suggesting that a fraction of Rac1 is activated downstream of RhoG, probably via ELMO-Dock180. This reduction in Rac1 activity may be responsible for the decrease in number of cells that form CDRs when RhoG is silenced.
Inhibition of either Src or PI3K also inhibited CDR formation, as previously shown (Wennstrom et al., 1994; Wymann and Arcaro, 1994; Scaife et al., 2003; Veracini et al., 2006). The fact that inhibiting Src or PI3K has a significantly stronger effect on CDR than silencing RhoG is not surprising because they both regulate a wide variety of signaling pathways downstream PDGFR. It also makes it difficult to analyze the specific roles of Src and PI3K on RhoG activation downstream of PDGF. In the case of PI3K inhibition, it most likely affects CDR formation through Rac1, which was shown to be activated downstream of PI3K in PDGF-treated cells (Hawkins et al., 1995).
The mechanisms that control ring size in CDRs are not completely understood. Hasegawa et al. (2012) showed that ARAP1, a GAP for Arf and Rho GTPases, can regulate ring size. In ARAP1 KD cells, CDR rings are smaller, whereas overexpression of ARAP1, as well as of dominant-negative forms of Arf1 and Arf5, resulted in larger rings (Hasegawa et al., 2012). Catalytic dead mutants in the Arf-GAP domain but not in the Rho-GAP domain abolished the overexpression effect, suggesting that it is mediated via the function of Arf1/Arf5 and not Rho proteins (Hasegawa et al., 2012). It is possible that RhoG function on CDR area may occur in conjunction Arf1/5 inactivation by ARAP1. Cross-talk was found between Arf and Rho GTPases (Boshans et al., 2000; White et al., 2010). However, further experiments are necessary to determine whether these two pathways are connected.
We can only speculate about the mechanism by which RhoG controls CDR size. One possible explanation is that RhoG regulates the initial expansion of the CDRs. As mentioned earlier, CDRs form at or close to the maximum size and then start closing rapidly. This quick expansion occurs rarely and is extremely fast, and so we were not able to measure the expansion rate of the CDRs accurately in CTRL and RhoG KD cells. Another possibility is that RhoG regulates the turnover in the CDR machinery required for the CDRs to open/close. Modeling work showed that CDRs behavior can be described as propagation of a wave in an excitable medium (Zeng et al., 2011). This process involves spatiotemporal regulation of actin polymerization/depolymerization, myosin-mediated contractility, and regulation of membrane curvature (Allard and Mogilner, 2013). Each of these processes can be controlled by Rho GTPases. We recently described a role for RhoG in the turnover of invadopodia in breast cancer cells (Goicoechea et al., 2017). Invadopodia have many components in common with CDRs (Goicoechea et al., 2006), and it is possible that RhoG plays a similar role in CDRs, regulating the turnover required during the expansion or closing of CDRs. In contrast to RhoG, Rac1 overexpression had no significant effect in the area of CDR, suggesting a more fundamental role in CDR formation. These results agree with reports that RhoG may function independently of Rac1 via specific effectors (Wennerberg et al., 2002), and that Rac1 activation alone is not sufficient for CDR formation (Suetsugu et al., 2003; Lanzetti et al., 2004). The identity of the RhoG effectors involved in the regulation of CDRs is not known and is the focus of our future studies.
Cdc42 was associated with CDR function in studies that characterized the role of its downstream effectors, including N-WASP/Arp2/3, CIP4, and PAK1/2 (Jimenez et al., 2000; Legg et al., 2007; Toguchi et al., 2010). In addition, the BAR domain–containing Cdc42 GEF Tuba was shown to recruit N-WASP and dynamin to CDRs and stimulate their formation (Kovacs et al., 2006; Peleg et al., 2011). Of interest, we show in this study that silencing Cdc42 expression reduces both the number of cells forming CDRs and the area of CDRs in a manner similar to that of silencing RhoG, suggesting that there may be cross-talk between RhoG and Cdc42. Supporting these results, when the Cdc42 effector N-WASP is knocked out in cells, the area of CDRs is approximately threefold smaller than in control cells (Legg et al., 2007). RhoG was previously shown to influence Cdc42 function, both positively and negatively, via yet-uncharacterized mechanisms (Gauthier-Rouviere et al., 1998; Franke et al., 2012). Our results suggest that RhoG and Cdc42 regulate the CDR ring size independently and that CDR formation and ring size may be controlled by different pathways. First, Cdc42 was robustly activated after PDGF treatment, as shown in other cell types (Jimenez et al., 2000; Yu et al., 2009; Toguchi et al., 2010), but its activation was not affected by RhoG KD. Second, the effects of silencing RhoG and Cdc42 were additive in relation to the number of cells forming CDRs but not on CDR area. Finally, overexpression of Cdc42 did not increase CDR area as observed with the overexpression of RhoG.
A number of GEFs have been described that stimulate nucleotide exchange of RhoG, including SGEF, PLEKHG6, Ephexin 4, Trio, and P-REX1 (Bellanger et al., 2000; Blangy et al., 2000; May et al., 2002; Wennerberg et al., 2002; Ellerbroek et al., 2004; Bustelo et al., 2007; D’Angelo et al., 2007; Krishna Subbaiah et al., 2012; Damoulakis et al., 2014). Here we focused on Trio because it was previously associated with the regulation of diverse actin structures at the dorsal surface of the cells. Trio and RhoG are involved in the formation of peripheral dorsal ruffles (Bellanger et al., 2000; Blangy et al., 2000), as well as in the regulation of phagocytosis (deBakker et al., 2004) and transendothelial migration (van Rijssel et al., 2012b). Trio and kalirin are the only members of the mammalian Dbl family that display two GEF domains of distinct specificity (Debant et al., 1996). The first GEF domain of Trio (Trio GEFD1) activates RhoG and Rac, and the second (Trio GEFD2) acts on RhoA, which suggest that Trio may link several GTPase pathways in vivo (Bellanger et al., 2000; Blangy et al., 2000).
Our immunofluorescence work shows that endogenous Trio localizes to CDRs. This result by itself should be taken with caution because staining three-dimensional structures such as CDRs often produces false positives. However, our results also show that Trio is the GEF responsible for the regulation of RhoG downstream of PDGF and that, just as we observe for RhoG, it affects the number of cells that make CDRs, as well as the size of the CDRs. Of interest, inhibiting Trio selectively reduced RhoG activation and not Rac1 downstream of PDGFR, suggesting that Rac1 activation downstream of PDGF may be mediated by a different GEF. These results agree with findings showing that Trio has a higher affinity for RhoG than Rac1 (Blangy et al., 2000) and that TrioD1 inhibition with ITX3 did not completely abolish Rac1 activation (van Rijssel et al., 2012a). Our results do not exclude the involvement of other RhoGEFs that may act in conjunction with Trio to regulate this pathway. Other signals upstream of RhoG and/or Rac1 and Cdc42 may include the adaptor protein Nck, which was shown to play a role in the regulation of Rho GTPases and actin dynamics downstream of the PDGF receptor and is also required for the formation of CDRs (Ruusala et al., 2008). We plan to investigate the role of Nck in the regulation of RhoG and other GTPases during CDR formation.
One of the most important cellular functions associated with CDR formation is macropinocytosis. The formation of CDRs has been associated with internalization of both fluid-phase components (Dowrick et al., 1993) and growth factor receptors and other transmembrane proteins (Orth et al., 2006; Gu et al., 2011). RhoG has also been associated with macropinocytosis (Ellerbroek et al., 2004), internalization of integrins (Bass et al., 2011), and phagocytosis (Tzircotis et al., 2011). Trio has been associated with phagocytosis of apoptotic cells by activating RhoG and ELMO-DOCK180-Rac complex (deBakker et al., 2004) but not directly with macropinocytosis. Our results show that silencing RhoG or Trio inhibits both macropinocytosis, as measured by dextran uptake, and internalization of the PDGF receptor, suggesting that it may be playing a role in receptor recycling. This decrease in dextran uptake and receptor internalization may result from the observed decrease in CDR size when RhoG or Trio is silenced, as CDR ring size is correlated with a decrease in macropinocytosis in ARAP1-knockdown cells (Hasegawa et al., 2012). In migrating cells, macropinocytosis contributes to the recycling of growth factor receptors and integrins from disassembling focal adhesion to the leading edge (Gu et al., 2011). Both RhoG and Trio were previously associated with roles in migration (Katoh et al., 2006; Hiramoto-Yamaki et al., 2010; Bass et al., 2011; van Rijssel et al., 2012a) and also play a role in regulating PDGF-mediated migration in our system.
In summary, our results describe a novel role for Trio and RhoG in the regulation of CDR size, which affects macropinocytosis, receptor recycling and internalization, and cell migration.
MATERIALS AND METHODS
Antibodies and other reagents
The following antibodies were used: mouse monoclonals anti-RhoG and anti-myc (9E10), rabbit anti-cortactin and anti-RhoGDI1, goat anti-Trio, and rabbit anti-fibronectin (Santa Cruz Biotechnology); mouse anti–α-tubulin and anti-vinculin (Sigma-Aldrich); total-Akt, rabbit anti–phospho-Akt (S473), rabbit anti-Src, rabbit anti–phospho Src (Y416), rabbit anti p44/42 MAPK, rabbit anti–phospho-p44/42 (Thr-202/Tyr-204), and rabbit monoclonal anti–PDGFR β (Cell Signaling); mouse monoclonals anti-Rac1 and anti-Cdc42 (BD Biosciences); mouse anti-GFP (Thermo Fisher); mouse anti-actin (Abcam); Alexa Fluor 488 and Alexa Fluor 594 anti-mouse immunoglobulin G (IgG) and anti-rabbit IgG-conjugated secondary antibodies and Alexa 488 and Alexa 594–phalloidin (Life Technologies); and horseradish peroxidase (HRP)–conjugated anti-mouse, anti-rabbit, and anti-goat secondary antibodies (Jackson ImmunoResearch).
Cell culture
Rat embryonic thoracic aorta smooth muscle–derived cells (A7r5) were obtained from the American Type Culture Collection and cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 1 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies) at 37°C in a humidified atmosphere of 95% air and 5% CO2. After the attainment of 70–80% confluence, the cells were incubated in serum-free DMEM during 2 h. In some experiments, cells were preincubated with the Src inhibitors SU6656 (Calbiochem) at 2.5 μM or PP2 (Sigma-Aldrich) at 10 μM, PI3K inhibitor (LY-294002; Calbiochem) at 25 μM, MEK1/2 inhibitor U0126 (Sigma-Aldrich) at 10 μM, Trio inhibitor (ITX-3; Tocris) at 100 μM, or the equivalent amount of dimethyl sulfoxide (DMSO) during 30 min and then stimulated with 20 ng/ml PDGF-BB (Invitrogen) during 1 min for GTPase activity assays or 2.5 min for immunofluorescence assay.
Rho GTPase activity assays
To determine the levels of active RhoG, Rac1, and Cdc42, A7r5 cells were growth until 70–80% confluence and serum starved for 2 h. The cells were then washed twice with ice-cold Tris-buffered saline (50 mM Tris, pH 7.4, 5 mM MgCl2, 150 mM NaCl) and then lysed in lysis buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, EZBlock Protease Inhibitor Cocktail [BioVision]). Lysates were cleared by centrifugation at 16,000 × g for 3 min, and protein concentrations of the supernatants were determined using the DC Protein Assay (Bio-Rad). Equal amounts of total protein were incubated during 30 min with rotation at 4°C with 50 μg of glutathione-Sepharose beads (GE Healthcare) loaded with glutathione S-transferase (GST)–ELMO (full-length ELMO for RhoG) or GST-PBD (Pak1-binding domain for Rac and Cdc42). The beads were washed three times with lysis buffer, and pull downs and lysates were immunoblotted using antibodies for RhoG, Rac1, or Cdc42, respectively.
Cell transfection and adenoviruses
A7r5 cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. The siRNA was used to a final concentration of 25 nM, with the sequences specific for rat RhoG, 5-CGUCUUCGUCAUCUGUUUCUU-3; Cdc42 and Rac (ON-TARGETplus SMARTpool); and Trio, 5-GAACAUGAUUGACGAGCAU[dT][dT]-3. After 72 h of transfection, cells were serum starved for 2 h and then stimulated with 20 ng/ml PDGF-BB during 1 min for pull-down assays or 2.5 min for immunofluorescence assays.
The ViraPower Adenoviral Expression System (Life Technologies) was used for transient expression of human mycRhoG, mycRhoG Q61L, mycRac, mycRac Q61L, mycCdc42, and mycCdc42 Q61L, which were subcloned into pAd/CMV/V5-DEST using Gateway recombination according to the manufacturer’s instructions (Life Technologies). pAdeno-TrioD1-GFP was a gift of Jaap vanBuul (Sanquin, Netherlands). Adenovirus was prepared according to manufacturer’s recommendations (Life Technologies).
For the overexpression experiments or rescue, cells were infected for 48 h with the indicated viruses. The efficiency of the overexpression was further determined for each assay by immunoblotting using specific antibodies as indicated in the respective figures.
Cell lysis and immunoblotting
Cells cultured on 100-mm tissue culture dishes were briefly rinsed with phosphate-buffered saline (PBS) and then scraped into a lysis buffer containing 50 mM Tris/HCl, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and EZBlock Protease Inhibitor Cocktail. The supernatant was collected after centrifugation at 14,000 rpm for 10 min. For immunoblotting, lysates were boiled in 2× Laemmli buffer, and 20 μg of protein was resolved by SDS–PAGE in each lane of a 13% gel. The proteins were transferred to polyvinylidene fluoride, immunoblotted with the indicated antibodies, and visualized using the Immobilon Western Millipore Chemiluminescence HRP substrate (Millipore). Blots were developed using either film or an Azure c600 chemiluminescence detector (Azure Biosystems).
Immunofluorescence
A7r5 cells were seeded in six-well plates, and after 16 h, they were either transfected with siRNA against RhoG, Rac1, or Cdc42 or infected with adenoviruses encoding myc-Rac, myc-Cdc42, or myc-RhoG (or their constitutive active forms) for 48 h (empty virus as control). After that, cells were trypsinized, seeded on coverslips precoated with 10 μg/ml fibronectin, and allowed to attach for 16 h. Cells were then serum starved for 2 h and stimulated with 20 ng/ml PDGF-BB for indicated times. Immunofluorescence assay was performed as described previously (García-Mata et al., 2003). Briefly, cells were fixed for 10 min with 3.7% paraformaldehyde and quenched with 10 mM ammonium chloride. Cells were then permeabilized with 0.1% Triton X-100 in PBS. The coverslips were then washed with PBS and blocked in PBS, 2.5% goat serum, and 0.2% Tween 20 for 5 min, followed by 5 min of blocking in PBS, 0.4% fish skin gelatin, and 0.2% Tween 20. Cells were incubated with primary antibody for 1 h at room temperature. Coverslips were washed with PBS and 0.2% Tween 20 and incubated with secondary antibodies for 45 min, washed as described, and mounted on glass slides in Mowiol mounting solution. Images were acquired on an Olympus IX81 inverted microscope using a PlanApo N 60×/1.42 oil objective lens and a XM10 camera (Olympus). All experiment were repeated three independent times, and from each one, at least 100 cells/condition were analyzed by counting the percentage of cells with CDR, the number of CDRs per cell, and the area of each CDR. The area was calculated by drawing the outline of each ruffle and measuring it using ImageJ software (National Institutes of Health, Bethesda, MD).
Live imaging
Dynamics of CDR formation was imaged by differential interference contrast (DIC) on an Olympus VivaView FL microscope using a UPLSAPO 20×, numerical aperture 0.75, DIC objective and a Hamamatsu ORCA II camera. Cells were stabilized for 30 min in the incubator before the addition of PDGF. Images were acquired every 35 s. Six different samples (three CTRL and three RhoG KD) were analyzed in parallel for every experiment, and five fields were imaged simultaneously for each of the samples. CDR properties were measured in every frame for each ruffle using ImageJ as described.
PDGF receptor internalization by trypsinization
PDGFR internalization was determined by evaluating the amount of receptor resistant to trypsin digestion (Ceresa et al., 1998). In brief, cells were transfected with siRNA against RhoG or control as described, serum starved for 2 h, and stimulated with 20 ng/ml PDGF-BB for 0.5, 2, and 6 h. Cells were washed twice with PBS and incubated for 8 min on ice with ice-cold 20 mM sodium acetate, pH 3.7. Afterward, cells were washed with ice-cold PBS and incubated with 0.05% trypsin on ice for 30 min. The reaction was stopped by adding soybean trypsin inhibitor (1 mg/ml), and then cells were washed with PBS and scraped in lysis buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, EZBlock Protease Inhibitor Cocktail). Samples were analyzed by SDS–PAGE 10%, followed by immunoblots for PDGFR (Cell Signaling) and RhoG (Millipore) to evaluate the efficiency of knockdown, with GDI as a loading control.
Wound-healing assay
A7r5 cells were seeded to confluence and transfected with siRNA control or siRNA against RhoG as described. After 48 h, cells were serum starved for 2 h before the monolayer was scratched using a micropipette tip. Cells were then incubated with medium containing 20 ng/ml PDGF-BB for 16 h. Pictures were captured at 0 and 16 h, and the wound area was measured using ImageJ. Results are expressed as percentage of wound area relative to the initial area at time zero.
Dextran assay
To determine the levels of dextran uptake, A7r5 cells were transfected with siRNA against RhoG, Trio, or control. After 24 h of transfection, the expression of RhoG was rescued by infecting the cells with an adenovirus coding myc-RhoG. Cells were then serum starved for 2 h using DMEM without phenol red and 0.15% lipid-free bovine serum albumin (BSA; 48 h after transfection/24 h after infection). Cells were then incubated with Ringer’s buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM d-glucose, pH 7.2, plus 0.05% lipid-free BSA) containing 250 μg/ml dextran, 10,000 molecular weight, conjugated to Alexa Fluor 594 (Invitrogen) with or without 20 ng/ml PDGF-BB for 30 min. Plates were rinsed three times with iced-cold Ringer’s buffer and washed by immersion in 1 l of iced-cold PBS three times for 5 min each time. After that, cells were lysed with 500 μl of 0.5% Triton X-100 in PBS and incubated at 37°C. Fluorescence was determined in duplicate at 590 nm (excitation) and 617 nm (emission) with a Spectramax M5 microplate reader (Molecular Devices). Dextran concentration in each sample was determined by a dextran standard curve, normalized by the amount of protein in each condition, and expressed as nanograms of dextran/microgram of protein.
Real-time cell analysis experiments
Experiments of real-time migration were performed with the xCELLigence RTCA DP Instrument (Roche) using the CIM-plate 16. Briefly, cells lines either infected with lentivirus encoding short hairpin RNA targeting RhoG or with a noncoding virus as control (CTRL; Open Biosystems) were serum starved for 2 h. A total of 25,000 cells were seeded on the upper part of the chamber in DMEM 0.5% lipid free-BSA and allowed to migrate toward a lower chamber containing DMEM 0.1% FBS or DMEM 0.1% FBS and 20 ng/ml PDGF-BB. In other assays, A7r5 cells were allowed to migrate as described in the presence of 100 μM ITX3 or the equivalent volume of DMSO. Changes in the impedance were assessed every 5 min for 16 h. A representative graph from three independent experiments shows the first 6 h of migration expressed as delta cell index, with each point corresponding to the average of three of four wells (±SD).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7. One-way analysis of variance was used to compare multiple-condition assays and unpaired t test to compare the means of two independent groups.
Supplementary Material
Acknowledgments
We thank Carol Otey (University of North Carolina at Chapel Hill) for support during the early phases of this project and for comments and valuable discussion. We also thank Congying (Emma) Wu and James Bear (University of North Carolina at Chapel Hill) for assistance with live imaging and Jaap van Buul (Sanquin Institute, Netherlands) for the Trio-D1-GFP construct.
Abbreviations used:
- CDR
circular dorsal ruffle
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- PDGF
platelet-derived growth factor
- PR
peripheral ruffle
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0412) on May 3, 2017.
REFERENCES
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. II. “Ruffling.”. Exp Cell Res. 1970;60:437–444. doi: 10.1016/0014-4827(70)90537-9. [DOI] [PubMed] [Google Scholar]
- Allard J, Mogilner A. Traveling waves in actin dynamics and cell motility. Curr Opin Cell Biol. 2013;25:107–115. doi: 10.1016/j.ceb.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell. 2011;21:681–693. doi: 10.1016/j.devcel.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol. 2000;2:888–892. doi: 10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- Blangy A, Vignal E, Schmidt S, Debant A, Gauthier-Rouviere C, Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J Cell Sci. 2000;113:729–739. doi: 10.1242/jcs.113.4.729. [DOI] [PubMed] [Google Scholar]
- Boshans RL, Szanto S, van Aelst L, D’Souza-Schorey C. ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol Cell Biol. 2000;20:3685–3694. doi: 10.1128/mcb.20.10.3685-3694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquier N, Vignal E, Charrasse S, Weill M, Schmidt S, Leonetti JP, Blangy A, Fort P. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem Biol. 2009;16:657–666. doi: 10.1016/j.chembiol.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- Buchanan FG, Elliot CM, Gibbs M, Exton JH. Translocation of the Rac1 guanine nucleotide exchange factor Tiam1 induced by platelet-derived growth factor and lysophosphatidic acid. J Biol Chem. 2000;275:9742–9748. doi: 10.1074/jbc.275.13.9742. [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. BioEssays. 2007;29:356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresa BP, Kao AW, Santeler SR, Pessin JE. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol. 1998;18:3862–3870. doi: 10.1128/mcb.18.7.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesio CL, Perrin BJ, Bennin DA, Huttenlocher A. Actin-binding protein-1 interacts with WASp-interacting protein to regulate growth factor-induced dorsal ruffle formation. Mol Biol Cell. 2010;21:186–197. doi: 10.1091/mbc.E09-02-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, Welch HC, Der CJ, Stephens LR, Hawkins PT. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci. 2014;127:2589–2600. doi: 10.1242/jcs.153049. [DOI] [PubMed] [Google Scholar]
- D’Angelo R, Aresta S, Blangy A, Del Maestro L, Louvard D, Arpin M. Interaction of ezrin with the novel guanine nucleotide exchange factor PLEKHG6 promotes RhoG-dependent apical cytoskeleton rearrangements in epithelial cells. Mol Biol Cell. 2007;18:4780–4793. doi: 10.1091/mbc.E06-12-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deBakker CD, Haney LB, Kinchen JM, Grimsley C, Lu M, Klingele D, Hsu PK, Chou BK, Cheng LC, Blangy A, et al. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol. 2004;14:2208–2216. doi: 10.1016/j.cub.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Debant A, Serra-Pages C, Seipel K, O’Brien S, Tang M, Park SH, Streuli M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA. 1996;93:5466–5471. doi: 10.1073/pnas.93.11.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmawardhane S, Schurmann A, Sells MA, Chernoff J, Schmid SL, Bokoch GM. Regulation of macropinocytosis by p21-activated kinase-1. Mol Biol Cell. 2000;11:3341–3352. doi: 10.1091/mbc.11.10.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowrick P, Kenworthy P, McCann B, Warn R. Circular ruffle formation and closure lead to macropinocytosis in hepatocyte growth factor/scatter factor-treated cells. Eur J Cell Biol. 1993;61:44–53. [PubMed] [Google Scholar]
- Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15:3309–3319. doi: 10.1091/mbc.E04-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrach S, Schmidt S, Diriong S, Penna A, Blangy A, Fort P, Debant A. The human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr Biol. 2002;12:307–312. doi: 10.1016/s0960-9822(02)00658-9. [DOI] [PubMed] [Google Scholar]
- Franke K, Otto W, Johannes S, Baumgart J, Nitsch R, Schumacher S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J. 2012;31:2908–2921. doi: 10.1038/emboj.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabunia K, Jain S, England RN, Autieri MV. Anti-inflammatory cytokine interleukin-19 inhibits smooth muscle cell migration and activation of cytoskeletal regulators of VSMC motility. Am J Physiol Cell Physiol. 2011;300:C896–C906. doi: 10.1152/ajpcell.00439.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mata R, Szul T, Alvarez C, Sztul E. ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol Biol Cell. 2003;14:2250–2261. doi: 10.1091/mbc.E02-11-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier-Rouviere C, Vignal E, Meriane M, Roux P, Montcourier P, Fort P. RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol Biol Cell. 1998;9:1379–1394. doi: 10.1091/mbc.9.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea S, Arneman D, Disanza A, Garcia-Mata R, Scita G, Otey CA. Palladin binds to Eps8 and enhances the formation of dorsal ruffles and podosomes in vascular smooth muscle cells. J Cell Sci. 2006;119:3316–3324. doi: 10.1242/jcs.03076. [DOI] [PubMed] [Google Scholar]
- Goicoechea SM, Zinn A, Awadia SS, Snyder K, Garcia-Mata R. A RhoG-mediated signaling pathway that modulates invadopodia dynamics in breast cancer cells. J Cell Sci. 2017;130:1064–1077. doi: 10.1242/jcs.195552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Noss EH, Hsu VW, Brenner MB. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J Cell Biol. 2011;193:61–70. doi: 10.1083/jcb.201007003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Tsujita K, Takenawa T, Itoh T. ARAP1 regulates the ring size of circular dorsal ruffles through Arf1 and Arf5. Mol Biol Cell. 2012;23:2481–2489. doi: 10.1091/mbc.E12-01-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PT, Eguinoa A, Qiu RG, Stokoe D, Cooke FT, Walters R, Wennstrom S, Claesson-Welsh L, Evans T, Symons M, et al. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Hiramoto-Yamaki N, Takeuchi S, Ueda S, Harada K, Fujimoto S, Negishi M, Katoh H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J Cell Biol. 2010;190:461–477. doi: 10.1083/jcb.201005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshmand-Rad R, Claesson-Welsh L, Wennström S, Yokote K, Siegbahn A, Heldin C-H. Involvement of phosphatidylinositide 3′-kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp Cell Res. 1997;234:434–441. doi: 10.1006/excr.1997.3636. [DOI] [PubMed] [Google Scholar]
- Huynh J, Bordeleau F, Kraning-Rush CM, Reinhart-King CA. Substrate stiffness regulates PDGF-induced circular dorsal ruffle formation through MLCK. Cell Mol Bioeng. 2013;6 doi: 10.1007/s12195-013-0278-7. doi: 10.1007/s12195-013-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez C, Portela RA, Mellado M, Rodriguez-Frade JM, Collard J, Serrano A, Martinez AC, Avila J, Carrera AC. Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J Cell Biol. 2000;151:249–262. doi: 10.1083/jcb.151.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Hiramoto K, Negishi M. Activation of Rac1 by RhoG regulates cell migration. J Cell Sci. 2006;119:56–65. doi: 10.1242/jcs.02720. [DOI] [PubMed] [Google Scholar]
- Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature. 2003;424:461–464. doi: 10.1038/nature01817. [DOI] [PubMed] [Google Scholar]
- Katoh H, Yasui H, Yamaguchi Y, Aoki J, Fujita H, Mori K, Negishi M. Small GTPase RhoG is a key regulator for neurite outgrowth in PC12 cells. Mol Cell Biol. 2000;20:7378–7387. doi: 10.1128/mcb.20.19.7378-7387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada K, Upadhyay G, Ferandon S, Janarthanan S, Hall M, Vilardaga J-P, Yajnik V. Cell migration is regulated by platelet-derived growth factor receptor endocytosis. Mol Cell Biol. 2009;29:4508–4518. doi: 10.1128/MCB.00015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SJ, Worth DC, Scales TM, Monypenny J, Jones GE, Parsons M. Beta1 integrins regulate fibroblast chemotaxis through control of N-WASP stability. EMBO J. 2011;30:1705–1718. doi: 10.1038/emboj.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EM, Makar RS, Gertler FB. Tuba stimulates intracellular N-WASP-dependent actin assembly. J Cell Sci. 2006;119:2715–2726. doi: 10.1242/jcs.03005. [DOI] [PubMed] [Google Scholar]
- Krishna Subbaiah V, Massimi P, Boon SS, Myers MP, Sharek L, Garcia-Mata R, Banks L. The invasive capacity of HPV transformed cells requires the hDlg-dependent enhancement of SGEF/RhoG activity. PLoS Pathog. 2012;8:e1002543. doi: 10.1371/journal.ppat.1002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger EW, Orth JD, Cao H, McNiven MA. A dynamin-cortactin-Arp2/3 complex mediates actin reorganization in growth factor-stimulated cells. Mol Biol Cell. 2003;14:1085–1096. doi: 10.1091/mbc.E02-08-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP. Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature. 2004;429:309–314. doi: 10.1038/nature02542. [DOI] [PubMed] [Google Scholar]
- Legg JA, Bompard G, Dawson J, Morris HL, Andrew N, Cooper L, Johnston SA, Tramountanis G, Machesky LM. N-WASP involvement in dorsal ruffle formation in mouse embryonic fibroblasts. Mol Biol Cell. 2007;18:678–687. doi: 10.1091/mbc.E06-06-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machuy N, Campa F, Thieck O, Rudel T. c-Abl-binding protein interacts with p21-activated kinase 2 (PAK-2) to regulate PDGF-induced membrane ruffles. J Mol Biol. 2007;370:620–632. doi: 10.1016/j.jmb.2007.04.080. [DOI] [PubMed] [Google Scholar]
- May V, Schiller MR, Eipper BA, Mains RE. Kalirin Dbl-homology guanine nucleotide exchange factor 1 domain initiates new axon outgrowths via RhoG-mediated mechanisms. J Neurosci. 2002;22:6980–6990. doi: 10.1523/JNEUROSCI.22-16-06980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller J, Vidali L, Schwartz MA. Endogenous RhoG is dispensable for integrin-mediated cell spreading but contributes to Rac-independent migration. J Cell Sci. 2008;121:1981–1989. doi: 10.1242/jcs.025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellstrom K, Heldin CH, Westermark B. Induction of circular membrane ruffling on human fibroblasts by platelet-derived growth factor. Exp Cell Res. 1988;177:347–359. doi: 10.1016/0014-4827(88)90468-5. [DOI] [PubMed] [Google Scholar]
- Monypenny J, Zicha D, Higashida C, Oceguera-Yanez F, Narumiya S, Watanabe N. Cdc42 and Rac Family GTPases regulate mode and speed but not direction of primary fibroblast migration during platelet-derived growth factor-dependent chemotaxis. Mol Cell Biol. 2009;29:2730–2747. doi: 10.1128/MCB.01285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga C, Zohar M, Teramoto H, Gutkind JS. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-kappaB. Oncogene. 2002;21:207–216. doi: 10.1038/sj.onc.1205036. [DOI] [PubMed] [Google Scholar]
- Orth JD, Krueger EW, Weller SG, McNiven MA. A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res. 2006;66:3603–3610. doi: 10.1158/0008-5472.CAN-05-2916. [DOI] [PubMed] [Google Scholar]
- Patel JC, Galan JE. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol. 2006;175:453–463. doi: 10.1083/jcb.200605144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg B, Disanza A, Scita G, Gov N. Propagating cell-membrane waves driven by curved activators of actin polymerization. PLoS One. 2011;6:e18635. doi: 10.1371/journal.pone.0018635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruusala A, Pawson T, Heldin CH, Aspenstrom P. Nck adapters are involved in the formation of dorsal ruffles, cell migration, and Rho signaling downstream of the platelet-derived growth factor beta receptor. J Biol Chem. 2008;283:30034–30044. doi: 10.1074/jbc.M800913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, Matsui O, Takuwa Y. Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ Res. 2002;90:325–332. doi: 10.1161/hh0302.104455. [DOI] [PubMed] [Google Scholar]
- Samson T, Welch C, Monaghan-Benson E, Hahn KM, Burridge K. Endogenous RhoG is rapidly activated after epidermal growth factor stimulation through multiple guanine-nucleotide exchange factors. Mol Biol Cell. 2010;21:1629–1642. doi: 10.1091/mbc.E09-09-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaife RM, Courtneidge SA, Langdon WY. The multi-adaptor proto-oncoprotein Cbl is a key regulator of Rac and actin assembly. J Cell Sci. 2003;116:463–473. doi: 10.1242/jcs.00244. [DOI] [PubMed] [Google Scholar]
- Schmees C, Villasenor R, Zheng W, Ma H, Zerial M, Heldin CH, Hellberg C. Macropinocytosis of the PDGF beta-receptor promotes fibroblast transformation by H-RasG12V. Mol Biol Cell. 2012;23:2571–2582. doi: 10.1091/mbc.E11-04-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sero JE, Thodeti CK, Mammoto A, Bakal C, Thomas S, Ingber DE. Paxillin mediates sensing of physical cues and regulates directional cell motility by controlling lamellipodia positioning. PLoS One. 2011;6:e28303. doi: 10.1371/journal.pone.0028303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- Toguchi M, Richnau N, Ruusala A, Aspenström P. Members of the CIP4 family of proteins participate in the regulation of platelet-derived growth factor receptor-β-dependent actin reorganization and migration. Biol Cell. 2010;102:215–230. doi: 10.1042/BC20090033. [DOI] [PubMed] [Google Scholar]
- Tzircotis G, Braga VM, Caron E. RhoG is required for both FcgammaR- and CR3-mediated phagocytosis. J Cell Sci. 2011;124:2897–2902. doi: 10.1242/jcs.084269. [DOI] [PubMed] [Google Scholar]
- van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–1293. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijssel J, Hoogenboezem M, Wester L, Hordijk PL, Van Buul JD. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS One. 2012a;7:e29912. doi: 10.1371/journal.pone.0029912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FP, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell. 2012b;23:2831–2844. doi: 10.1091/mbc.E11-11-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veracini L, Franco M, Boureux A, Simon V, Roche S, Benistant C. Two distinct pools of Src family tyrosine kinases regulate PDGF-induced DNA synthesis and actin dorsal ruffles. J Cell Sci. 2006;119:2921–2934. doi: 10.1242/jcs.03015. [DOI] [PubMed] [Google Scholar]
- Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigorito E, Billadeu DD, Savoy D, McAdam S, Doody G, Fort P, Turner M. RhoG regulates gene expression and the actin cytoskeleton in lymphocytes. Oncogene. 2003;22:330–342. doi: 10.1038/sj.onc.1206116. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Ellerbroek SM, Liu RY, Karnoub AE, Burridge K, Der CJ. RhoG signals in parallel with Rac1 and Cdc42. J Biol Chem. 2002;277:47810–47817. doi: 10.1074/jbc.M203816200. [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Hawkins P, Cooke F, Hara K, Yonezawa K, Kasuga M, Jackson T, Claesson-Welsh L, Stephens L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr Biol. 1994;4:385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- White DT, McShea KM, Attar MA, Santy LC. GRASP and IPCEF promote ARF-to-Rac signaling and cell migration by coordinating the association of ARNO/cytohesin 2 with Dock180. Mol Biol Cell. 2010;21:562–571. doi: 10.1091/mbc.E09-03-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann M, Arcaro A. Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem J. 1994;298:517–520. doi: 10.1042/bj2980517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki N, Negishi M, Katoh H. RhoG regulates anoikis through a phosphatidylinositol 3-kinase-dependent mechanism. Exp Cell Res. 2007;313:2821–2832. doi: 10.1016/j.yexcr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Zaal K, Hailey D, Presley J, Lippincott-Schwartz J, Samelson LE. Role of Grb2 in EGF-stimulated EGFR internalization. J Cell Sci. 2002;115:1791–1802. doi: 10.1242/jcs.115.9.1791. [DOI] [PubMed] [Google Scholar]
- Yu JA, Deakin NO, Turner CE. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol Biol Cell. 2009;20:4706–4719. doi: 10.1091/mbc.E09-07-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Lai T, Koh CG, LeDuc PR, Chiam KH. Investigating circular dorsal ruffles through varying substrate stiffness and mathematical modeling. Biophys J. 2011;101:2122–2130. doi: 10.1016/j.bpj.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Lin F, Brown DA, Clark RA. A fibronectin peptide redirects PDGF-BB/PDGFR complexes to macropinocytosis-like internalization and augments PDGF-BB survival signals. J Invest Dermatol. 2014;134:921–929. doi: 10.1038/jid.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.