

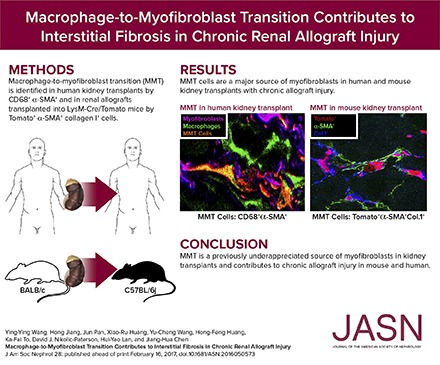
Abstract
Interstitial fibrosis is an important contributor to graft loss in chronic renal allograft injury. Inflammatory macrophages are associated with fibrosis in renal allografts, but how these cells contribute to this damaging response is not clearly understood. Here, we investigated the role of macrophage-to-myofibroblast transition in interstitial fibrosis in human and experimental chronic renal allograft injury. In biopsy specimens from patients with active chronic allograft rejection, we identified cells undergoing macrophage-to-myofibroblast transition by the coexpression of macrophage (CD68) and myofibroblast (α–smooth muscle actin [α-SMA]) markers. CD68+/α-SMA+ cells accounted for approximately 50% of the myofibroblast population, and the number of these cells correlated with allograft function and the severity of interstitial fibrosis. Similarly, in C57BL/6J mice with a BALB/c renal allograft, cells coexpressing macrophage markers (CD68 or F4/80) and α-SMA composed a significant population in the interstitium of allografts undergoing chronic rejection. Fate-mapping in Lyz2-Cre/Rosa26-Tomato mice showed that approximately half of α-SMA+ myofibroblasts in renal allografts originated from recipient bone marrow–derived macrophages. Knockout of Smad3 protected against interstitial fibrosis in renal allografts and substantially reduced the number of macrophage-to-myofibroblast transition cells. Furthermore, the majority of macrophage-to-myofibroblast transition cells in human and experimental renal allograft rejection coexpressed the M2-type macrophage marker CD206, and this expression was considerably reduced in Smad3-knockout recipients. In conclusion, our studies indicate that macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. Moreover, the transition of bone marrow–derived M2-type macrophages to myofibroblasts in the renal allograft is regulated via a Smad3-dependent mechanism.
Keywords: macrophage myofibroblast transition, chronic allograft rejection, interstitial fibrosis, lineage tracing, Smad3, M2 macrophage
Interstitial fibrosis is a major feature in chronic renal allograft injury. It is induced by a wide range of immune and nonimmune insults and is a predictor of allograft loss.1–3 It is well accepted that myofibroblasts are the major collagen-producing cell type during active fibrosis, but the origin of myofibroblasts continues to be a subject of intense debate.4 Accumulating evidence indicates a diverse cellular origin of myofibroblasts during active fibrosis,4,5 including bone marrow–derived fibrocytes and fibroblasts.6–11 Recent studies in the mouse model of unilateral ureteric obstruction (UUO) and in patients with progressive CKD have shown that bone marrow–derived monocytes/macrophages are capable of transition into myofibroblasts as identified by coexpression of macrophage markers (F4/80 and CD68) and α–smooth muscle actin (α-SMA) in conjunction with production of collagen I.12–14 These observations suggest that macrophage-to-myofibroblast transition (MMT) may be another pathway leading to myofibroblast accumulation during renal fibrosis.13,14 However, it remains unknown whether MMT contributes to chronic renal allograft rejection and what the mechanisms are that regulate the process of MMT.
TGF-β1/Smad3 signaling is a pivotal pathway in renal fibrosis.15 This is supported by the finding that mice lacking Smad3 are protected against tissue fibrosis in models of CKD.16,17 Smad3 is also required for the transition of cultured tubular epithelial cells, endothelial cells, and mesothelial cells into myofibroblasts in response to stimulation with TGF-β1, angiotensin II, and advanced glycation end products.18–20 In addition, studies in the UUO model of interstitial fibrosis and in cultured macrophages have identified that TGF-β1–induced MMT operates via a Smad3-dependent mechanism.14 However, little is known of the potential role of MMT or Smad3 signaling in the development of fibrosis in renal allograft injury.
This study investigated MMT and the functional role of Smad3 signaling in interstitial fibrosis in chronic renal allograft rejection. Phenotype analysis and lineage tracing were used to identify MMT in active interstitial fibrosis in human and experimental renal allograft rejection. In addition, a gene deletion approach demonstrated that the MMT response and the development of interstitial fibrosis in experimental renal allograft rejection operate via a Smad3-dependent mechanism.
Results
Identification of MMT in Human Chronic Active Renal Allograft Injury
Immunostaining was performed to identify cells undergoing MMT on the basis of coexpression of macrophage (CD68) and myofibroblast (α-SMA) markers. Biopsy samples taken immediately after transplantation (control) showed the presence of both CD68+ macrophages and resident α-SMA+ cells, but no double labeled cells were evident (Figure 1A). Acute renal allograft rejection (n=7, Table 1) exhibited a marked infiltrate of CD68+ macrophages and only a few coexpressing CD68+α-SMA+ cells were apparent (Figure 1, B–E). Biopsy specimens from patients with chronic active renal allograft rejection (n=13) contained large numbers of both CD68+ macrophages and α-SMA+ myofibroblasts in areas of severe interstitial fibrosis, with many double positive cells evident (Figure 1C). These CD68+α-SMA+ MMT cells accounted for approximately 50% of the total α-SMA+ myofibroblast population (Figure 1E). Three-color confocal microscopy, including Z-stack analysis, identified collagen I production by CD68+α-SMA+ cells in patients with chronic active renal allograft rejection (Figure 2, Supplemental Video 1). In comparison, patients with chronic renal allograft fibrosis without clear evidence of rejection (n=12) showed higher numbers of α-SMA+ myofibroblasts, but fewer macrophages and CD68+α-SMA+ MMT cells, compared with chronic active renal allograft rejection (Figure 1, D and E). In patients with chronic active rejection, the number of CD68+ macrophages and of CD68+α-SMA+ MMT cells correlated with allograft function, whereas the number of MMT cells correlated with α-SMA+ myofibroblasts (Figure 1, F–H). In addition, the number of α-SMA+ myofibroblasts correlated with Banff scores of interstitial fibrosis and tubular atrophy in patients with chronic active allograft rejection and in those patients with chronic renal allograft fibrosis without evidence of rejection (Supplemental Figure 1). By contrast, CD68+ macrophages and CD68+α-SMA+ MMT cells correlated with interstitial fibrosis in patients with chronic active allograft rejection but not in patients with chronic renal allograft fibrosis without evidence of rejection (Supplemental Figure 1). In addition, nuclear staining for phosphorylated Smad3 was detected in rejecting allografts indicating active TGF-β signaling (Supplemental Figure 2A). These findings suggest an active role for MMT cells in chronic active renal allograft rejection.
Figure 1.

MMT cells in patients with chronic active renal transplantation rejection. Two-color immunofluorescence identifies MMT cells that coexpress macrophage (CD68, green) and myofibroblast (α-SMA, red) markers in biopsy tissues. (A) Allograft immediately after transplantation (control). (B) Acute rejection. (C) Chronic active rejection. Examples of CD68+ α-SMA+ MMT cells are indicated by arrows and insert shows a high-power view of a single MMT cell. (D) Chronic allograft injury with no evidence of active rejection. Nuclei were stained with DAPI in blue. (E) Quantification of the number of CD68+ cells, α-SMA+ cells, and CD68+ α-SMA+ cells. Data are mean±SEM. ***P<0.001 versus acute rejection. (F–I) Correlation analysis of cell populations and allograft function in patients with chronic active transplant rejection. SCr, serum creatinine.
Table 1.
Patient demographics
| Characteristics | Acute Rejection (n=7) | Chronic Active Rejection (n=13) | Chronic Rejection (n=12) |
|---|---|---|---|
| Age, yr | 51±8.16 | 48.69±9.35 | 50.92±8.13 |
| Men/women, n | 6/1 | 8/5 | 10/2 |
| Timing of biopsy post-transplantation, mo | 0.51±0.21 | 79.47±66.11 | 104±76.26 |
| SCr, μmol/L | 259.14±140.19 | 267.67±86.01 | 184.36±55.60 |
| Hypertensiona (Y/N), n | 1/6 | 2/11 | 3/9 |
| Proteinuriab (Y/N), n | 2/5 | 2/11 | 3/9 |
| eGFR<60 ml/min (Y/N), n | 6/1 | 12/1 | 10/2 |
| Immunosuppressive, n | |||
| Tac+MMF+steroid | 4 | 6 | 4 |
| CyA+MMF+steroid | 2 | 4 | 4 |
| CyA+Aza+steroid | 0 | 1 | 2 |
| CyA+steroid | 1 | 2 | 2 |
| Banff scores | |||
| Interstitial infiltrate (i) | 0.86±0.64 | 1.62±0.62 | 0.50±0.50 |
| Tubulitis (t) | 0.57±0.49 | 0.92±0.62 | 0.17±0.37 |
| Intimal arteritis (v) | 0.43±0.49 | 0.54±0.63 | 0.42±0.49 |
| Glomerulitis (g) | 0.29±0.45 | 1.23±0.97 | 0.92±0.76 |
| Interstitial fibrosis (ci) | 0.00±0.00 | 1.31±0.72 | 1.50±0.65 |
| Tubular atrophy (ct) | 0.00±0.00 | 1.38±0.62 | 1.42±0.76 |
| Vascular fibrous intimal thickening (cv) | 0.00±0.00 | 0.23±0.42 | 1.08±0.49 |
| Allograft glomerulopathy (cg) | 0.00±0.00 | 0.85±0.86 | 1.17±0.69 |
The data are presented as mean±SEM. SCr, serum creatinine; Y, yes; N, no; Tac, tacrolimus; MMF, mycophenolate mofetil; CyA, cyclosporine A; Aza, azathioprine; i, interstitial infiltrate; t, tubulitis; v, intimal arteritis; g, glomerulitis; ci, interstitial fibrosis; ct, tubular atrophy; cv, vascular fibrous intimal thickening; cg, allograft glomerulopathy.
BP≥140/80 mmHg at time of biopsy.
Proteinuria≥0.5 g/24 h at time of biopsy.
Figure 2.

MMT cells produce collagen in human chronic active renal allograft rejection. (A) Three-color confocal microscopy identifies cells coexpressing CD68 (green), α-SMA (red), and collagen I (blue). One example of collagen-producing MMT is shown in high power in the insert. (B) Z-stack images illustrate the coexpression of CD68 (green), α-SMA (red), and collagen I (blue) in an MMT cell, which is also shown in the Supplemental Material.
Lineage Tracing in Mice Identifies That a Subset of Myofibroblasts in Chronic Renal Allograft Rejection Are Derived from Recipient Bone Marrow Macrophages
We used a genetic approach to determine the origin of MMT cells and myofibroblasts in chronic renal allograft rejection. BALB/c kidneys were transplanted ectopically into Lyz2-Cre/26-Tomato C57BL/6J mice which express the Tomato fluorescent protein exclusively in cells of the myeloid lineage. Thus, any Tomato-expressing myofibroblasts would be derived from recruited bone marrow–derived macrophages rather than from resident tissue macrophages. Native kidneys in recipient mice were left in place. In addition, recipient mice received tacrolimus to blunt the acute rejection response thereby allowing the development of chronic renal allograft rejection with interstitial fibrosis by the day 28 endpoint (Supplemental Figure 3). As a control, we confirmed Tomato expression by macrophage populations in kidney, liver, and lung in Lyz2-Cre/Rosa26-Tomato mice (Supplemental Figure 4).
Analysis of BALB/c renal allografts by three-color confocal microscopy showed that most F4/80+ macrophages infiltrating renal allograft were Tomato+ (Figure 3A). In addition, many of these F4/80+ Tomato+ cells coexpressed α-SMA and collagen I, indicating that bone marrow–derived collagen-producing MMT cells make a substantial contribution to progressive renal fibrosis in chronic renal allograft rejection (Figure 3, A and B). Examples of Tomato+ α-SMA+ collagen I+ MMT cells in the renal allograft are clearly illustrated by Z-stack confocal images (Figure 3C, Supplemental Video 2). Quantitative three-color flow cytometry confirmed that >80% of CD68+ macrophages in the renal allograft were derived from recipient bone marrow on the basis of Tomato expression (Figure 3D). In addition, around 90% of CD68+α-SMA+ MMT cells expressed Tomato (Figure 3D), with Tomato expression seen in 37% of the total α-SMA+ myofibroblast population in the renal allograft (Figure 3D), indicating that MMT makes a substantial contribution to the development of fibrosis in chronic renal allograft rejection in the mouse.
Figure 3.

Fate-mapping identifies a bone marrow macrophage origin for myofibroblasts in mouse chronic renal allograft rejection. Kidney allografts were transplanted into Lyz2-Cre/Rosa26-Tomato recipient mice and examined 28 days later. (A) Confocal microscopy shows that most F4/80+ macrophages (blue) express the Tomato marker, confirming their origin from recipient bone marrow macrophages. In addition, F4/80+Tomato+ cells can be seen coexpressing α-SMA (green), indicating MMT (insert shows high power of an MMT cell). (B) Identification of Tomato+ cells expressing α-SMA (green) and collagen I (blue). Insert shows an example of a Tomato+α-SMA+collagen I+ MMT cell. (C) Z-stack images show individual MMT cells coexpressing Tomato (red), α-SMA (green), and collagen (blue) in the rejecting allograft. Note that an elongated Tomato+ F4/80+α-SMA+ MMT cell with rich submembranous bundles of α-SMA+ actin filaments is clearly shown, which is further illustrated in the Supplemental Material. (D) Two-color flow cytometry analysis shows most Tomato+ cells in the rejecting allograft coexpress the CD68 macrophage marker. In addition, 90% of the CD68+α-SMA+ cells in the rejecting allograft express Tomato, demonstrating a bone marrow macrophage origin. Graph shows quantification (mean±SEM) of these populations from a group of six transplant recipients.
Recipient Mice Lacking Smad3 Are Protected from MMT and Interstitial Fibrosis in Chronic Renal Allograft Rejection
Because TGF-β/Smad3 signaling plays a critical role in renal fibrosis,15 we investigated whether Smad3 regulates MMT and the development of interstitial fibrosis during chronic renal allograft rejection. BALB/c kidneys were transplanted into either Smad3 wild type (WT) or knockout (KO) C57BL/6J mice. Recipient mice received tacrolimus from the time of transplantation until being euthanized on day 28. Smad3 activation was prominent in allografts as determined by C-terminal phosphorylation and its nuclear localization in glomerular and tubulointerstitial cells (Supplemental Figure 2B). Renal allografts in Smad3 WT mice developed chronic allograft injury with marked interstitial fibrosis and a substantial infiltration of F4/80+ macrophages by day 28 as shown by Masson trichrome staining and immunostaining (Figure 4). By contrast, allografts in Smad3 KO mice showed significantly less fibrosis and a partial reduction in macrophage accumulation (Figure 4) without significantly affecting T cell infiltration (Supplemental Figure 5). The isograft controls showed no evidence of graft fibrosis or increased macrophage infiltration at day 28 (Figure 4), confirming that the fibrosis seen in the allograft model was due to rejection rather than ischemia/reperfusion injury or toxicity from tacrolimus treatment. Western blot and mRNA analysis confirmed that the increase in α-SMA and collagen I seen in renal allografts in Smad3 WT recipient mice was substantially reduced in Smad3 KO recipients (Figure 5).
Figure 4.

Deletion of Smad3 in the recipient alleviates interstitial fibrosis in mouse chronic renal allograft rejection. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. An isograft group was used as a control. (A) Masson trichrome stain. (B) Immunohistochemistry stain for collagen I deposition. (C) Immunohistochemistry stain for F4/80+ macrophage infiltration. Graphs show quantification of the area of collagen staining using Masson trichrome and collagen I staining and the number of F4/80+ macrophages. Data are mean±SEM for groups of 6–8 mice. *P<0.05, ***P<0.001 versus isograft control; ###P<0.001 versus Smad3 WT mice.
Figure 5.

Deletion of Smad3 in the recipient inhibits fibrosis in mouse chronic renal allograft rejection. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. An isograft group was used as a control. (A) α-SMA protein levels in renal allografts shown by Western blotting and α-SMA mRNA levels shown by real-time PCR. (B) Collagen I protein levels in renal allografts shown by Western blotting and collagen I mRNA levels shown by real-time PCR. Data are mean±SEM for groups of 6–8 mice. *P<0.05, ***P<0.001 versus isograft controls; #P<0.05, ##P<0.01, ###P<0.001 versus Smad3 WT mice.
Confocal microscopy identified the presence of many F4/80+α-SMA+ MMT cells in fibrosing renal allografts in Smad3 WT recipient mice, whereas only small numbers of F4/80+α-SMA+ MMT cells were seen in allografts in Smad3 KO recipients (Figure 6, AD). In addition, many F4/80+α-SMA+ MMT cells coexpressed collagen I, indicating that MMT cells contribute to the development of interstitial fibrosis in chronic renal allograft rejection (Figure 6B). An example of collagen I expression by a CD68+α-SMA+ MMT cell is shown by Z-stack images (Figure 6C, Supplemental Video 3). This histologic analysis was confirmed by flow cytometry. Two-color analysis found few CD68+α-SMA+ or CD68+collagen I+ cells in allograft controls (Figure 7, A and B). However, a significant population of CD68+α-SMA+ and CD68+collagen I+ cells was identified in allografts from Smad3 WT recipients (Figure 7, A and B), accounting for >55% of total α-SMA+ myofibroblasts and 60% of total collagen I+ cells (Figure 7, C and D). Interestingly, around 40%–50% of CD68+ macrophages infiltrating the kidney were capable of transitioning into collagen-producing MMT cells (Figure 7, E and F). In contrast, renal allografts in recipient mice lacking Smad3 were protected from interstitial fibrosis with a significant reduction in collagen-producing MMT cells (Figure 7).
Figure 6.

Deletion of Smad3 in the recipient inhibits MMT in mouse chronic renal allograft rejection. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. (A) Immunofluorescence shows many F4/80+ α-SMA+ MMT cells in renal allografts from Smad3 WT recipients. This was substantially inhibited in Smad3 KO recipients. (B) Three-color confocal microscopy identifies MMT cells in an allograft from a Smad3 WT recipient on the basis of coexpression of F4/80 (green), α-SMA (red), and collagen I (blue) as indicated by arrows. (C) Z-stack image shows collagen-producing MMT cells, which is also further illustrated in the Supplemental Material. Note that a CD68+α-SMA+ collagen I+ MMT cell is indicated by arrow. (D) Quantitative data for counting of the F4/80+, α-SMA+, and F4/80+α-SMA+ cell populations. Data are mean±SEM for groups of 6–8 mice. ##P<0.01, ###P<0.001 versus Smad3 WT mice.
Figure 7.

Flow cytometric analysis shows that deletion of Smad3 from recipient mice inhibits collagen-producing MMT cells in grafted kidneys with chronic renal allograft rejection. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. An isograft group was used as a control. (A) Analysis of macrophage (CD68) and myofibroblast (α-SMA) antigen expression identifies CD68+α-SMA+ MMT cells in renal allografts in Smad3 WT or KO recipients. (B) Identification of CD68+collagen I+ MMT cells in renal allografts in Smad3 WT or KO recipients. (C–F) Quantitative analysis of MMT cells as a percentage of the total α-SMA+ (C), collagen I+ (D), or CD68+ macrophage (E and F) populations. Data are mean±SEM for groups of six mice. #P<0.05, ##P<0.01, ###P<0.001 versus Smad3 WT mice.
MMT Cells in Chronic Renal Allograft Rejection Have a Predominant M2 Phenotype
Previous studies of interstitial fibrosis in the obstructed kidney identified that MMT cells are largely derived from M2 macrophages.14 In this study, we also found that MMT cells had a predominant M2 phenotype in both human and experimental chronic renal allograft rejection. Analysis by two-color immunofluorescence showed that the majority of CD68+ macrophages (75%) were M2 phenotype identified by coexpressing CD206, whereas a few CD68+iNOS+ cells were detected in patients with active chronic allograft rejection (Figure 8, A–C). Further analysis revealed that up to 40% of the M2 macrophages coexpressed α-SMA (CD206+α-SMA+ cells) (Figure 8, D and F), which was further illustrated by a Z-stack image (Supplemental Figure 6, Supplemental Video 4). In contrast, only a few iNOS+α-SMA+ cells were detected (Figure 8, E and F). In the mouse model of chronic renal allograft rejection, we found that >80% of infiltrating CD68+ macrophages in Smad3 WT recipients coexpressed CD206 whereas only a minority expressed iNOS (Figure 9). In addition, although there were fewer MMT cells in renal allografts from Smad3 KO recipient mice, there was also a reduction in CD206 expression in MMT cells in these allografts (Figure 10). This finding was confirmed by flow cytometry which showed that >80% of F4/80+α-SMA+ MMT cells in renal allografts from Smad3 WT recipients coexpressed CD206, and this CD206 expression was significantly reduced in MMT cells from renal allografts from Smad3 KO recipient mice (Figure 11A). Real-time PCR analysis of renal allograft tissue showed substantial increases in mRNA levels for both M1 (iNOS, MCP-1, IL-1β, TNF-α) and M2 (CD206, Arg-1, TGF-β1, IL-10, IL-4, IL-13) markers in renal allografts from Smad3 WT recipient mice and these were largely reduced in allografts from Smad3 KO recipient mice (Figure 11, B and C, Supplemental Figure 5B), although inhibition of IL-13 expression did not reach statistically significant level.
Figure 8.

The majority of MMT cells in human chronic active transplant rejection have an M2 phenotype. (A and B) Immunofluorescence shows many CD206+CD68+ M2 cells but few iNOS+CD68+ M1 cells in human chronic active transplant rejection. (C) Quantitative analysis of CD206+CD68+ cells and iNOS+CD68+ cells as a percentage of total CD68+ cells in human chronic active transplant rejection. (D and E) Two-color immunofluorescence shows that many α-SMA+ myofibroblasts (red) coexpress CD206 (M2 marker), but lack expression of iNOS (M1 marker) in human chronic active transplant rejection. Insert shows an example of a CD206+F4/80+α-SMA+ MMT cell (D). (F) Quantitative analysis of CD206+α-SMA+ cells and iNOS+α-SMA+ cells as a percentage of total α-SMA+ cells in human chronic active transplant rejection. Data are mean±SEM for six patients with chronic active allograft rejection. ###P<0.001 versus CD206+CD68+ cells/CD68+ cells; ***P<0.001 versus CD206+α-SMA+ cells/α-SMA+ cells.
Figure 9.

Renal allografts in Smad3-deficient mice have reduced M2 macrophage infiltration. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. (A) Immunofluorescence staining for total macrophages (CD68, green) and the M1 marker (iNOS). (B) Immunofluorescence staining for total macrophages (CD68, green) and the M2 marker (CD206). (C and D) Quantitative data analysis of the number of M1 (iNOS+CD68+) and M2 (CD206+CD68+) macrophages in renal allografts from Smad3 WT and Smad3 KO recipients. Data are mean±SEM. ##P<0.01, ###P<0.001 versus Smad3 WT mice.
Figure 10.

The majority of MMT cells in allografts in Smad3 WT recipients have an M2 phenotype. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. Immunofluorescence staining shows that (A) many α-SMA+ myofibroblasts (red) coexpress CD206 (M2 marker), but (B) lack expression of iNOS (M1 marker). (C) Quantitative analysis of CD206+α-SMA+ cells and iNOS+α-SMA+ cells as a percentage of total α-SMA+ cells in renal allografts from Smad3 WT and Smad3 KO recipients. Data are mean±SEM. ##P<0.01, ###P<0.001 versus Smad3 WT mice.
Figure 11.

Analysis of macrophage phenotype in grafted kidneys with chronic renal allograft rejection in Smad3 WT or KO recipient mice. Kidney allografts were transplanted into Smad3 WT or Smad3 KO recipient mice and examined 28 days later. An isograft group was used as a control. (A) Two-color flow cytometry analysis identifies F4/80+α-SMA+ MMT cells followed by analysis of CD206 expression by the gated MMT cells. Graph shows a significant reduction in the percentage of MMT cells expressing CD206 in renal allografts in Smad3 KO recipient mice. (B) Real-time PCR analysis of RNA extracted from whole renal allograft tissue for M1 macrophage markers (iNOS, MCP-1, IL-1β, and TNF-α). (C) Real-time PCR analysis for M2 macrophage markers (CD206, arginase-1, TGF-β1, and IL-10). Data are mean±SEM for groups of six mice. **P<0.01, ***P<0.001 versus isograft controls; #P<0.05, ##P<0.01 versus Smad3 WT mice.
Discussion
This study has identified that macrophages are an important source of myofibroblasts and contribute to the development of interstitial fibrosis in human and experimental chronic renal allograft rejection. Fate-mapping studies in mice clearly established bone marrow–derived macrophages as a source of myofibroblasts in chronic renal allograft rejection through a process of MMT. This MMT process occurred predominantly in M2 phenotype macrophages. In addition, the MMT process and development of interstitial fibrosis in renal allografts was mediated by TGF-β/Smad3 signaling.
It has been well recognized that inflammation is a key process leading to progressive renal fibrosis.21 Increasing evidence shows that intragraft macrophages, particularly activated macrophages, correlate with a poor outcome in renal transplantation in humans and animal models.22–29 In addition, α-SMA+ myofibroblast accumulation has been recognized as an early marker of chronic renal allograft dysfunction.30,31 Of note, CD68+ macrophages and α-SMA+ myofibroblasts colocalize in areas of active interstitial fibrosis in renal allografts.25,27,29 Although macrophages may stimulate renal fibrosis in an indirect fashion by producing proinflammatory and profibrotic cytokines and growth factors,13 a direct link between inflammatory macrophages and myofibroblast accumulation during chronic allograft rejection has remained undefined. In this study, we confirmed that intragraft CD68+ macrophages correlated with loss of allograft function and the development of interstitial fibrosis in patients with chronic active allograft rejection, consistent with previous observations.23–29 The new finding in this study was the identification that macrophages can transform into collagen-producing myofibroblasts, providing a novel mechanism for the direct involvement of macrophages in interstitial fibrosis during chronic renal allograft rejection. This MMT process was delineated by fate-mapping in combination with confocal microscopy and flow cytometry which demonstrated that 37% of the total α-SMA+ myofibroblast population in the renal allograft was derived from recipient bone marrow myeloid cells. The fate-mapping also confirmed the specificity of detecting MMT cells coexpressing macrophage and myofibroblast antigens using these techniques. Interestingly, CD68+ macrophages and CD68+α-SMA+ MMT cells, but not the total α-SMA+ myofibroblast population, correlated significantly with allograft function in patients with chronic active renal allograft rejection, suggesting that MMT is an important component of the link between inflammation and fibrosis in active fibrotic disease. This finding may explain the previous observations that fibrosis with inflammation predicts loss of renal transplant function better than the fibrosis index alone.30
The identification of MMT as an important pathway of myofibroblast accumulation during chronic renal allograft rejection confirms and extends our previous demonstration of MMT during the development of interstitial fibrosis in the mouse model of UUO.14 This study used fate-mapping to confirm the myeloid lineage of MMT cells and α-SMA+ myofibroblasts in chronic renal allograft rejection. In both studies, MMT was found to account for a substantial component of the myofibroblast population, supporting the argument that MMT is a common mechanism of interstitial fibrosis despite very different underlying causes of renal injury.
Macrophages undergoing MMT in both human and experimental chronic renal allograft rejection had a predominant M2 phenotype. Macrophages can promote both damage and repair in renal allografts depending upon the underlying insults and effectiveness of immunosuppression therapy.28–34 In kidney transplant rejection, proinflammatory M1 macrophages contribute to graft loss and are characterized by production of proinflammatory cytokines and chemokines (IL-1β, MCP-1, and TNF-α) and the generation of reactive oxygen species.32–36 By contrast, M2 macrophages are generally considered to be reparative and are induced upon exposure to IL-10 or TGF-β and express CD206 and Arg-1.28–33 Ongoing inflammation leads to chronic renal allograft rejection with fibrogenic M2 macrophages.28–33,37,38 However, macrophage heterogeneity and plasticity are major obstacles to determining whether the profibrotic M2 macrophages that undergo MMT are derived from M1 proinflammatory macrophages—thereby representing a change in function from acute inflammation to fibrosis during chronic rejection—or if they are derived from a distinct macrophage population.
It is well documented that TGF-β1 is an important mediator in renal fibrosis after renal transplantation.39 Smad3 is directly activated by the TGF-β receptor and binds directly to specific sites in promoter regions to induce transcription of genes involved in the fibrotic response, such as collagen I.15,40 In the context of renal fibrosis, Smad3 is necessary for the transition of tubular epithelial and endothelial cells into mesenchymal cells.16–20,41–43 In this study, we demonstrated that MMT and interstitial fibrosis in chronic renal allograft rejection depended upon Smad3 expression in recipient cells, whereas Smad3 expression in the intrinsic cells of the allograft (i.e., resident macrophages, epithelial cells, endothelial cells, pericytes, and resident fibroblasts) did not make a major contribution to interstitial fibrosis. The demonstration that Smad3 is required for the differentiation and transition of bone marrow macrophages into myofibroblasts and the development of interstitial fibrosis in chronic allograft rejection is consistent with our previous finding that TGF-β/Smad3 signaling plays a critical role in MMT during renal fibrosis in the obstructed kidney.14
In summary, the MMT process may be an important pathway, giving rise to myofibroblasts and contributing to interstitial fibrosis in chronic active renal allograft rejection. This transition involves macrophages with a predominant M2 phenotype. Finally, TGF-β/Smad3 signaling is a key regulatory mechanism promoting MMT and interstitial fibrosis during chronic renal allograft rejection.
Concise Methods
Human Renal Allograft Biopsy Samples
Renal biopsy samples and clinical data were collected from a total of 59 patients in the Prince of Wales Hospital, Hong Kong in 2013. Four-month and 2-year protocol biopsies were performed on all consenting patients. Clinically indicated allograft biopsies were performed if the serum creatinine rose ≥30% from baseline in 1 month, or proteinuria was ≥0.5 g/d, or the serum creatinine rose <30% from baseline for 3 months without a known cause. A pathologist scored the biopsy specimens according to Banff 2011. Among these, we selected patients with confirmed acute or chronic active rejection or chronic allograft injury (details in Table 1). Exclusion criteria were as follows: no sign of acute or chronic allograft injury, the presence of another solid organ transplant, the presence of other forms of CKD such as GN, and lack of available clinical data. Biopsy samples from 32 renal transplant recipients were examined, which included seven cases of acute rejection (including acute antibody-mediated rejection, acute T cell–mediated rejection, and borderline changes), 13 cases of chronic active rejection (including chronic active antibody-mediated rejection and chronic active T cell–mediated rejection), and 12 cases of interstitial fibrosis/tubular atrophy without evidence of rejection (details in Table 1). All biopsy specimens were scored according to the Banff 1997 classification.44 In addition, ten samples from biopsies performed immediately after transplantation were used as normal controls. Snap-frozen sections were fixed with acetone before being used for immunofluorescence or confocal microscopy. The serum creatinine level (micromoles per liter) was taken as the average of the three most recent measurements taken during the month before biopsy. The study was approved by The Joint Chinese University of Hong Kong–New Territories East Cluster Clinical Research Ethics Committee.
Mouse Kidney Transplantation
Kidneys from BALB/c mice were transplanted into C57Bl6/J recipients. Ectopic kidney transplantation was performed as previously described.45 In brief, the donated renal artery and vein were anastomosed to the recipient’s abdominal aorta and vena cava, respectively. The donated ureter was attached to the recipient’s bladder. Both native kidneys of the recipient mouse were left in place. The technical success rate was 80%. A chronic model of renal allograft rejection was created by treating all recipient mice with FK506 (1 mg/kg per day by intraperitoneal injection from the day of transplantation until being euthanized 4 weeks later).46 Lineage tracing of recipient myeloid cells was investigated by transplanting BALB/c donor kidneys into mice hemizygous for Lyz2-Cre and heterozygous for Rosa26-Tomato on the C57BL/6 strain (H-2b, both sexes, age 8 weeks, obtained from Jackson Laboratory).47 The role of Smad3 signaling was investigated by transplanting WT type BALB/c donor kidneys into Smad3 WT or Smad3 KO C57Bl6/J mice.48 Isograft controls involved transplantation of BABL/c donor kidneys into BALB/c recipient mice (which also received FK506 treatment). All transplant experiments were performed using sex-matched animals (n=7 in Smad3 WT and Smad3 KO groups, n=6 in Tomato groups and isografts). Animal studies were performed according to the Department of Health (Hong Kong) guidelines in Care and Use of Animals and the experimental protocol was approved by the Animal Experimentation Ethics Committee at the Chinese University of Hong Kong.
Histology and Immunohistochemistry in Human and Mouse Tissues
Kidney tissue was fixed in formalin and embedded in paraffin for Masson trichrome stain and immunohistochemistry staining as described previously.14,46 Periodate-lysine-paraformaldehyde–fixed cryostat sections of human and mouse tissues were stained with antibodies against CD68 (Dako, Glostrup, Denmark), α-SMA (Sigma, St Louis, MO), collagen I (South Biotech, Birmingham, AL), CD206 (Biolegend, CA), iNOS (BD Biosciences, San Jose, CA), and mouse F4/80 (Serotec, Oxford, UK). Slides were examined by fluorescence microscopy (Axioplan2 imaging; Carl Zeiss, Oberkoche, Germany) and confocal microscopy (LSM 510 META and LSM 880; Carl Zeiss, Oberkoche, Germany). Z-stack images were collected on the confocal microscope from single MMT cells coexpressing CD68 (F4/80), α-SMA, and collagen I. Positive-stained cells and the area of staining were quantified in at least ten consecutive high-power fields using Image-Pro Plus 7.0 software and expressed as number of positive cells per square centimeter or percent positive area, as previously described.14
Flow Cytometry
Mouse kidney graft tissue was digested using Blenzyme 4 (Roche Inc., Indianapolis, IN), sieved, and a cell suspension prepared. Cells were fixed for 30 minutes in IC Fixation Buffer (eBioscience), permeabilized in 1× Permeabilization Buffer (eBioscience) for 30 minutes, and then labeled with various combinations of the following antibodies: FITC-conjugated antibodies (α-SMA, collagen I, or CD206), APC-conjugated F4/80, Alexa647-conjugated CD68, and PE-Cy7–conjugated α-SMA in the dark for 1 hour at 4°C. Cells incubated with isotype-matched antibodies and unstained cells were used as negative controls. After being extensively washed, single cells were analyzed by FACSCalibur flowcytometer (BD Biosciences, San Jose, CA) as previously described.14 The initial gating, gating on single cells, and negative controls for all flow plots are shown in Supplemental Figure 7.
Real-Time PCR
RNA from whole tissue samples was isolated using the RNeasy kit (Qiagen Inc., Valencia, CA). mRNA levels of α-SMA, collagen I, iNOS, IL-1β, MCP-1, TNF-α, CD206, Arg-1, IL-10, TGF-β1, IL-4, IL-13, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were detected by real-time PCR with the Opticon 2 Real-Time PCR detector (Bio-Rad) using the primers as previously described14,49,50 and as follows: iNOS: forward 5′-CAGCTGGGTCGTACAAAC-3′, reverse 5′-CATTGGAAGTGAAGCGTTT-3′; CD206: forward 5′-GTCAGAACAGACTGCGTGGA-3′, reverse 5′-AGGGATCGCCTGTTTTCCAG-3′; IL-13: forward 5′-GGAGCTGAGCAACATCACACA-3′, reverse 5′-GGTCCTGTAGATGGTGGCATTGCA-3′.
Western Blot Analysis
Protein from renal graft tissues was extracted with RIPA lysis buffer for Western blot analysis as described previously.46,49 Briefly, after blocking, membranes were incubated overnight at 4°C with primary antibodies against α-SMA (Sigma), collagen I (South Biotech), or GAPDH. After washing, membranes were incubated with LI-COR IRDye 800–labeled secondary antibodies (Rockland). The signal was detected with Odyssey Infrared Imaging System (Li-COR Biosciences, Lincoln, NE) and quantified against the internal loading control GAPDH with ImageJ version 1.48 (National Institutes of Health, Bethesda, MD).
Statistical Analyses
Data are expressed as mean±SEM. Statistical analyses using the student t test and one-way ANOVA followed by Newman–Keuls multiple comparison test were performed with GraphPad Prism 5.0. Correlation analysis used the Pearson correlation coefficient. A P value <0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
The authors thank Dr. Hao Yang and Dr. Ying-Ying Lu from the Kidney Disease Center, The First Affiliated Hospital, Zhejiang University for assistance in revising the manuscript and Dr. Jun Xiao for technical help in confocal microscopy.
This study was supported by the Research Grants Council of Hong Kong (GRF 468711, CUHK3/CRF/12R), the Focused Investment Scheme A from the Chinese University of Hong Kong, the Major State Basic Research Development Program of China (973 program, No. 2012CB517705), the National Nature Science Foundation of China (2011BAI10B07, 2012CB517603, 2012AA02A512, 2014KYA057), and the National Fund Committee of China (81470938). D.J.N.-P. is supported by the National Health Medical Research Council of Australia.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016050573/-/DCSupplemental.
References
- 1.Boor P, Floege J: Renal allograft fibrosis: Biology and therapeutic targets. Am J Transplant 15: 863–886, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Racusen LC, Regele H: The pathology of chronic allograft dysfunction. Kidney Int Suppl 78: S27–S32, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Haas M: Chronic allograft nephropathy or interstitial fibrosis and tubular atrophy: What is in a name? Curr Opin Nephrol Hypertens 23: 245–250, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Eddy AA: The origin of scar-forming kidney myofibroblasts. Nat Med 19: 964–966, 2013 [DOI] [PubMed] [Google Scholar]
- 5.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ: Diverse origins of the myofibroblast—implications for kidney fibrosis. Nat Rev Nephrol 11: 233–244, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS: Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broekema M, Harmsen MC, van Luyn MJ, Koerts JA, Petersen AH, van Kooten TG, van Goor H, Navis G, Popa ER: Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol 18: 165–175, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Chen G, Lin SC, Chen J, He L, Dong F, Xu J, Han S, Du J, Entman ML, Wang Y: CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol 22: 1876–1886, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan J, Zhang Z, Yang J, Mitch WE, Wang Y: JAK3/STAT6 stimulates bone marrow-derived fibroblast activation in renal fibrosis. J Am Soc Nephrol 26: 3060–3071, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Deane JA, Campanale NV, Bertram JF, Ricardo SD: The contribution of bone marrow-derived cells to the development of renal interstitial fibrosis. Stem Cells 25: 697–706, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Lin SC, Chen G, He L, Hu Z, Chan L, Trial J, Entman ML, Wang Y: Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol 24: 1644–1659, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikolic-Paterson DJ, Wang S, Lan HY: Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl (2011) 4: 34–38, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, Yang C, Huang XR, Xiao J, Wang YY, Ka SM, Tang YJ, Chung AC, To KF, Nikolic-Paterson DJ, Lan HY: TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 7: 8809–8822, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng XM, Tang PM, Li J, Lan HY: TGF-β/Smad signaling in renal fibrosis. Front Physiol 6: 82, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A: Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112: 1486–1494, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Huang XR, Lan HY: Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am J Physiol Renal Physiol 302: F986–F997, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF: Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59: 2612–2624, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY: Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol 221: 390–401, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Duan WJ, Yu X, Huang XR, Yu JW, Lan HY: Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. Am J Pathol 184: 2275–2284, 2014 [DOI] [PubMed] [Google Scholar]
- 21.Meng XM, Nikolic-Paterson DJ, Lan HY: Inflammatory processes in renal fibrosis. Nat Rev Nephrol 10: 493–503, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Matheson PJ, Dittmer ID, Beaumont BW, Merrilees MJ, Pilmore HL: The macrophage is the predominant inflammatory cell in renal allograft intimal arteritis. Transplantation 79: 1658–1662, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Toki D, Zhang W, Hor KLM, Liuwantara D, Alexander SI, Yi Z, Sharma R, Chapman JR, Nankivell BJ, Murphy B, O’Connell PJ: The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant 14: 2126–2136, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Baek J-H, Zeng R, Weinmann-Menke J, Valerius MT, Wada Y, Ajay AK, Colonna M, Kelley VR: IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J Clin Invest 125: 3198–3214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guillén-Gómez E, Guirado L, Belmonte X, Maderuelo A, Santín S, Juarez C, Ars E, Facundo C, Ballarín JA, Vidal S, Díaz-Encarnación MM: Monocyte implication in renal allograft dysfunction. Clin Exp Immunol 175: 323–331, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan HY, Yang N, Brown FG, Isbel NM, Nikolic-Paterson DJ, Mu W, Metz CN, Bacher M, Atkins RC, Bucala R: Macrophage migration inhibitory factor expression in human renal allograft rejection. Transplantation 66: 1465–1471, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Brown FG, Nikolic-Paterson DJ, Metz C, Bucala R, Atkins RC, Lan HY: Up-regulation of macrophage migration inhibitory factor in acute renal allograft rejection in the rat. Clin Exp Immunol 118: 329–336, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikezumi Y, Suzuki T, Yamada T, Hasegawa H, Kaneko U, Hara M, Yanagihara T, Nikolic-Paterson DJ, Saitoh A: Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatr Nephrol 30: 1007–1017, 2015 [DOI] [PubMed] [Google Scholar]
- 29.Grimm PC, McKenna R, Nickerson P, Russell ME, Gough J, Gospodarek E, Liu B, Jeffery J, Rush DN: Clinical rejection is distinguished from subclinical rejection by increased infiltration by a population of activated macrophages. J Am Soc Nephrol 10: 1582–1589, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Park WD, Griffin MD, Cornell LD, Cosio FG, Stegall MD: Fibrosis with inflammation at one year predicts transplant functional decline. J Am Soc Nephrol 21: 1987–1997, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badid C, Desmouliere A, Babici D, Hadj-Aissa A, McGregor B, Lefrancois N, Touraine JL, Laville M: Interstitial expression of alpha-SMA: An early marker of chronic renal allograft dysfunction. Nephrol Dial Transplant 17: 1993–1998, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Salehi S, Reed EF: The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant 20: 446–453, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwan T, Wu H, Chadban SJ: Macrophages in renal transplantation: Roles and therapeutic implications. Cell Immunol 291: 58–64, 2014 [DOI] [PubMed] [Google Scholar]
- 34.Ma FY, Woodman N, Mulley WR, Kanellis J, Nikolic-Paterson DJ: Macrophages contribute to cellular but not humoral mechanisms of acute rejection in rat renal allografts. Transplantation 96: 949–957, 2013 [DOI] [PubMed] [Google Scholar]
- 35.Jose MD, Ikezumi Y, van Rooijen N, Atkins RC, Chadban SJ: Macrophages act as effectors of tissue damage in acute renal allograft rejection. Transplantation 76: 1015–1022, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Qi F, Adair A, Ferenbach D, Vass DG, Mylonas KJ, Kipari T, Clay M, Kluth DC, Hughes J, Marson LP: Depletion of cells of monocyte lineage prevents loss of renal microvasculature in murine kidney transplantation. Transplantation 86: 1267–1274, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Tinckam KJ, Djurdjev O, Magil AB: Glomerular monocytes predict worse outcomes after acute renal allograft rejection independent of C4d status. Kidney Int 68: 1866–1874, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Pilmore HL, Painter DM, Bishop GA, McCaughan GW, Eris JM: Early up-regulation of macrophages and myofibroblasts: A new marker for development of chronic renal allograft rejection. Transplantation 69: 2658–2662, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Campistol JM, Iñigo P, Larios S, Bescos M, Oppenheimer F: Role of transforming growth factor-beta1 in the progression of chronic allograft nephropathy. Nephrol Dial Transplant 16[Suppl 1]: 114–116, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB, Mauviel A: SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc Natl Acad Sci USA 95: 14769–14774, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM, Tang MJ: Transforming growth factor-beta1 induces Smad3-dependent beta1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am J Pathol 177: 1743–1754, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y, Mao H, Li S, Cao S, Li Z, Zhuang S, Fan J, Dong X, Borkan SC, Wang Y, Yu X: HSP72 inhibits Smad3 activation and nuclear translocation in renal epithelial-to-mesenchymal transition. J Am Soc Nephrol 21: 598–609, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN, Lan HY: Mechanism of chronic aristolochic acid nephropathy: Role of Smad3. Am J Physiol Renal Physiol 298: F1006–F1017, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, Croker BP, Demetris AJ, Drachenberg CB, Fogo AB, Furness P, Gaber LW, Gibson IW, Glotz D, Goldberg JC, Grande J, Halloran PF, Hansen HE, Hartley B, Hayry PJ, Hill CM, Hoffman EO, Hunsicker LG, Lindblad AS, Marcussen N, Mihatsch MJ, Nadasdy T, Nickerson P, Olsen TS, Papadimitriou JC, Randhawa PS, Rayner DC, Roberts I, Rose S, Rush D, Salinas-Madrigal L, Salomon DR, Sund S, Taskinen E, Trpkov K, Yamaguchi Y: The Banff 97 working classification of renal allograft pathology. Kidney Int 55: 713–723, 1999 [DOI] [PubMed] [Google Scholar]
- 45.Kalina SL, Mottram PL: A microsurgical technique for renal transplantation in mice. Aust N Z J Surg 63: 213–216, 1993 [DOI] [PubMed] [Google Scholar]
- 46.Moffatt SD, McAlister V, Calne RY, Metcalfe SM: Potential for improved therapeutic index of FK506 in liposomal formulation demonstrated in a mouse cardiac allograft model. Transplantation 67: 1205–1208, 1999 [DOI] [PubMed] [Google Scholar]
- 47.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I: Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277, 1999 [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C: Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J 18: 1280–1291, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lan HY, Mu W, Nikolic-Paterson DJ, Atkins RC: A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: Use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem 43: 97–102, 1995 [DOI] [PubMed] [Google Scholar]
- 50.Huang XR, Chung AC, Zhou L, Wang XJ, Lan HY: Latent TGF-beta1 protects against crescentic glomerulonephritis. J Am Soc Nephrol 19: 233–242, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.