

Abstract
Purpose
Ibrutinib inhibits Bruton tyrosine kinase (BTK) by irreversibly binding to the Cys-481 residue in the enzyme. However, ibrutinib also inhibits several other enzymes that contain cysteine residues homologous to Cys-481 in BTK. Patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia (CLL) demonstrate a high overall response rate to ibrutinib with prolonged survival. Acalabrutinib, a selective BTK inhibitor developed to minimize off-target activity, has shown promising overall response rates in patients with relapsed/refractory CLL. A head-to-head comparison of ibrutinib and acalabrutinib in CLL cell cultures and healthy T cells is needed to understand preclinical biologic and molecular effects.
Experimental Design
Using samples from patients with CLL, we compared the effects of both BTK inhibitors on biologic activity, chemokine production, cell migration, BTK phosphorylation, and downstream signaling in primary CLL lymphocytes and on normal T-cell signaling to determine effects on other kinases.
Results
Both BTK inhibitors induced modest cell death accompanied by cleavage of PARP and caspase 3. Production of CCL3 and CCL4 chemokines and pseudoemperipolesis were inhibited by both drugs to a similar degree. These drugs also showed similar inhibitory effects on phosphorylation of BTK and downstream S6 and ERK kinases. By contrast, off-target effects on SRC-family kinases were more pronounced with ibrutinib than acalabrutinib in healthy T lymphocytes.
Conclusion
Both BTK inhibitors show similar biological and molecular profile in primary CLL cells but appear different on their effect on normal T-cells.
Keywords: ibrutinib, acalabrutinib, chronic lymphocytic leukemia, apoptosis, Bruton tyrosine kinase
Introduction
The B-cell receptor (BCR) pathway is critical for the proliferation, maintenance, and survival of B cells (1). Bruton tyrosine kinase (BTK) is pivotal in the BCR axis (2) and is activated when BCR is stimulated. BTK is a cytoplasmic protein that is expressed in hematopoietic cells in general and B-lymphoid cells in particular. Because of its critical role in BCR axis signaling, importance of the BCR pathway in B-cell proliferation and maintenance, and its selective expression in B-cells, BTK is an attractive therapeutic target. The concept of inhibiting BTK to treat B-cell malignancies stems from observations that agammaglobulinemia (3) and immunodeficiency diseases (4) in pediatric patients are associated with decreased number of B-cells and their function (5). These attributes were due to loss of activity of a cytoplasmic kinase, which was cloned and termed BTK (2,6). Similar to humans, mice lacking BTK activity develop X chromosome–linked immunodeficiency syndrome (7).
BTK is expressed in normal and malignant B-cells. Furthermore, upon crosslinking and activation of BCR, BTK protein levels have been demonstrated to increase (8) in murine normal B cells and through CXCR4 and CXCL12 signaling in murine chronic lymphocytic leukemia (CLL) lymphocytes (9). In human specimens, BTK mRNA levels are higher in CLL lymphocytes compared with normal B cells, although protein levels vary (2,10). An increase in BCR signaling pathway proteins, such as LYN, SYK, and BTK, was observed in CLL cells derived from lymph node tissue samples that had activated NF-κB signatures (11). BTK plays a pivotal role in the BCR pathway, promoting proliferation, survival, maintenance, and migration of malignant B cells. Collectively, these observations provide strong rationale for targeting BTK in B-cell malignancies, including CLL.
Ibrutinib is a first-in-class BTK inhibitor that irreversibly binds to cysteine (Cys)-481 in the kinase domain and potently blocks its enzymatic activity (12). Ibrutinib decreases proliferation, moderately increases apoptosis, attenuates survival signals in stroma or nurse-like cells, and reduces cell adhesion and chemokine production in preclinical models (10,12,13). These effects reflect BCR pathway inhibition, which has also been demonstrated by reduced levels of phospho-BTK, diminished downstream ERK-MAP kinase pathway signaling, and altered expression of antiapoptotic proteins (10,12,14,15). Clinical and laboratory investigations of ibrutinib during phase I (16) and phase II (17) studies demonstrated clinical efficacy of ibrutinib, identified a dose, and defined favorable pharmacodynamics, with more than 95% occupancy of the cellular BTK protein at 4 and 24 hours after intake of ibrutinib.
Ibrutinib demonstrated compelling activity in the clinic. Even during phase I investigations (16), it became clear that this oral once-a-day drug is highly effective. Responses and overall and progression-free survival durations were clearly superior to standard treatment with limited toxicities observed during phase II and phase III trials (17) and long-term follow-up clinical investigations (18). Even elderly patients with CLL tolerated continuous therapy with ibrutinib (19). Ibrutinib also demonstrated clinical activity in previously treated patients (n=144) with CLL and del(17p) in the phase II clinical trial RESONATE-17, achieving high overall response rate (ORR) and progression free survival (PFS) rates (20). These responses resulted in approval of ibrutinib for previously treated CLL patients (21). In phase III setting, compared with ofatumumab, ibrutinib demonstrated an improved response rates, overall survival (OS) rate in previously treated patients with CLL (n=391) (22). Consistent with these data, even in previously untreated patients, improved response rate continued when ibrutinib was compared with chlorambucil (n=269) during the randomized phase III clinical trial RESONATE-2 (23).
Overall, ibrutinib is well tolerated with limited untoward toxicity. Limited patients, generally 5%, had bleeding and atrial fibrillation. While these toxicities were low, they were higher than the comparator arm of ofatumumab (22) or chlorambucil (23). Furthermore, for CLL patients with poor prognosis such as 17p deletion (n=144), the incidence of major bleeding increased to 9% and atrial fibrillation to 7% (20) .
On-target ibrutinib binds to Cys-481 of the BTK. However, off-target ibrutinib also binds covalently to several other homologous cysteine-containing kinases and non-covalently to other kinases which may play a role in toxicities, such as bleeding (24). For example, SRC family kinases play an important role in platelet activation (25) (26) and aggregation (27). In fact, phosphorylation of Src at tyrosine 418 is reported in aggregated platelets (28) and platelet signaling through phospho-SRC (and SYK) is required for stable platelet adhesion to lymphatic endothelial cells (29). Even in whole cells, the impact of ibrutinib on other cysteine-containing kinases, such as ITK/EGFR and down-stream signaling, has been observed (30,31). T-cell receptor–mediated signal transducers downstream of ITK and CLEC-2 receptor mediated aggregation and Syk-phosphorylation in platelets were inhibited by ibrutinib (32,33). Similarly, phosphorylation of TEC was decreased by ibrutinib (32). The contributions of these off-target effects to ibrutinib-mediated activity and toxicity (34) remain unclear.
Overall, these clinical response data with ibrutinib strongly validated BTK as a target and ibrutinib as a therapeutic intervention for CLL. Toxicity, albeit limited, and inhibition of other cysteine containing kinases, underscored the need for more selective BTK inhibitor (35).
Acalabrutinib (ACP-196) is a selective BTK inhibitor in clinical development for the treatment of hematologic malignancies. Acalabrutinib has a unique reactive butynamide group, which forms a covalent bond with the Cys-481 residue in BTK (Supplemental Figure 1A), as does the acrylamide group of ibrutinib (Supplemental Figure 1B). The half maximal inhibitory concentration (IC50) of ibrutinib for the BTK protein is 1.5 nM compared with 5.1 nM for acalabrutinib, indicating stronger BTK inhibition with ibrutinib in biochemical assays. However, acalabrutinib is a more selective BTK inhibitor. For example, the IC50 values for inhibition of 8 of 9 kinases that contain cysteine residues that align with Cys-481 in BTK were less than 10 nM for ibrutinib; however, none of the 9 kinases had IC50 values less than 10 nM for acalabrutinib. Indeed, a recent clinical report demonstrated that toxicities, including atrial fibrillation and bleeding, which are associated with ibrutinib, were not observed in patients with relapsed/refractory CLL treated with acalabrutinib for a median of 14 months. Acalabrutinib monotherapy achieved an ORR of 95% in patients with relapsed/refractory CLL (n=60 evaluable), including patients with del(17p) (n = 18) (32).
In the current study, we compared ibrutinib and acalabrutinib to determine their effects on biologic and molecular activities, including inhibition of BCR/BTK axis and downstream signaling, induction of apoptosis, chemokine production, and migration in CLL cells, and off-target signaling in healthy T cells. Our data suggest that acalabrutinib and ibrutinib demonstrate similar molecular and biologic consequences in primary CLL cells but different effects on signaling events, including LCK and SRC kinase phosphorylation, in primary T-lymphocytes. While ibrutinib inhibited LCK and SRC phosphorylation in normal T cells, acalabrutinib did not reach IC50 values for these enzymes at physiologic relevant concentrations. Collectively, our data establish a similar preclinical activity profile of acalabrutinib and ibrutinib in primary CLL cells but a differentiated one in T-cells.
Methods
Patient sample collection and cell culture
Peripheral blood was obtained from patients with CLL who provided written informed consent as part of a protocol approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center in accordance with the Declaration of Helsinki. Baseline characteristics of patients are summarized in Supplemental Table 1. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density centrifugation (Atlanta Biologicals, Norcross, GA) and suspended in Roswell Park Memorial Institute 1640 (RPMI 1640) media supplemented with 10% human serum (Sigma-Aldrich, St. Louis, MO). PBMCs consisted primarily (>90%) of CLL lymphocytes based on CD19 and CD5 positivity. BCR signaling was stimulated by incubating CLL cells with goat F(ab’)2 fragments of human immunoglobulin M (IgM; MP Biomedicals, Santa Ana, CA).
Drugs
Acalabrutinib was provided by Acerta Pharma (Redwood City, CA). Ibrutinib was purchased from Selleck Chemicals (Houston, TX). Stock solutions of both drugs were made in dimethyl sulfoxide (DMSO) and used generally at equimolar concentrations. Time-matched, DMSO-treated (i.e., vehicle-treated) cells were used as controls.
Cytotoxicity assays
CLL cells were treated with 1 μM or 3 μM acalabrutinib or ibrutinib for 24, 48, or 72 hours. Cells were then stained with annexin V and propidium iodide and counted by flow cytometry as described previously (36). Cell death in drug-treated samples was normalized by subtracting the cell death of control vehicle-treated, time-matched CLL samples, which ranged from 5% to 20%.
Chemokine assays
CCL3 (Mip-1α) and CCL4 (Mip-1β) levels in the media of CLL cell cultures exposed to α-IgM stimulation and BTK inhibitors were measured using a Quantikine enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN) (37). Concentrations were extrapolated from a standard curve and expressed in pg/mL.
Migration assays
For pseudoemperipolesis, NKTert stromal cells were seeded onto collagen-coated plates. The next day, CLL cells were incubated with or without drug and layered on stroma. After four hours, cells that had not migrated into the stromal cell layer were removed by vigorously washing with RPMI 1640. The stromal cell layer containing transmigrated cells was detached with trypsin/ethylenediaminetetraacetic acid. CLL cells were resuspended in media and counted by flow cytometry for 20 seconds at 60 μL/min (37).
Immunoblot analyses
Immunoblots were performed with cellular protein extracts and visualized with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) (37). Antibodies for specific proteins are listed in Supplemental Table 2.
Effect on Phospho-LCK and SRC in healthy T-cells
PBMCs from healthy patients were treated with each drug for 2 hours and stimulated with 3.3 mM H2O2 for 10 minutes at 37°C. The cells were then fixed with 1.6% paraformaldehyde solution for 10 minutes at 37°C and permeabilized with 100% methanol overnight at –80°C. Cells were stained with antibodies against phospho-LCK (#558552; BD Biosciences, Franklin Lakes, NJ), phospho-SRC (#560096; BD Biosciences), cleaved Poly(ADP-ribose) polymerase (PARP) (#560640; BD Biosciences) and CD3 (#562426; BD Biosciences). A similar assay was previously described for T-cell phosphoflow analysis using CD3/CD28 and H2O2 stimuli (38,39). Data were acquired on a FACSVerse flow cytometer and analyzed with FCS Express (version 4; BD Biosciences).
Effect on phospho-ITK and PLC γ1 in T-cell line
Jurkat cells were treated with acalabrutinib, ibrutinib, or DMSO control for 2 hours and stimulated with 3.3mM H2O2 for 10 min. Following stimulation, cells were lysed with Cell Extraction Buffer (Thermo Fisher #FNN0011) containing protease inhibitors on ice for 30 min. Lysates were pre-cleared and incubated with Protein A sepharose beads (GE Healthcare #17-5138-01) and ITK antibody (Abcam #ab32507) overnight at 4C. The next day, supernatant from the beads was collected, and a Western blot for phospho-PLCγ1 (Cell Signaling #14008S) was performed on the supernatant. ITK bound to the beads was eluted with Sample Buffer (Life Technologies #B0007) containing reducing reagent (Life Technologies #B0009), and a Western blot was performed with 4G10 antibody (Millipore #05-321) to detect phospho-ITK, with total ITK (Abcam #ab32507) as control. Ponceau staining was used to verify even loading after transfer. Densitometry was performed using GelQuant software (version 1.8.2).
Statistical analysis
Paired two-tailed Student t-tests were performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA) to compare DMSO-treated cells with drug-treated cells. Similarly, paired two-tailed students t-tests were used to compare ibrutinib-treated cells with acalabrutinib-treated cells at either 1 μM or 3 μM concentration of the drug.
Results
Ibrutinib and acalabrutinib induce apoptosis
In vitro experiments were performed with 1 μM or 3 μM ibrutinib or acalabrutinib. The peak plasma concentrations of ibrutinib in CLL patients following an oral dose of 560 mg ranges from 150-200 ng/mL (340 nM – 450 nM total ibrutinib levels. The peak plasma concentrations of acalabrutinib following a single 100 mg dose are 520 +/- 286 ng/mL (1118 nM total acalabrutinib. Hence, the concentration of drug in the experiments (1 μM or 3 μM total) were selected to span a range similar to, or a half-log greater than, total concentrations of ibrutinib or acalabrutinib achieved during clinical trials (17,32).
Without stimulation of the BCR pathway, at 1 μM and 3 μM, compared with controls, ibrutinib and acalabrutinib induced modest yet statistically significant (p values range from 0.05 to <0.0001) increases in apoptosis rates in primary CLL cells at 24, 48, and 72 hours of treatment (Figure 1A-C). Median cell viability for samples treated with 1 μM ibrutinib were 95%, 90%, and 88% at 24, 48, and 72 hours, respectively. Median cell viability rates for samples treated with 1 μM acalabrutinib were 98%, 96%, and 93% at 24, 48, and 72 hours, respectively. While the differences between treatment groups were only 3% to 6% at each time point, they were statistically significant. For example, the p values were 0.0048, 0.0041, and 0.0065 at 24, 48, and 72 hours. Similar small differences between ibrutinib- and acalabrutinib-induced apoptosis were also observed at 3 μM of the inhibitors. In general, at each concentration and time point, ibrutinib induced consistently and significantly higher apoptosis of CLL cells than acalabrutinib. As expected, IgM stimulation resulted in a survival advantage, with moderate cell death due to both inhibitors (Figure 1D-F). Prognostic factors such as IGVH mutation status (9 mutated versus 5 unmutated), ZAP-70 positivity (6 positive and 9 negative), B2M level (9 less than 2.5 and 8 more than 2.5) and other characteristics such as prior therapy (7 treated and 12 previously untreated), absolute lymphocyte count (11 less than 100,000 and 7 more than 100,000 ALC/μl) , age (10 less than 60 and 10 more than 60 years old), and gender (13 male and 7 female), did not appear to affect acalabrutinib-mediated cell death (p value always >0.2; data not shown).
Figure 1. Comparison of ibrutinib and acalabrutinib-induced apoptosis and influence of BCR pathway stimulation.
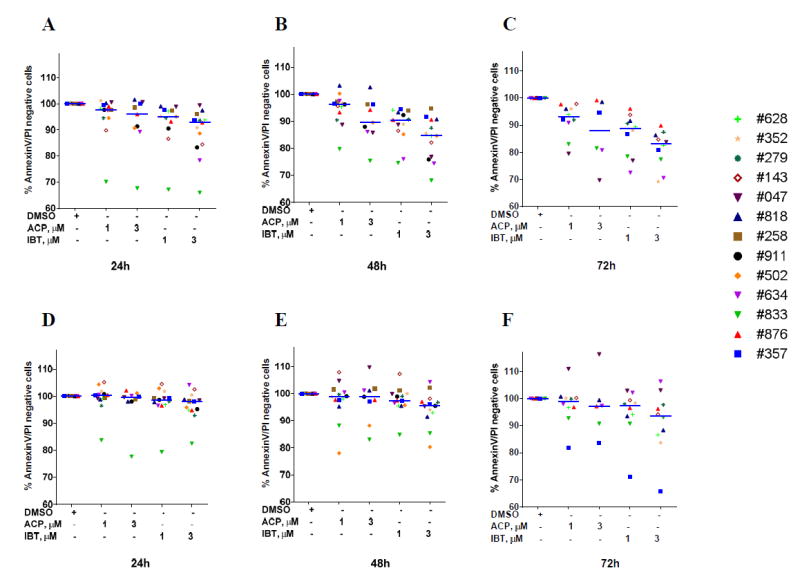
A-C. Dose- and time-dependent induction of apoptosis of CLL primary lymphocytes treated with ibrutinib (IBT) or acalabrutinib (ACP). Freshly isolated CLL cells from 13 patients were incubated with DMSO alone (control) or 1 μM or 3 μM IBT or ACP for 24 (A), 48 (B), or 72 (C) hours. Cells were washed and stained with annexin V and propidium iodide and analyzed by flow cytometry. To determine p values, either treated cells were compared with controls or IBT-treated were compared with ACP-treated. D-F. Impact of BCR pathway stimulation by IgM on dose- and time-dependent induction of apoptosis of CLL primary lymphocytes treated with IBT or ACP. Freshly isolated CLL cells from 13 patients were incubated with IgM followed by incubation with DMSO alone (control) or 1 μM or 3 μM IBT or ACP for 24, 48, or 72 hours. Cells were washed and stained with annexin V and propidium iodide and analyzed by flow cytometry. Each data point represents an individual patient and is denoted by a three-digit number, as shown in Supplemental Table 1.
PARP cleavage increased from 0.8- to 5.3-fold in CLL samples (n = 5) treated with 1 μM or 3 μM ibrutinib or acalabrutinib compared with time-matched vehicle treated control (Figure 2A). There were not significant differences in PARP cleavage between cells treated with 1 μM (p = 0.14) or 3 μM (p = 0.57) ibrutinib vs acalabrutinib (Figure 2B). Compared with time-matched, vehicle-treated controls, cleaved caspase 3 levels increased from 0.8- to almost 3-fold without significant differences between ibrutinib- and acalabrutinib-treated cells (p = 0.50 and p = 0.08 at 1 μM and 3 μM, respectively) (Figure 2C). Collectively, these data indicate that the BTK inhibitors ibrutinib and acalabrutinib induced moderate levels of apoptotic cell death (<15%).
Figure 2. Cell death induced by ibrutinib and acalabrutinib was associated with PARP and caspase 3 cleavage.
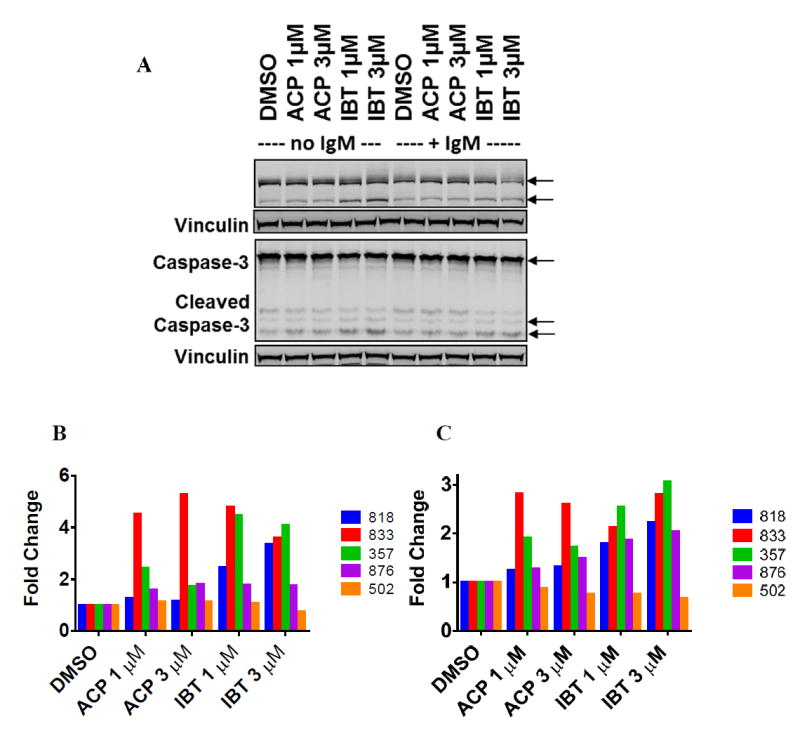
A. Increase in PARP and caspase 3 cleavage after treatment with BTK inhibitors without IgM stimulation (lanes 1-5) and with IgM stimulation (lanes 6-10). Leukemic lymphocytes were treated with DMSO only (control) or with 1 μM or 3 μM ibrutinib or acalabrutinib for 48 hours with or without BCR pathway stimulation with IgM. The protein lysates were immunoblotted with total and cleaved PARP or caspase 3 antibodies as described in the Methods. Vinculin was used as a loading control. B and C. Immunoblots were analyzed for five patients, and cleaved PARP (B) and caspase 3 (C) bands were quantitated for CLL cells treated with BTK inhibitors without IgM stimulation. Values represent fold change relative to control.
Ibrutinib and acalabrutinib inhibit chemokine production and migration
BCR activation increases production of the chemokines CCL3 and CCL4 in CLL cells (37,40). Conversely, inhibition of BCR signaling by ibrutinib decreases levels of these chemokines (12). As shown in Figure 3, IgM-induced production of CCL3 was inhibited by ibrutinib and acalabrutinib (Figure 3A-D). CCL3 levels did not significantly differ between ibrutinib-treated and acalabrutinib-treated cells without IgM (p = 0.355) or with IgM (p = 0.170). CCL4 levels (Figure 3E-H) were reduced in cells treated with ibrutinib or acalabrutinib compared with vehicle control without significant differences between drug treatment groups (p = 0.246). Both drugs showed comparable inhibition (p = 0.747) of IgM-stimulated CCL4 production.
Figure 3. Comparison of ibrutinib and acalabrutinib-mediated inhibition of chemokine production and pseudoemperipolesis.

A-D. Effect of BTK inhibitors on CCL3 production and secretion from CLL cells. Freshly isolated CLL cells from four patients were either untreated (A-B) or stimulated with IgM (C-D). Cells were treated with DMSO or 1 μM ibrutinib (IBT) or acalabrutinib (ACP) for 24 hours. CCL3 concentrations in media were measured by enzyme-linked immunosorbent assay as described in the Methods section. Statistical analyses were performed to compare ibrutinib with acalabrutinib without IgM (p = 0.355) or with IgM (p = 0.170). E-F. Effect of BTK inhibitors on CCL4 production and secretion from CLL cells. Freshly isolated CLL cells from four patients were either untreated (E-F) or stimulated with IgM (G-H). Cells were treated with DMSO or 1 μM ibrutinib (IBT) or acalabrutinib (ACP) for 24 hours. CCL4 concentrations in media were measured by enzyme-linked immunosorbent assay as described in the Methods section. Statistical analyses were performed to compare ibrutinib with acalabrutinib without IgM (p = 0.246) or with IgM (p = 0.747). I. Impact of BTK inhibitors on pseudoemperipolesis. Primary CLL cells were treated with DMSO, acalabrutinib, or ibrutinib at different concentrations and placed on stromal cells for 4 hours. Cells were removed from the suspension. Cells that migrated under the stroma were counted. Values represent the percentage of cells that migrated relative to DMSO controls. Statistical analyses were performed to compare 1 μM ibrutinib with 1 μM acalabrutinib (p = 0.678) or 3 μM ibrutinib with 3 μM acalabrutinib (p = 0.887).
Activation of BCR signaling by chemokines promotes cell migration beneath the stroma, also known as pseudoemperipolesis (13,40-42). BTK inhibition impairs the expression and function of the chemokine CXCR4, resulting in retention of CLL cells in niches (9); similarly, chemokine-controlled adhesion and migration of CLL lymphocytes are inhibited by ibrutinib (15). In the present study, CLL cells (from seven patients) cultured with stroma showed maximum pseudoemperipolesis, which was inhibited by acalabrutinib and ibrutinib (Figure 3I) without significant differences between drug treatments at 1 μM (p = 0.678) or 3 μM (p = 0.887).
Ibrutinib and acalabrutinib inhibit BTK phosphorylation and downstream signaling in CLL cells
Ibrutinib and acalabrutinib reduced phosphorylation of BTK protein levels, as shown by immunoblots (Figure 4A). Both drugs appear to impede the BTK signaling pathway by decreasing phosphorylation of ERK and S6 (Figure 4A). However, phosphorylation of AKT at Thr308, was not affected by ibrutinib or acalabrutinib. Quantitation of immunoblots representing five patient samples demonstrated that in the absence of IgM stimulation BTK phosphorylation was significantly (p<0.0001 with ACP at both concentrations; p=0.033 and p=0.0005 with IBT at 1 and 3 μM, respectively) reduced by both drugs relative to control and to similar extents (Figure 4B). Stimulation with IgM mitigated the BTK inhibitory effects of these drugs (not shown). ERK phosphorylation was reduced by 50% to 60% by both drugs with both concentrations (p<0.002). Similarly, S6 phosphorylation, which was measured in samples from three patients, was decreased by 50% upon drug treatment. In conclusion, the inhibitory signature of ibrutinib and acalabrutinib on BTK pathway was similar in CLL cells.
Figure 4. Comparison of ibrutinib- and acalabrutinib-mediated inhibition of BTK phosphorylation and downstream signaling.
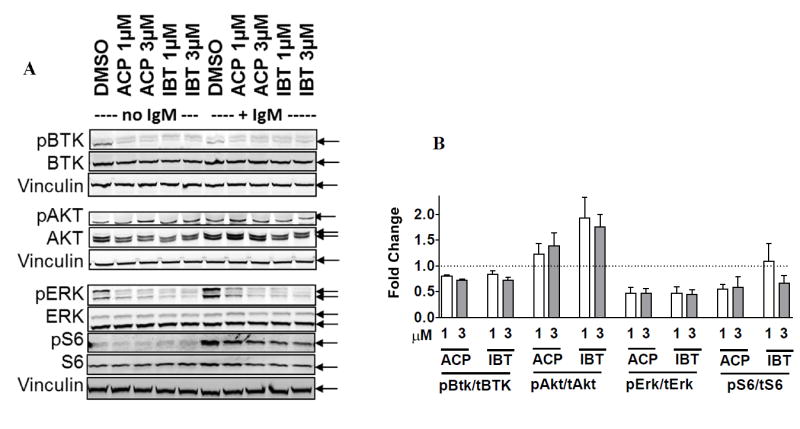
A. CLL cells were incubated with DMSO only (controls) or with 1 μM or 3 μM ibrutinib or acalabrutinib for 48 hours without IgM stimulation (lanes 1-5) or with IgM stimulation (lanes 6-10). The protein lysates were immunoblotted for total and phosphorylated BTK, AKT, ERK, and S6 proteins. Vinculin was used as a loading control. B. Immunoblots representing five different patient samples without IgM stimulation were quantitated. Values represent fold change in phosphorylated protein relative to total protein.
Ibrutinib and acalabrutinib treatment decrease Bcl-2 and Mcl-1 total protein levels
We previously reported that ibrutinib decreases Mcl-1 protein levels in primary CLL cells without changing Bcl-2 protein expression (14). Consistent with those data, ibrutinib and acalabrutinib decreased Mcl-1 (p = <0.002) without IgM stimulation (Figure 5A and 5B). The decrease in Mcl-1 total protein was also significant when culture conditions included IgM stimulation (p = <0.008) (Figure 5A and 5C). In contrast, Bcl-2 total protein level did not change in either culture conditions (p = >0.11) protein levels (Figure 5A – 5C). Collectively, these data establish that acalabrutinib is similar to ibrutinib in its action on Bcl-2 and Mcl-1 proteins in CLL cells.
Figure 5. Comparison of ibrutinib and acalabrutinib-mediated changes in the levels of Bcl-2 and Mcl-1 antiapoptotic proteins.
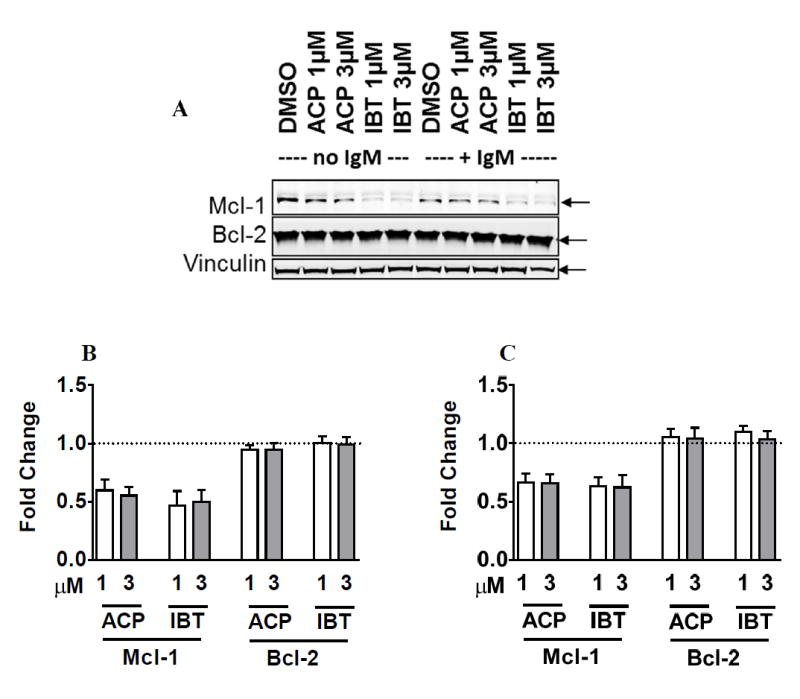
A. CLL cells were incubated with DMSO only (controls) or with 1 μM or 3 μM ibrutinib or acalabrutinib for 48 hours without IgM stimulation (lanes 1-5) or with IgM stimulation (lanes 6-10). The protein lysates were immunoblotted for total Bcl-2 and Mcl-1 proteins. Vinculin was used as a loading control. B-C. Immunoblots were analyzed from five different patients, and changes in Mcl-1 and Bcl-2 in CLL cells treated with BTK inhibitors without (B) or with (C) IgM stimulation were quantitated. Values represent fold change relative to DMSO controls.
Ibrutinib and acalabrutinib treatment differ in inhibiting SRC family kinases
Next, we examined the effects of these two BTK inhibitors on SRC family kinases in T cells obtained from healthy donors. SRC family kinases play an important role in platelet activation, and their inhibition may contribute to adverse bleeding events (26). To help ensure that any observed dephosphorylation was due to the BTK inhibitors, and not to endogenous phosphatase activity, a low dose of the phosphatase inhibitor H2O2 was added during cell stimulation (38). While both drugs decreased levels of phospho-LCK (Y505) (Figure 6A) and phospho-SRC (Y418) (Figure 6B) in a dose-dependent manner, the extent of inhibition was very different; with ibrutinib demonstrating a more potent inhibitory effect on phosphorylation of LCK and SRC. The EC50 for ibrutinib was less than 0.2 μM, whereas the EC50 for acalabrutinib was not reached at 10 μM. These data suggest that ibrutinib may have greater off-target effects on SRC kinase inhibition in healthy T cells than acalabrutinib.
Figure 6. Comparison of ibrutinib and acalabrutinib-induced signaling in T cells.
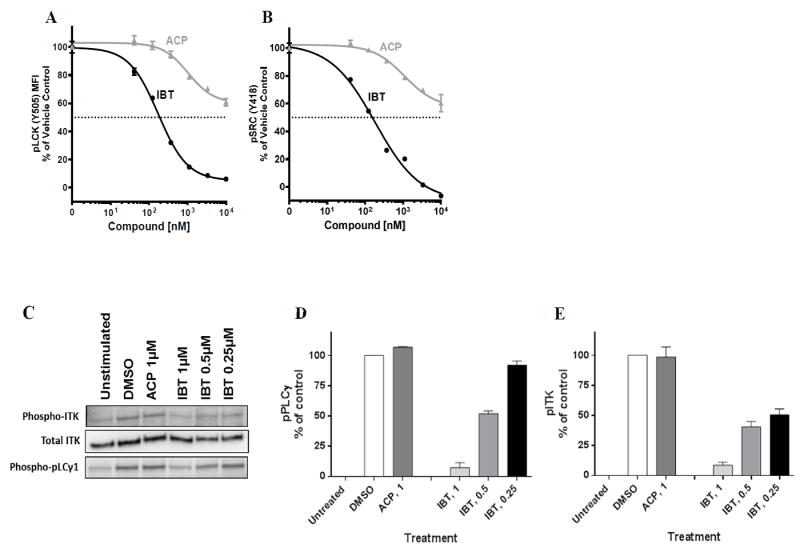
Peripheral blood mononuclear cells from healthy donors were treated with indicated concentrations of BTK inhibitors for 2 hours and then stimulated with 3.3 mM H2O2 for 10 minutes at 37°C. Phosphoflow analysis for A. phospho-LCK (Y505) and B. phospho-SRC (Y418) were performed with gated CD3+ T cells. Grey lines with solid triangles represent acalabrutinib treatment, and black lines with solid circles represent ibrutinib treatment. Jurkat cells were either unstimulated, stimulated and treated with vehicle (DMSO), or stimulated and treated with acalabrutinib or ibrutinib at indicated concentrations. (C) Western blot showing phospho-ITK, total ITK, and phospho-PLCγ1 expression in Jurkat cell line before and after treatment with acalabrutinib or ibrutinib at indicated concentrations. (D-E) Quantification of immunoblots for phospho-PLCγ1 and phospho-ITK. Immunoblots in Figure 6C were quantified three times using GelQuant software and normalized to DMSO control. Abbreviations; ACP, acalabrutinib; IBT, ibrutinib. Concentrations of both drugs are in nM for Figures 6A and B and μM for Figures 6 C-E.
Ibrutinib and acalabrutinib show differential inhibition of ITK
To further characterize the effects of both drugs in T-cells, we measured the phosphorylation of ITK and its substrate, PLCγ1 in stimulated Jurkat T-cells treated with ibrutinib or acalabrutinib. We observed a dramatic difference in phospho-ITK and phospho- PLCγ1 at 1 µM concentrations of each drug, with neither enzyme affected by acalabrutinib compared with >90% inhibition with ibrutinib treatment (Figure 6C-E). Additionally, ibrutinib reduced phosphorylation of both enzymes even at lower (0.25 and 0.5 μM) concentrations. These data show that ibrutinib, but not acalabrutinib, inhibits ITK and its downstream target PLCγ1 in T-cells as an off-target effect.
Discussion
The overall aim of this project was to compare ibrutinib and acalabrutinib in CLL patient samples to determine similarities and differences of these two agents. We focused on four different approaches. First, we compared the biological effect of each drug in CLL patient samples. Second, we evaluated the impact of these BTK inhibitors on chemokine production and migration. Third, we investigated the impact on substrate (BTK) phosphorylation and down-stream events after on-target inhibition in CLL cells. Finally, we compared the effect of these agents on signaling in healthy T-cells, which could be considered off-target effects.
Similar to what has been reported for ibrutinib (10,12); acalabrutinib treatment resulted in cell death in less than 15% of CLL lymphocytes. The extent of cell death was 3% to 6% higher with ibrutinib vs acalabrutinib, which could be due to off-target activities of ibrutinib (32). Patient characteristics, including prognostic factors and prior therapy, were not associated with the degree of cell death induced by acalabrutinib and ibrutinib. These observations suggest that at equimolar free drug concentrations, both acalabrutinib and ibrutinib would be expected to have similar efficacy in patients with CLL.
BCR activation promotes the secretion of pro-inflammatory chemokines, including CCL3 and CCL4, in CLL cells (40). Ibrutinib treatment has been shown to decrease CCL3 and CCL4 levels in CLL cell cultures. This finding of decreased chemokine levels due to BTK inhibition by ibrutinib was also observed during separate clinical trials with either ibrutinib (12) or acalabrutinib (32). Consistent with those data, the current study showed that ibrutinib and acalabrutinib similarly reduced CCL3 and CCL4 production in the presence of BCR pathway stimulation (Figure 3). This was also reflected in the migration of CLL cells toward stroma, presumably mediated in part by CXCL12 (SDF1), consistent with a previous study (15).
BTK mRNA levels have been reported to be higher in CLL cells than in normal B cells; however, BTK protein levels vary among patients (10). Because ibrutinib and acalabrutinib are potent, covalent, and irreversible inhibitors of BTK, both drugs reduced phosphorylation of BTK. Consistent with this inhibition of the kinase, phosphorylation of downstream proteins such as ERK and S6 was similarly blunted with ibrutinib and acalabrutinib (Figure 4B). In our samples, at this 24-hour time point, phosphorylation of Akt was not inhibited, possibly due to sustained activation of other signaling pathways (43). Downstream of BCR signaling pathway is an effect on the NFkB activity. We were not able to observe a clear effect on downstream phospho-p65 levels in this study (data not shown).
Our prior preclinical and clinical investigations demonstrated that ibrutinib decreased Mcl-1 protein levels while Bcl-2 levels remain the same or increase (14). This finding was also recently reported by other investigators (44). Our current data indicate that acalabrutinib and ibrutinib cause similar reductions in Mcl-1 protein expression even in the presence of BCR stimulation with IgM (Figure 5). CLL cell survival is associated with expression of Bcl-2 family anti-apoptotic proteins, such as Mcl-1 and Bcl-2. Hence, a decline in Mcl-1 protein levels in response to treatment with BTK inhibitors provides rationale for combining ibrutinib or acalabrutinib with the Bcl-2 antagonist venetoclax (45,46). Indeed, such combination strategies have been tested and found to augment ibrutinib-induced cytotoxicity in CLL (14,44) and other B-cell malignancies (47).
BTK belongs to the Tec family of tyrosine kinases, which has five members. While BTK is expressed specifically in B cells and not in T cells, other Tec family members, such as ITK and Txk, are expressed in T cells. T-cell receptor–mediated signal transducers downstream of ITK were inhibited by ibrutinib (30,48) but not acalabrutinib in the Jurkat T-cell line (32). Similarly, phosphorylation of TEC was decreased by ibrutinib (32). Such off-target effects have the potential to lead to toxicities, such as bleeding (24,49). The SRC family, mostly FYN and LYN, plays a critical role in platelet activation via the collagen receptor GPVI–FcRγ complex.(50) Autophosphorylation of Y418 and dephosphorylation of Y530 on SRC are required to switch the kinase from the inactive closed formation to the active open formation. The two main phosphorylation sites on LCK are tyrosines 394 and 505. The former is an autophosphorylation site and is linked to activation of the protein. The latter is phosphorylated by CSK, which inhibits LCK because the protein folds up and binds its own SH2 domain. LCK (Y505) was inhibited only by ibrutinib (Figure 6A). Our data demonstrate that ibrutinib reduces phosphorylation of SRC and LCK. In contrast to ibrutinib, acalabrutinib showed minimal inhibition of SRC and LCK. Differential effect of these two drugs on T-cells was further evidenced in Jurkat T-lymphoblastic cell line for ITK and PLCγ1 (Figure 6C-E). Consistent with these data, previous studies with acalabrutinib demonstrated minimal effects on EGFR, Tec, or ITK signaling, and no inhibition of thrombus formation in vivo at clinically relevant concentrations (32,51).
In conclusion, our investigations demonstrate that the selective BTK inhibitor acalabrutinib produces biological effects in CLL cells that are comparable to those exerted by ibrutinib but that the molecular impact on healthy T cells appeared different.
Supplementary Material
Translational Relevance.
Ibrutinib is an oral, first-in-class Bruton tyrosine kinase (BTK) inhibitor that is approved by the US Food and Drug Administration for the treatment of patients with previously treated and untreated chronic lymphocytic leukemia (CLL). This drug irreversibly binds to the Cys-481 residue of BTK, however, ibrutinib also binds to several other cysteine-containing kinases, which could result in off-target toxicities. Acalabrutinib, a more selective BTK inhibitor developed to minimize off-target effects, has shown promising clinical activity in patients with relapsed/refractory CLL. Preclinical comparisons of ibrutinib and acalabrutinib are needed to better understand potential differences between the biological and molecular effects of these BTK inhibitors in primary CLL cells and normal T cells.
Acknowledgments
This work was supported in part by grant P01-CA81534 of the CLL Research Consortium from the National Cancer Institute, a CLL Global Research Foundation Alliance grant, and a Sponsored Research Agreement from Acerta Pharma. Authors are thankful to Sarah Bronson for critically editing the manuscript and Mark Nelson for coordinating CLL blood sample distribution. The authors received editorial support in the preparation of this manuscript from Team 9 Science, LLC, funded by Acerta Pharma. The authors directed development of the manuscript and were fully responsible for all content and editorial decisions for this manuscript.
Footnotes
Authorship contribution
V.K.P. designed and performed the experiments, analyzed the results, and wrote the manuscript. K.B. directed V.K.P., designed experiments, and reviewed the manuscript. E.B. performed T-cell signaling experiments. M.A. assisted in experimental planning and performing immunoblots. M.J.K. and W.G.W. identified patients to obtain peripheral blood samples, provided clinical and patient-related input, and reviewed the manuscript. V.G. conceptualized and supervised the research, obtained funding, analyzed the data, and wrote and reviewed the manuscript.
Conflict-of-interest disclosure
V.G. and W.G.W. received research and clinical trial funding from Acerta Pharma. E.B. is an employee of Acerta Pharma. The other authors do not have conflicts of interest.
References
- 1.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103(12):4389–95. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 2.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72(2):279–90. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 3.Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–8. [PubMed] [Google Scholar]
- 4.Buckley RH. Primary immunodeficiency diseases due to defects in lymphocytes. The New England journal of medicine. 2000;343(18):1313–24. doi: 10.1056/NEJM200011023431806. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Q, Zhang M, Winkelstein J, Chen SH, Ochs HD. Unique mutations of Bruton’s tyrosine kinase in fourteen unrelated X-linked agammaglobulinemia families. Human molecular genetics. 1994;3(10):1899–900. doi: 10.1093/hmg/3.10.1899. [DOI] [PubMed] [Google Scholar]
- 6.Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–33. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 7.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261(5119):358–61. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 8.Nisitani S, Satterthwaite AB, Akashi K, Weissman IL, Witte ON, Wahl MI. Posttranscriptional regulation of Bruton’s tyrosine kinase expression in antigen receptor-stimulated splenic B cells. Proceedings of the National Academy of Sciences. 2000;97(6):2737–42. doi: 10.1073/pnas.050583597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016;30(4):833–43. doi: 10.1038/leu.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez A, Martinez N, Camacho FI, Ruiz-Ballesteros E, Algara P, Garcia JF, et al. Variability in the degree of expression of phosphorylated IkappaBalpha in chronic lymphocytic leukemia cases with nodal involvement. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(20):6796–806. doi: 10.1158/1078-0432.CCR-04-0753. [DOI] [PubMed] [Google Scholar]
- 12.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26(1):93–104. doi: 10.1016/j.immuni.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(16):3705–15. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119(11):2590–4. doi: 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- 16.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765 has significant activity in patients with relapsed/refractory B-cell malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(1):88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497–506. doi: 10.1182/blood-2014-10-606038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. The Lancet Oncology. 2014;15(1):48–58. doi: 10.1016/S1470-2045(13)70513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien S, Jones JA, Coutre SE, Mato AR, Hillmen P, Tam C, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. The Lancet Oncology. 2016;17(10):1409–18. doi: 10.1016/S1470-2045(16)30212-1. [DOI] [PubMed] [Google Scholar]
- 21.de Claro RA, McGinn KM, Verdun N, Lee SL, Chiu HJ, Saber H, et al. FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(16):3586–90. doi: 10.1158/1078-0432.CCR-14-2225. [DOI] [PubMed] [Google Scholar]
- 22.Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–37. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829–30. doi: 10.1182/blood-2014-10-604272. [DOI] [PubMed] [Google Scholar]
- 25.Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124(26):3991–5. doi: 10.1182/blood-2014-06-583294. [DOI] [PubMed] [Google Scholar]
- 26.Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124(13):2013–24. doi: 10.1182/blood-2014-01-453134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–7. doi: 10.1038/leu.2014.247. [DOI] [PubMed] [Google Scholar]
- 28.Saitoh Y, Terada N, Ohno N, Hamano A, Okumura N, Jin T, et al. Imaging of thrombosis and microcirculation in mouse lungs of initial melanoma metastasis with in vivo cryotechnique. Microvascular research. 2014;91:73–83. doi: 10.1016/j.mvr.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Knapen MH, Braam LA, Drummen NE, Bekers O, Hoeks AP, Vermeer C. Menaquinone-7 supplementation improves arterial stiffness in healthy postmenopausal women. A double-blind randomised clinical trial. Thrombosis and haemostasis. 2015;113(5):1135–44. doi: 10.1160/TH14-08-0675. [DOI] [PubMed] [Google Scholar]
- 30.Dubovsky JA, Chappell DL, Harrington BK, Agrawal K, Andritsos LA, Flynn JM, et al. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood. 2013;122(19):3308–16. doi: 10.1182/blood-2013-05-504597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao W, Wang M, Wang L, Lu H, Wu S, Dai B, et al. Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. J Natl Cancer Inst. 2014;106(9) doi: 10.1093/jnci/dju204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2016;374(4):323–32. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manne BK, Badolia R, Dangelmaier C, Eble JA, Ellmeier W, Kahn M, et al. Distinct pathways regulate Syk protein activation downstream of immune tyrosine activation motif (ITAM) and hemITAM receptors in platelets. The Journal of biological chemistry. 2015;290(18):11557–68. doi: 10.1074/jbc.M114.629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA oncology. 2015;1(1):80–7. doi: 10.1001/jamaoncol.2014.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(29):13075–80. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balakrishnan K, Burger JA, Fu M, Doifode T, Wierda WG, Gandhi V. Regulation of Mcl-1 expression in context to bone marrow stromal microenvironment in chronic lymphocytic leukemia. Neoplasia. 2014;16(12):1036–46. doi: 10.1016/j.neo.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balakrishnan K, Peluso M, Fu M, Rosin NY, Burger JA, Wierda WG, et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29(9):1811–22. doi: 10.1038/leu.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haas A, Weckbecker G, Welzenbach K. Intracellular phospho-flow cytometry reveals novel insights into TCR proximal signaling events. A comparison with Western blot. Cytometry A. 2008;73(9):799–807. doi: 10.1002/cyto.a.20598. [DOI] [PubMed] [Google Scholar]
- 39.Nyakeriga AM, Garg H, Joshi A. TCR-induced T cell activation leads to simultaneous phosphorylation at Y505 and Y394 of p56(lck) residues. Cytometry A. 2012;81(9):797–805. doi: 10.1002/cyto.a.22070. [DOI] [PubMed] [Google Scholar]
- 40.Burger JA, Quiroga MP, Hartmann E, Burkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113(13):3050–8. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–67. [PubMed] [Google Scholar]
- 42.Mohle R, Failenschmid C, Bautz F, Kanz L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1) Leukemia. 1999;13(12):1954–9. doi: 10.1038/sj.leu.2401602. [DOI] [PubMed] [Google Scholar]
- 43.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nature reviews Cancer. 2014;14(4):219–32. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 44.Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Ibrutinib therapy increases BCL-2 dependence and enhances sensitivity to venetoclax in CLL. Blood. 2015;126(23):490. doi: 10.1038/leu.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19(2):202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 46.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2016;374(4):311–22. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Portell CA, Axelrod M, Brett LK, Gordon VL, Capaldo B, Xing JC, et al. Synergistic cytotoxicity of ibrutinib and the BCL2 antagonist, ABT-199 (GDC-0199) in mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL): molecular analysis reveals mechanisms of target interactions. Blood. 2014;124(21):509. [Google Scholar]
- 48.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alberelli MA, Innocenti I, Sica S, Laurenti L, De Candia E. PO-54 - Clinical and laboratory characterization of platelet dysfunction caused by ibrutinib treatment in patients with chronic lymphocytic leukemia. Thromb Res. 2016;140(Suppl 1):S196. doi: 10.1016/S0049-3848(16)30187-6. [DOI] [PubMed] [Google Scholar]
- 50.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor gamma chain complex on human platelets. The Journal of experimental medicine. 1998;188(2):267–76. doi: 10.1084/jem.188.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Covey TBT, Gulrajani M, et al. ACP-196: a novel covalent Bruton’s tyrosine kinase (Btk) inhibitor with improved selectivity and in vivo target coverage in chronic lymphocytic leukemia (CLL) patients. Annual meeting of the American Association for Cancer Research; 2015. Abstract 2596. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.