

Abstract
Intense exercise may cause heart remodeling to compensate increases in blood pressure or volume by increasing muscle mass. Cardiac changes do not involve only the left ventricle, but all heart chambers. Physiological cardiac modeling in athletes is associated with normal or enhanced cardiac function, but recent studies have documented decrements in left ventricular function during intense exercise and the release of cardiac markers of necrosis in athlete’s blood of uncertain significance. Furthermore, cardiac remodeling may predispose athletes to heart disease and result in electrical remodeling, responsible for arrhythmias. Athlete’s heart is a physiological condition and does not require a specific treatment. In some conditions, it is important to differentiate the physiological adaptations from pathological conditions, such as hypertrophic cardiomyopathy, arrhythmogenic dysplasia of the right ventricle, and non-compaction myocardium, for the greater risk of sudden cardiac death of these conditions. Moreover, some drugs and performance-enhancing drugs can cause structural alterations and arrhythmias, therefore, their use should be excluded.
Keywords: Athlete’s heart, Cardiac damage, Fibrosis, Intense exercise, Arrhythmogenic dysplasia of the right ventricle, Atrial fibrillation, Doping, Anabolic-androgenic steroids, Hypertrophic cardiomyopathy
Core tip: Athlete’s heart is a physiological condition that in some cases can simulate pathological disease, sometimes due to the use of doping drugs. Furthermore, exercise can induce atrial dilation and arrhythmias. Our objective is to analyze the current literature and to review the most important changes in the heart of athletes, from the different molecular pathways to the structural anomalies.
INTRODUCTION
High-intensity exercise training leads to morphological, functional, and electrical remodeling of the heart, which are included in the ‘‘athlete’s heart’’, characterized by increased left ventricular mass (LVM), cavity dimensions and wall thickness[1]. Athletes with left ventricular (LV) hypertrophy generally have normal cardiac function and normal systolic and diastolic function[2]. Athletes exhibit an improvement in myocardial diastolic indices and supernormal LV diastolic function[3]. Recent studies have documented decrements, especially in right ventricular (RV) function, during intense endurance exercise[4,5]. Actual evidence suggests that there may be some overlap between physiological and pathological conditions, such that a modest amount of fibrosis may be present in cardiac remodeling associated with lifelong endurance training and then acts as a substrate for arrhythmias[6].
Our objective is to describe the mechanisms of cardiac remodeling in athletes and to delineate the most important differences, from the molecular mechanisms to the structural changes, between athlete’s heart and pathological conditions, taking into account the most important cardiomyopathy, arrhythmias and the abuse of performance-enhancing drugs.
PHYSIOLOGICAL VS PATHOLOGICAL CARDIAC HYPERTROPHY: FROM PHYSICAL PRINCIPLES TO MOLECULAR MECHANISMS
Cardiac hypertrophy is an adaptive response to the increased cardiac loading that normalizes wall stress, according to Laplace relationship (Figure 1). Unfortunately, long-term maladaptive remodeling of reactive hypertrophy in various cardiovascular diseases (e.g., valvular heart disease, myocardial ischemia, coronary artery disease, hypertension, and cardiomyopathy) is associated with gradual ventricular dilation, due to loss of myocytes and cardiac fibrosis[7]. Physiological hypertrophy, such as athlete’s heart, is typically not associated with myocyte death, although recent studies have shown myocardial damage during intense exercise and RV inflammation and fibrosis in long term endurance athletes[4,8]. The shift from compensated pathological hypertrophy to failure of myocardium includes cellular and molecular events, such as myocyte death, with three different mechanisms: Apoptosis, necrosis and autophagic cell death[9,10]. Cardiomyocyte replacement and myocardial fibrosis are representative of all types of pathological hypertrophy and proceed along with functional decompensation. To explain fibrosis and necrosis in athlete’s heart, interesting is the “ischemic core” hypothesis: Hypertrophic cardiomyocyte becomes ischemic when his surface exceeds the distance across which oxygen can diffuse down its concentration gradient from adjacent capillaries, with contractile depression and cellular death[11]. Physiological hypertrophy is associated with a normal or increased number of myocardial capillaries, due to the activation of VEGF pathway[9]. Akt is a serine/threonine protein kinase responsible for the cellular growth in multiple cell types, which can be activated by exercise. Recent data suggest that Akt pathway might be implicated both in physiological and pathological cardiac growth. In animal models, myocardial expression of Akt pathway caused reversible hypertrophy after 2 wk of strenuous exercise, but an irreversible cardiomyopathy with decreased capillary density and fibrosis after 6 wk of intense training. It seems that myocardial angiogenesis is more intense in the acute phase of heart hypertrophy but insufficient in the advanced phase: Excessive “physiological” hypertrophy might be associated with poor angiogenesis and consequently with heart failure[12].
Figure 1.
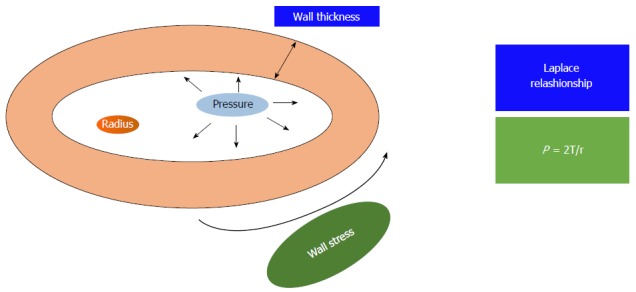
The figure shows the Laplace relationship: The pressure (P) generated in a sphere is directly proportional to the wall tension (T) and inversely related to the radius of the sphere (r).
Autocrine and paracrine triggers are released in response to hemodynamic overload, and definite substances are preferentially released for pathological or physiological stimuli. Insulin like growth factor 1 (IGF1) is released in the course of postnatal development and during exercise training and is increased in swim-trained rats and in veteran athletes compared with controls[13], whereas elevated levels of angiotensin II (Ang II), catecholamine and endothelin-1 (ET-1) were observed in pathological hypertrophy and in heart failure subjects[14]. IGF1 promotes the PI3K-Akt molecular pathway to induce physiological cardiac hypertrophy, whilst the mitogen activated protein kinase (MAPK) pathway and calcineurin system are activated by Ang II and ET-1 in pathological hypertrophy (Table 1)[15].
Table 1.
Differences in between physiological and pathological hypertrophy
| Physiological hypertrophy | Pathological hypertrophy |
| Angiogenesis, release of VEGF | Perivascular fibrosis and inflammation |
| Activation of IGF-1 pathway (IGF-1- > PI3K- > Akt) | Activation of Angiotensin II, Catecholamine and Endotelin-1 |
| No fibrosis | MAPK and Calcineurin pathway |
| Normal gene expression | Fibrosis, myocyte necrosis and apoptosis |
| Proportional chamber enlargement | Cardiac dysfunction |
The table summarizes the differences in the cellular and molecular pattern between physiological and pathological hypertrophy. VEGF: Vascular endothelial growth factor; IGF-1: Insulin like growth factor; PI3K: Phosphoinositide 3-kinase; MAPK: Mitogen-activated protein kinase.
In conclusion, familial hypertrophic cardiomyopathy[16] is associated with sarcomeric protein mutations, such as cardiac troponin I or T, β-MHC, α-MHC, myosin light chain, α-tropomyosin, titin, and actin, with loss of contractile filaments and proteins of sarcomeric skeleton[17].
CARDIOMYOCYTE DAMAGE DURING INTENSE EXERCISE
After intense exercise, acute increases in troponin (cTn) and B-type natriuretic peptide have been detected in athletes[18]. These are specific markers of myocyte injury and strain, but do not indicate a permanent injury. Potential mechanisms have been showed to elucidate cTn elevation after intense exercise, but actually, the elevation of cTn levels in healthy individuals cannot be explained by any of these theories[18]. It is possible that exercise induces an increase in myocardial sarcolemma permeability, due to mechanical stress on the cardiomyocytes and to increased production of oxidative radicals or altered acid base balance exercise, with passive diffusion of cTn from the intra- to extra-cellular compartment[18]. Cellular stretching might cause transient disruption of the myocardial plasma membrane and then, the release of cTn[19]. Furthermore, it can stimulate integrins, mediating the transport of entire cTn molecules out of viable cardiomyocytes[20]. Increased levels of cTn are more common in cycling or triathlon and depend on the exercise intensity[21].
Case reports have shown myocardial fibrosis and late gadolinium enhancement, associated with cTn elevation post-exercise, in a small number of veteran athletes, but the pathogenesis of these cases remains unclear[18].
EXERCISE-INDUCED MYOCARDIAL FIBROSIS
Despite the widely recognized benefits of regular physical activity, high-level exercise training may be associated with increased arrhythmia risk and even with sudden cardiac death[22].
The athlete’s heart is a benign condition, representing a normal adaptation to chronic exercise, in which loss of myocytes and abnormal deposition of collagen do not usually occur[15]. Pathological hypertrophy is associated with apoptosis and necrosis; in this case, the loss of myocytes is replaced with excessive collagen deposition. Excessive collagen deposition increases the stiffness of the ventricles, with consequent impaired contraction and relaxation, electrical conduction system fibrosis and reduced capillary density, leading to myocardial ischemia and the transition from hypertrophy to failure[15]. Interestingly, recent studies have shown myocardial inflammation and fibrosis in animal models of long term, intensive exercise. Chen et al[23] forced rats to swim strenuously and found histological evidence of localized myocyte damage, myocardial necrosis and inflammatory infiltrates. Benito et al[24] instituted an intensive treadmill running protocol in rats and demonstrated an increase in atrial and ventricular inflammation and fibrosis and a greater risk of ventricular arrhythmias in the “marathon rats”. Fibrosis and inflammatory infiltrates have been identified in well-trained athletes who underwent cardiac biopsy for high probability of identifying a cardiac pathology[25]. Histology offers the tangible evidence of fibrosis, but inflammatory infiltrates and fibrosis are non-specific and their etiology can be supposed only by other clinical factors. Furthermore, cardiac biopsy is an invasive procedure with significant risks and it is not applicable in the absence of high suspicion of heart disease. An accurate, non-invasive surrogate tool for detecting fibrosis is cardiac magnetic resonance (CMR) imaging with gadolinium contrast. Gadolinium-based extracellular paramagnetic contrast agents concentrate in areas of fibrosis and thus can be used to characterize injured myocardium. Using gradient-echo inversion recovery imaging, fibrosis appears as bright signal, with a prolonged wash-out time for gadolinium [delayed gadolinium enhancement (DGE)], contrasting with the normal myocardium, which looks black[6]. Several studies have identified the presence of DGE in the heart of extensively trained veteran athletes. In most cases, the patches of DGE were very small and sited in the septum and in RV insertion points, regions subjected to local stretching during exercise[6]. More recently, La Gerche et al[4] have shown myocardial fibrosis by CMR and a reduction in RV systolic function in athletes with long-term exercise, suggesting that the heart has a limited capacity to tolerate the overload exercise. The patches of cardiac fibrosis may be the substrate for ventricular tachycardia and sudden death, in predisposed individuals[4]. Some authors have recently suggested a new entity, the so called Phidippides cardiomyopathy: Long term strenuous exercise can induce cardiac dilation and also activates resident macrophages, pericytes, and fibroblasts, resulting in the deposition of collagen and fibrosis[26,27]. CMR can also specifically detect intra-myocardial fibrofatty infiltration of the RV wall, typical of the arrhythmogenic right ventricular cardiomyopathy (ARVC), which often leads to ventricular arrhythmias and usually appears in young adulthood and affected asymptomatic or minimally symptomatic individuals[27]. In conclusion, it is possible that the RV is more susceptible to fatigue than the left ventricle after prolonged exercise. It needs more studies to identify a probable effect of exercise “dose” and their implication in the development of heart failure.
IS CARDIAC REMODELING IN ATHLETES ALWAYS BENIGN?
Cardiac adaptations to exercise not involve only the left ventricle, but all the heart chambers. Often these changes are absolutely physiological, but in some cases, they can predispose to pathological conditions, such as arrhythmias. Below, we report the main morpho-functional changes of the different cardiac structures in athletes and their implications in the pathogenesis of cardiovascular diseases.
ATHLETE’S ATRIA FUNCTION AND DYSFUNCTION
Atrial abnormalities can be present in athletes, such as a mild increase in atrial volume and diameter, and may be considered a physiological adaptation to exercise[28]. The pathophysiological mechanisms are not well defined. Studies in animal models have shown that, in rats, prolonged and vigorous exercise resulted in eccentric hypertrophy and diastolic dysfunction with atrial dilatation and fibrosis, especially in the atria and the right ventricle, and increased fibrotic mRNA compared with controls[24]. A recent meta-analysis of 7189 adult elite athletes have shown that exercise causes an increase in left atrium (LA) dimensions, compared with controls, evaluating both diameter and volume, corrected for body surface area. The endurance athletes reported the largest average LA diameters[29]. Since pre-adolescence, the long-term endurance exercise results in considerable bi-atrial remodeling and enlargement compared with sedentary subjects of the same age, with a preserved cardiac function[30,31]. LA enlargement could be considered part of athlete’s heart, considering that the LA pressure rises during ventricular diastole more than in sedentary subjects, to maintain adequate filling whereas LV stiffness or pressure are increased[32]. On the other hand, there is evidence that the endurance exercise increases the risk of developing atrial fibrillation (AF) and atrial flutter in the middle age, in subjects without any clinical or echocardiographic signs of cardio-pulmonary pathologies or hypertension[33]. The mechanisms responsible for these arrhythmias might be: The major incidence of atrial ectopic beats in this population, as a consequence of physical activity; the influence of autonomic nervous systems, and in particular the vagal system, responsible for the “vagal AF”[34]; the atria dilatation, fibrosis, and inflammation induced by high exercise training and the atrial remodeling[33] (Table 2). In mice, exercise can induce TNFα-dependent activation of both NF-κB and p38MAPK, increasing inflammation and AF susceptibility[35].
Table 2.
Pathological mechanisms of atrial fibrillation in long-term athletes
| Pathological mechanism |
| Atrial ectopic beats |
| Vagal nervous system |
| Atrial fibrosis |
| Atrial dilatation |
| Myocardial injury |
| Inflammation |
| Redox imbalance |
The table shows the most important mechanisms involved in atrial fibrillation exercise related.
Moreover, AF might be closely connected with oxidative cellular changes and redox imbalance in the atrium. The oxidative species, generated from cardiomyocytes in stress conditions such as strenuous exercise, can increase inflammation and activate downstream molecular pathways, promoting morphological and electrical modeling. Recently, Mont et al[36] have shown that loss of Nrf2, a gene with antioxidant function in the atria, could be associated with atrial hypertrophy and AF, suggesting that the preservation of the redox state is essential for the atrium health.
Finally, in athletes AF appears as some symptomatic and paroxysmal episodes that could become more frequent and progress to persistent AF. The GIRAFA study has showed that the crisis appears in the night or after the meal, related to an increased vagal tone[36].
Data about right atrial (RA) function in top level athletes are lacking. Previously, our group has delineated the upper limits of RV and RA dimensions in highly-trained athletes and showed that right heart dimensions were greater in elite endurance-trained athletes than in age- and ex-matched strength athletes and controls[37]. Then, D’Ascenzi et al[38] investigated the RA function and dimension in 100 top level athletes by standard echocardiography and 2D speckle tracking echocardiography and showed that RA area, volume, volume index, and inferior vena cava were significantly greater in athletes than in controls and the peak atrial longitudinal strain and peak atrial contraction strain values were lower in athletes than in controls. This strain reduction should not represent a real dysfunction, but only a physiological phenomenon, and can be included in the “athlete’s heart”[38].
LV CHANGES IN EXERCISE RELATED AND PATHOLOGICAL CONDITIONS
In some highly-trained athletes, the LV wall thickness may be increased, mimicking a hypertrophic cardiomyopathy (HCM). The thickening is usually mild, but in some cases, it may be significant and creates difficulties to differentiate athlete’s heart and hypertrophic cardiomyopathy, especially in the ambiguous “gray zone”, when the wall thickness is of 13 to 15 mm (12 to 13 mm in women)[39]. This differential diagnosis is important, since most cases of sudden death in athletes are probably due to HCM[40]. Echocardiography plays an important role in the differential diagnosis: HCM is probable with LV end-diastolic cavity < 45 mm, evidence of pathogenic sarcomere mutation, family history of HCM, abnormal LV diastolic function, left atrial dilatation, and late gadolinium enhancement on contrast-enhanced CMR imaging. Usually athlete’s heart is characterized by LV cavity enlargement (> 55 mm), peak VO2 > 110% of expected, and thickness or mass decreases with short periods of detraining[41,42]. Pelliccia et al[43] have shown that LV wall thickness ≥ 13 mm is mostly present in elite rowers and cyclists, and the upper limit appeared to be 16 mm (Figure 2).
Figure 2.
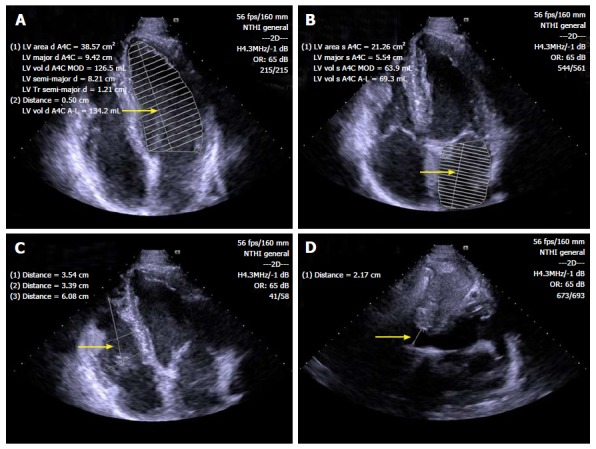
Standard B-mode echocardiography of endurance athlete showing enlargement of left ventricular (A), left atrial (B) and right ventricular (C) chambers, as well as inferior cavae vein dilatation (D) (arrows). LV: Left ventricular.
Other conditions that can cause cardiac hypertrophy include valve disease, hypertension and non-compaction myocardium. Despite the prevalence of hypertension is approximately 50% lower in athletes compared with the general population, it is also the most common cardiovascular condition in athletes. The pharmacological therapy can be difficult for the competition regulations and potential adverse effects[44]. It is important to diagnose this condition, because it is associated with an increased risk of developing heart failure. Recently, an elevated prevalence of LV noncompaction (LVNC) has been reported in athletes, phenotypically characterized by a more thick endocardial noncompact layer, increased trabeculations and deep recesses[45]. Caselli et al[46], in a recent study, have shown that in a large population of athletes, only a small subgroup presented LVNC. The increased trabeculations may represent a LV variant of athlete’s heart without any clinical significance[46].
Figure 3 summarizes the different characteristics of physiological remodeling and pathological condition of LV.
Figure 3.
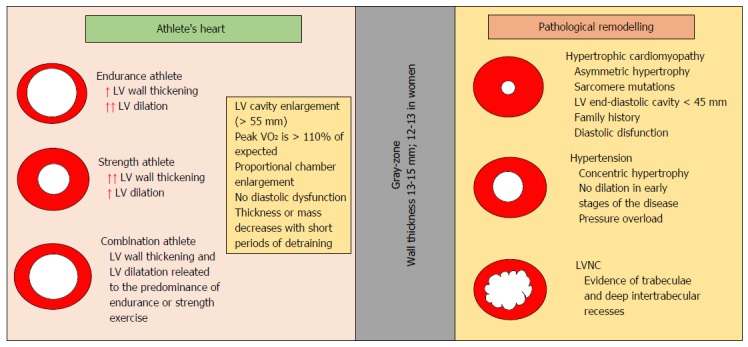
Different characteristics of physiological remodeling and pathological condition of the left ventricle. LV: Left ventricular; LVNC: LV noncompaction.
EXERCISE-RELATED RIGHT VENTRICLE REMODELING
Strenuous and prolonged exercise can cause RV dysfunction, usually transient, with evidence of increased biomarkers of cardiac damage. On the other hand, repeated bouts of exercise can lead to RV structural remodeling and arrhythmias and can lead to a syndrome similar to familial ARVC, without an identifiable genetic predisposition[47,48]. ARVC is present in 4% to 22% of athletes with sudden cardiac death[49,50]. As mentioned above, the RV function may be more interested by intense endurance training, therefore the diagnostic criteria for ARVC should be nonspecific in athletes with electrocardiographic anomalies and biventricular dilation.
Marcus et al[51] in a multi-center study of 108 probands with ARVD/C showed that 34% were athletes. Vigorous or long term athletic exercise might facilitate the phenotypic expression of ARVC due to the repetitive stretch of the RV with an underlying genetic desmosomal protein anomaly[51].
Signs of RV dysfunction seem to include: Syncope; Q waves in precordial leads; augmented QRS duration; three abnormal signal averaged electrocardiography parameters; delayed gadolinium enhancement; RV ejection fraction < 45% or wall motion anomalies at CMRI; > 1000 ventricular extra-systoles (or > 500 non-RV outflow tract) per 24 h; ventricular tachyarrhythmia or abnormal blood pressure response during exercise (Table 3)[52,53]. RV cavity size is not significantly larger in ARVC patients compared with athletes, whereas RV outflow tract is larger in ARVC subjects than in athletes[53]. The thickened and high reflective moderator band, commonly considered typical of ARVC, are present also in athletes and could be due to RV dilatation[53] (Figure 4). Further studies regarding the differential diagnosis between ARVC and physiological remodeling in athletes are needed to create useful clinical diagnostic algorithms.
Table 3.
Indicators of right ventricle pathology
| Episodes of syncope |
| > 1000 ventricular extra-systoles (or > 500 non-RV outflow tract) per 24 h; ventricular tachyarrhythmias; Q waves in precordial leads; augmented QRS duration |
| ≥ 3 abnormal signal averaged electrocardiography parameters |
| Delayed gadolinium enhancement; RV ejection fraction < 45%, or wall motion abnormalities at CMRI; impaired RV strain imaging |
| Attenuated blood pressure response during exercise |
| Dilatation of RV outflow tract |
The table shows the indicators of right ventricle pathology (ARVC vs athlete’s heart). CMRI: Cardiac magnetic resonance imaging; RV: Right ventricle; ARVC: Arrhythmogenic right ventricular cardiomyopathy.
Figure 4.
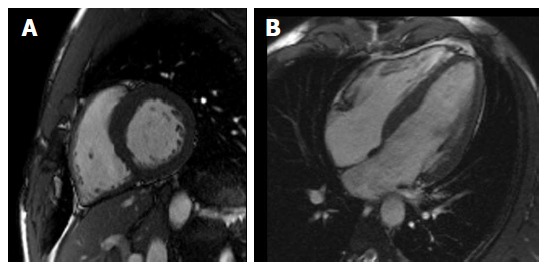
Cardiac magnetic resonance depicting in short-axis (A) and long-axis (B) view balanced biventricular enlargement in endurance athlete.
PERFORMANCE-ENHANCING DRUGS AND CARDIAC DAMAGE
Some banned athletic performance-enhancing drugs might have cardiac toxic effects, such as anabolic-androgenic steroids (AASs) and growth hormone (GH).
Healthy athletes abusing AASs may exhibit LV hypertrophy with both systolic and diastolic myocardial dysfunction and focal areas of DGE at CMR, with non-ischemic distribution[54] (Figure 5). AASs have direct toxicity on myocardial structures, with increased collagen deposition, fibrosis, and intimal hyperplasia of the intramural coronary vessels with chronic ischemic damage and microcirculation alterations. Moreover, testosterone might inhibit the extra-neuronal uptake of neuroamine and consequently result in vasospasm due to an abnormal vascular response to norepinephrine[55]. Post-mortem studies of athletes who used AASs have found infiltration of eosinophils into myocardial cells, as well as destruction of myofibrils. Endothelial dysfunction was also observed[56].
Figure 5.
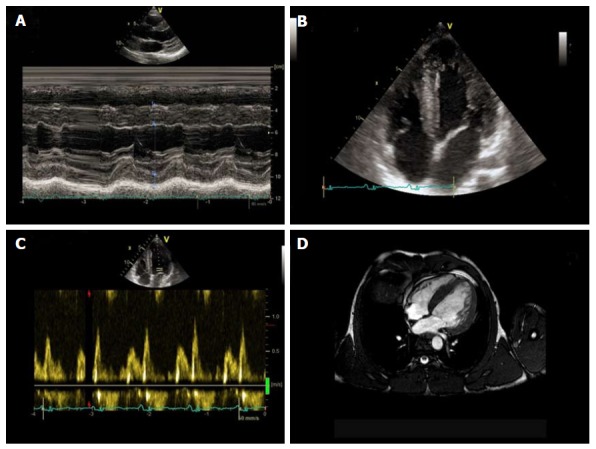
Non-invasive evaluation of power athlete abusing of steroids. Standard M-mode (A) and 4-chamber view B-mode (B) echocardiography, evidencing sever left ventricular hypertrophy, with diastolic dysfunction (C) underlined by Doppler transmitral flow pattern. Cardiac magnetic resonance confirmed severe left ventricular hypertrophy (D).
GH abuse has tainted many sports, including baseball, cycling, and track and field, for promoting an increase in muscle mass, though its effects on physical performance are not completely supported by the literature[57]. GH promotes cellular growth by stimulating protein synthesis, inhibiting catabolism, and inducing IGF-1 production. At the molecular level, GH binds its receptor and induces subsequent expression of growth-promoting molecules[58]. GH, both in excess or in deficient states, is related to increased cardiovascular mortality. The excess, like in acromegaly or in doping, results in cardiac hypertrophy and an increase in collagen, fibrosis, and cellular infiltration. In vivo studies on healthy mice demonstrated that increased GH levels induce significant LV hypertrophy and an increase in concentric anterior and posterior wall thicknesses, LV diastolic diameters and volumes, and cardiac output[59]. Unfortunately, the majority of conclusions about GH abuse and its cardiac effects result from data regarding acromegaly and not from direct data, which are lacking.
Also, erythropoietin (EPO), which increases hematocrit levels and thus improves aerobics capacity, may lead to cardiac dysfunction, increasing blood viscosity and cardiac afterload, and predisposes to hypertension and thromboembolism. Experimental studies have shown hypertension, cardiac hypertrophy and fibrosis after administration of high doses of EPO[60].
Thyroxine is used, generally, by athletes to promote weight loss. Thyroid hormones (TH) play an important role in cardiac growth and might cause cardiac hypertrophy and also heart failure if they are in excess. High levels of TH might result in elevated heart rate, decreased total peripheral resistance, widened pulse pressure, blood volume expansion, increased LVM and cardiac output, with improved contractile function and hemodynamic parameters in the short term. Longstanding hyperthyroidism can lead to dilatation of cardiac chambers and heart failure. Interestingly, the diminished cardiac function is often reversible when euthyroidism is re-established[61]. Weltman et al[61] showed that hyperthyroid rodents had important cardiac hypertrophy and adverse cardiac remodeling with chamber dilatation, LV systolic and diastolic dysfunction, decreased relative wall thickness, and fibrosis[61]. Few data in the literature are about the cardiac consequences of the prolonged use of thyroxine treatment. Thyroxine treatment, in high doses which suppress serum thyrotropin to below normal, has been associated with LV hypertrophy (in the absence of significant changes in heart rate, stroke volume, blood pressure, and LV systolic function), but untreated thyrotoxicosis resulted in more pronounced cardiovascular changes than thyroxin treatment[62]. Furthers studies are necessary to evaluate the cardiovascular risk in patients treated with thyroxine.
Many other drugs are responsible for heart failure in athletes, such as corticotrophin, beta 2 agonists, amphetamines and cocaine. Often athletes use combinations of different banned drugs, resulting in additive effects on cardiac remodeling. Cardiac alterations may lead to arrhythmias, heart failure and sudden death. It is important to exclude the abuse of these drugs, when athletes with heart dysfunction come to our attention. Figure 6 shows a flow chart to differentiate athlete’s heart from pathological conditions.
Figure 6.
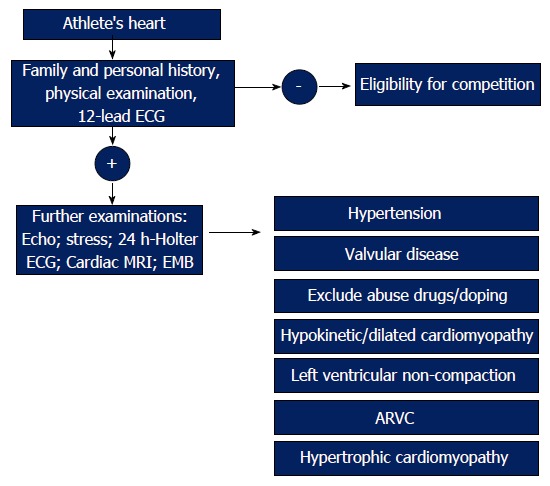
The management of athlete’s heart. The figure shows an algorithm to distinguish athlete’s heart from pathological conditions. ARVC: Arrhythmogenic right ventricular cardiomyopathy; ECG: Electrocardiograph; MRI: Magnetic resonance imaging.
ENERGY DRINK CONSUMPTION AND HEMODYNAMIC EFFECTS
A growing number of case reports of cardiovascular adverse events associated with energy drinks (EDs) are present in the literature. The use of EDs is more common in young students and in athletes. The consumption of EDs negatively affects the hemodynamic system. Important changes in arterial pressure and heart rate may occur with the ingestion of only one can (355 mL drink volume). Furthermore, it seems that EDs may diminish cerebral blood velocity, increasing breathing frequency[63]. Caffeine and sugar appear to be the ingredients underlying hemodynamic impact of EDs. Taurine and vitamin B complex play a minor role[64]. Genetic polymorphisms in cytochrome P-450 enzymes and variations of adenosine receptors play a role in the different responses to the caffeine[65]. Caffeine improves athletic performance in rowing[66], swimming[67,68], soccer[69] and hockey[70]. On the other hand, EDs can cause many cardiovascular adverse effects (Table 4), such as hypertension, palpitations, ischemic stroke, epileptic seizure[71] and myocardial ischemia, with no additional trigger[72]. The possible mechanism is related to the caffeine interaction with the G-protein coupled receptors on the cardiomyocytes that leads to an increase in intracellular cyclic-AMP and calcium concentrations with chronotropic and inotropic effects[73]. Large studies regarding EDs and their effects on the cardiovascular system are necessary, especially for the widespread consumption of these substances in recent years.
Table 4.
Adverse effects of energy drinks
| Adverse effect |
| Hypertension |
| Palpitations/arrhythmias (atrial fibrillation) |
| QTc prolongation |
| Myocardial ischemia |
| Ischemic stroke/Transient ischemic attack |
| Epileptic seizure |
| Anxiety, insomnia, irritability |
| Psychosis/Mania |
The table shows the most common adverse effect of consumption of energy drinks.
CONCLUSION
The exact clinical significance and prognostic value of cardiac injury and fibrosis in athletes are unknown. Physiological remodeling is characterized by specific molecular activation and gene expression. More large studies are needed to gain a better understanding of these conditions and the pathological changes in the heart structure in athletes and to investigate the cardiac effects of performing-enhanced drugs and EDs in this population.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): A, A, A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest exist.
Peer-review started: January 11, 2017
First decision: February 17, 2017
Article in press: April 10, 2017
P- Reviewer: Amiya E, de la Serna I, Kasai T, Ng TMH, Sicari R, Teragawa H S- Editor: Ji FF L- Editor: Wang TQ E- Editor: Li D
References
- 1.La Gerche A, Taylor AJ, Prior DL. Athlete’s heart: the potential for multimodality imaging to address the critical remaining questions. JACC Cardiovasc Imaging. 2009;2:350–363. doi: 10.1016/j.jcmg.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Lazzeroni D, Rimoldi O, Camici PG. From Left Ventricular Hypertrophy to Dysfunction and Failure. Circ J. 2016;80:555–564. doi: 10.1253/circj.CJ-16-0062. [DOI] [PubMed] [Google Scholar]
- 3.D’Andrea A, D’Andrea L, Caso P, Scherillo M, Zeppilli P, Calabrò R. The usefulness of Doppler myocardial imaging in the study of the athlete’s heart and in the differential diagnosis between physiological and pathological ventricular hypertrophy. Echocardiography. 2006;23:149–157. doi: 10.1111/j.1540-8175.2006.00186.x. [DOI] [PubMed] [Google Scholar]
- 4.La Gerche A, Burns AT, Mooney DJ, Inder WJ, Taylor AJ, Bogaert J, Macisaac AI, Heidbüchel H, Prior DL. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012;33:998–1006. doi: 10.1093/eurheartj/ehr397. [DOI] [PubMed] [Google Scholar]
- 5.D’Andrea A, La Gerche A, Golia E, Padalino R, Calabrò R, Russo MG, Bossone E. Physiologic and pathophysiologic changes in the right heart in highly trained athletes. Herz. 2015;40:369–378. doi: 10.1007/s00059-015-4220-8. [DOI] [PubMed] [Google Scholar]
- 6.La Gerche A. Can intense endurance exercise cause myocardial damage and fibrosis? Curr Sports Med Rep. 2013;12:63–69. doi: 10.1249/JSR.0b013e318287488a. [DOI] [PubMed] [Google Scholar]
- 7.Diwan A, Dorn GW. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 8.Galderisi M, Cardim N, D’Andrea A, Bruder O, Cosyns B, Davin L, Donal E, Edvardsen T, Freitas A, Habib G, et al. The multi-modality cardiac imaging approach to the Athlete’s heart: an expert consensus of the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:353. doi: 10.1093/ehjci/jeu323. [DOI] [PubMed] [Google Scholar]
- 9.Oka T, Komuro I. Molecular mechanisms underlying the transition of cardiac hypertrophy to heart failure. Circ J. 2008;72 Suppl A:A13–A16. doi: 10.1253/circj.cj-08-0481. [DOI] [PubMed] [Google Scholar]
- 10.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 11.Vatner SF. Reduced subendocardial myocardial perfusion as one mechanism for congestive heart failure. Am J Cardiol. 1988;62:94E–98E. doi: 10.1016/s0002-9149(88)80020-1. [DOI] [PubMed] [Google Scholar]
- 12.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neri Serneri GG, Boddi M, Modesti PA, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circ Res. 2001;89:977–982. doi: 10.1161/hh2301.100982. [DOI] [PubMed] [Google Scholar]
- 15.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;35:2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 17.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–666. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]
- 18.Shave R, Baggish A, George K, Wood M, Scharhag J, Whyte G, Gaze D, Thompson PD. Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol. 2010;56:169–176. doi: 10.1016/j.jacc.2010.03.037. [DOI] [PubMed] [Google Scholar]
- 19.Koller A, Schobersberger W. Post-exercise release of cardiac troponins. J Am Coll Cardiol. 2009;53:1341; author reply 1341–1342. doi: 10.1016/j.jacc.2008.12.046. [DOI] [PubMed] [Google Scholar]
- 20.Hessel MH, Atsma DE, van der Valk EJ, Bax WH, Schalij MJ, van der Laarse A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455:979–986. doi: 10.1007/s00424-007-0354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shave R, George KP, Atkinson G, Hart E, Middleton N, Whyte G, Gaze D, Collinson PO. Exercise-induced cardiac troponin T release: a meta-analysis. Med Sci Sports Exerc. 2007;39:2099–2106. doi: 10.1249/mss.0b013e318153ff78. [DOI] [PubMed] [Google Scholar]
- 22.Siscovick DS, Weiss NS, Fletcher RH, Lasky T. The incidence of primary cardiac arrest during vigorous exercise. N Engl J Med. 1984;311:874–877. doi: 10.1056/NEJM198410043111402. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Serfass RC, Mackey-Bojack SM, Kelly KL, Titus JL, Apple FS. Cardiac troponin T alterations in myocardium and serum of rats after stressful, prolonged intense exercise. J Appl Physiol (1985) 2000;88:1749–1755. doi: 10.1152/jappl.2000.88.5.1749. [DOI] [PubMed] [Google Scholar]
- 24.Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif JC, Brugada J, Nattel S, Mont L. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. 2011;123:13–22. doi: 10.1161/CIRCULATIONAHA.110.938282. [DOI] [PubMed] [Google Scholar]
- 25.Dello Russo A, Pieroni M, Santangeli P, Bartoletti S, Casella M, Pelargonio G, Smaldone C, Bianco M, Di Biase L, Bellocci F, et al. Concealed cardiomyopathies in competitive athletes with ventricular arrhythmias and an apparently normal heart: role of cardiac electroanatomical mapping and biopsy. Heart Rhythm. 2011;8:1915–1922. doi: 10.1016/j.hrthm.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 26.Trivax JE, McCullough PA. Phidippides cardiomyopathy: a review and case illustration. Clin Cardiol. 2012;35:69–73. doi: 10.1002/clc.20994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Limongelli G, Rea A, Masarone D, Francalanci MP, Anastasakis A, Calabro’ R, Giovanna RM, Bossone E, Elliott PM, Pacileo G. Right ventricular cardiomyopathies: a multidisciplinary approach to diagnosis. Echocardiography. 2015;32 Suppl 1:S75–S94. doi: 10.1111/echo.12399. [DOI] [PubMed] [Google Scholar]
- 28.D’Andrea A, Riegler L, Rucco MA, Cocchia R, Scarafile R, Salerno G, Martone F, Vriz O, Caso P, Calabrò R, et al. Left atrial volume index in healthy subjects: clinical and echocardiographic correlates. Echocardiography. 2013;30:1001–1007. doi: 10.1111/echo.12217. [DOI] [PubMed] [Google Scholar]
- 29.Iskandar A, Mujtaba MT, Thompson PD. Left Atrium Size in Elite Athletes. JACC Cardiovasc Imaging. 2015;8:753–762. doi: 10.1016/j.jcmg.2014.12.032. [DOI] [PubMed] [Google Scholar]
- 30.Rundqvist L, Engvall J, Faresjö M, Carlsson E, Blomstrand P. Regular endurance training in adolescents impacts atrial and ventricular size and function. Eur Heart J Cardiovasc Imaging. 2016 doi: 10.1093/ehjci/jew150. [DOI] [PubMed] [Google Scholar]
- 31.D’Ascenzi F, Solari M, Anselmi F, Maffei S, Focardi M, Bonifazi M, Mondillo S, Henein M. Atrial chamber remodelling in healthy pre-adolescent athletes engaged in endurance sports: A study with a longitudinal design. The CHILD study. Int J Cardiol. 2016;223:325–330. doi: 10.1016/j.ijcard.2016.08.231. [DOI] [PubMed] [Google Scholar]
- 32.D’Andrea A, Bossone E, Radmilovic J, Riegler L, Pezzullo E, Scarafile R, Russo MG, Galderisi M, Calabrò R. Exercise-Induced Atrial Remodeling: The Forgotten Chamber. Cardiol Clin. 2016;34:557–565. doi: 10.1016/j.ccl.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Calvo N, Brugada J, Sitges M, Mont L. Atrial fibrillation and atrial flutter in athletes. Br J Sports Med. 2012;46 Suppl 1:i37–i43. doi: 10.1136/bjsports-2012-091171. [DOI] [PubMed] [Google Scholar]
- 34.Wilhelm M, Roten L, Tanner H, Wilhelm I, Schmid JP, Saner H. Atrial remodeling, autonomic tone, and lifetime training hours in nonelite athletes. Am J Cardiol. 2011;108:580–585. doi: 10.1016/j.amjcard.2011.03.086. [DOI] [PubMed] [Google Scholar]
- 35.Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, Yang W, Dorian D, Simpson JA, Tuomi JM, et al. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFα. Nat Commun. 2015;6:6018. doi: 10.1038/ncomms7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mont L, Tamborero D, Elosua R, Molina I, Coll-Vinent B, Sitges M, Vidal B, Scalise A, Tejeira A, Berruezo A, et al. Physical activity, height, and left atrial size are independent risk factors for lone atrial fibrillation in middle-aged healthy individuals. Europace. 2008;10:15–20. doi: 10.1093/europace/eum263. [DOI] [PubMed] [Google Scholar]
- 37.D’Andrea A, Riegler L, Golia E, Cocchia R, Scarafile R, Salerno G, Pezzullo E, Nunziata L, Citro R, Cuomo S, et al. Range of right heart measurements in top-level athletes: the training impact. Int J Cardiol. 2013;164:48–57. doi: 10.1016/j.ijcard.2011.06.058. [DOI] [PubMed] [Google Scholar]
- 38.D’Ascenzi F, Cameli M, Padeletti M, Lisi M, Zacà V, Natali B, Malandrino A, Alvino F, Morelli M, Vassallo GM, et al. Characterization of right atrial function and dimension in top-level athletes: a speckle tracking study. Int J Cardiovasc Imaging. 2013;29:87–94. doi: 10.1007/s10554-012-0063-z. [DOI] [PubMed] [Google Scholar]
- 39.Pelliccia A, Maron MS, Maron BJ. Assessment of left ventricular hypertrophy in a trained athlete: differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog Cardiovasc Dis. 2012;54:387–396. doi: 10.1016/j.pcad.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–1075. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 41.Pelliccia A, Maron BJ, De Luca R, Di Paolo FM, Spataro A, Culasso F. Remodeling of left ventricular hypertrophy in elite athletes after long-term deconditioning. Circulation. 2002;105:944–949. doi: 10.1161/hc0802.104534. [DOI] [PubMed] [Google Scholar]
- 42.Caselli S, Maron MS, Urbano-Moral JA, Pandian NG, Maron BJ, Pelliccia A. Differentiating left ventricular hypertrophy in athletes from that in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2014;114:1383–1389. doi: 10.1016/j.amjcard.2014.07.070. [DOI] [PubMed] [Google Scholar]
- 43.Pelliccia A, Maron BJ, Spataro A, Proschan MA, Spirito P. The upper limit of physiologic cardiac hypertrophy in highly trained elite athletes. N Engl J Med. 1991;324:295–301. doi: 10.1056/NEJM199101313240504. [DOI] [PubMed] [Google Scholar]
- 44.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975. doi: 10.1002/ejhf.592. [DOI] [PubMed] [Google Scholar]
- 45.Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666–671. doi: 10.1136/heart.86.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caselli S, Ferreira D, Kanawati E, Di Paolo F, Pisicchio C, Attenhofer Jost C, Spataro A, Jenni R, Pelliccia A. Prominent left ventricular trabeculations in competitive athletes: A proposal for risk stratification and management. Int J Cardiol. 2016;223:590–595. doi: 10.1016/j.ijcard.2016.08.272. [DOI] [PubMed] [Google Scholar]
- 47.La Gerche A, Claessen G. Is exercise good for the right ventricle? Concepts for health and disease. Can J Cardiol. 2015;31:502–508. doi: 10.1016/j.cjca.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 48.Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42:1959–1963. doi: 10.1016/j.jacc.2003.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation. 2009;119:1085–1092. doi: 10.1161/CIRCULATIONAHA.108.804617. [DOI] [PubMed] [Google Scholar]
- 50.Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. doi: 10.1161/01.CIR.0000103130.33451.D2. [DOI] [PubMed] [Google Scholar]
- 51.Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, Estes NA, Marcus F, Scheinman MM. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 2009;54:609–615. doi: 10.1016/j.jacc.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaidi A, Sheikh N, Jongman JK, Gati S, Panoulas VF, Carr-White G, Papadakis M, Sharma R, Behr ER, Sharma S. Clinical Differentiation Between Physiological Remodeling and Arrhythmogenic Right Ventricular Cardiomyopathy in Athletes With Marked Electrocardiographic Repolarization Anomalies. J Am Coll Cardiol. 2015;65:2702–2711. doi: 10.1016/j.jacc.2015.04.035. [DOI] [PubMed] [Google Scholar]
- 53.Bauce B, Frigo G, Benini G, Michieli P, Basso C, Folino AF, Rigato I, Mazzotti E, Daliento L, Thiene G, et al. Differences and similarities between arrhythmogenic right ventricular cardiomyopathy and athlete’s heart adaptations. Br J Sports Med. 2010;44:148–154. doi: 10.1136/bjsm.2007.042853. [DOI] [PubMed] [Google Scholar]
- 54.D’Andrea A, Limongelli G, Morello A, Mattera Iacono A, Russo MG, Bossone E, Calabrò R, Pacileo G. Anabolic-androgenic steroids and athlete’s heart: When big is not beautiful....! Int J Cardiol. 2016;203:486–488. doi: 10.1016/j.ijcard.2015.10.186. [DOI] [PubMed] [Google Scholar]
- 55.Montisci M, El Mazloum R, Cecchetto G, Terranova C, Ferrara SD, Thiene G, Basso C. Anabolic androgenic steroids abuse and cardiac death in athletes: morphological and toxicological findings in four fatal cases. Forensic Sci Int. 2012;217:e13–e18. doi: 10.1016/j.forsciint.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 56.Pope HG, Wood RI, Rogol A, Nyberg F, Bowers L, Bhasin S. Adverse health consequences of performance-enhancing drugs: an Endocrine Society scientific statement. Endocr Rev. 2014;35:341–375. doi: 10.1210/er.2013-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu H, Bravata DM, Olkin I, Friedlander A, Liu V, Roberts B, Bendavid E, Saynina O, Salpeter SR, Garber AM, et al. Systematic review: the effects of growth hormone on athletic performance. Ann Intern Med. 2008;148:747–758. doi: 10.7326/0003-4819-148-10-200805200-00215. [DOI] [PubMed] [Google Scholar]
- 58.Palmeiro CR, Anand R, Dardi IK, Balasubramaniyam N, Schwarcz MD, Weiss IA. Growth hormone and the cardiovascular system. Cardiol Rev. 2012;20:197–207. doi: 10.1097/CRD.0b013e318248a3e1. [DOI] [PubMed] [Google Scholar]
- 59.Cittadini A, Strömer H, Katz SE, Clark R, Moses AC, Morgan JP, Douglas PS. Differential cardiac effects of growth hormone and insulin-like growth factor-1 in the rat. A combined in vivo and in vitro evaluation. Circulation. 1996;93:800–809. doi: 10.1161/01.cir.93.4.800. [DOI] [PubMed] [Google Scholar]
- 60.Noakes TD. Tainted glory--doping and athletic performance. N Engl J Med. 2004;351:847–849. doi: 10.1056/NEJMp048208. [DOI] [PubMed] [Google Scholar]
- 61.Weltman NY, Wang D, Redetzke RA, Gerdes AM. Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. PLoS One. 2012;7:e46655. doi: 10.1371/journal.pone.0046655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ching GW, Franklyn JA, Stallard TJ, Daykin J, Sheppard MC, Gammage MD. Cardiac hypertrophy as a result of long-term thyroxine therapy and thyrotoxicosis. Heart. 1996;75:363–368. doi: 10.1136/hrt.75.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grasser EK, Yepuri G, Dulloo AG, Montani JP. Cardio- and cerebrovascular responses to the energy drink Red Bull in young adults: a randomized cross-over study. Eur J Nutr. 2014;53:1561–1571. doi: 10.1007/s00394-014-0661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miles-Chan JL, Charrière N, Grasser EK, Montani JP, Dulloo AG. The blood pressure-elevating effect of Red Bull energy drink is mimicked by caffeine but through different hemodynamic pathways. Physiol Rep. 2015;3:pii: e12290. doi: 10.14814/phy2.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang A, Palmer AA, de Wit H. Genetics of caffeine consumption and responses to caffeine. Psychopharmacology (Berl) 2010;211:245–257. doi: 10.1007/s00213-010-1900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruce CR, Anderson ME, Fraser SF, Stepto NK, Klein R, Hopkins WG, Hawley JA. Enhancement of 2000-m rowing performance after caffeine ingestion. Med Sci Sports Exerc. 2000;32:1958–1963. doi: 10.1097/00005768-200011000-00021. [DOI] [PubMed] [Google Scholar]
- 67.MacIntosh BR, Wright BM. Caffeine ingestion and performance of a 1,500-metre swim. Can J Appl Physiol. 1995;20:168–177. doi: 10.1139/h95-012. [DOI] [PubMed] [Google Scholar]
- 68.Collomp K, Ahmaidi S, Chatard JC, Audran M, Préfaut C. Benefits of caffeine ingestion on sprint performance in trained and untrained swimmers. Eur J Appl Physiol Occup Physiol. 1992;64:377–380. doi: 10.1007/BF00636227. [DOI] [PubMed] [Google Scholar]
- 69.Lara B, Gonzalez-Millán C, Salinero JJ, Abian-Vicen J, Areces F, Barbero-Alvarez JC, Muñoz V, Portillo LJ, Gonzalez-Rave JM, Del Coso J. Caffeine-containing energy drink improves physical performance in female soccer players. Amino Acids. 2014;46:1385–1392. doi: 10.1007/s00726-014-1709-z. [DOI] [PubMed] [Google Scholar]
- 70.Del Coso J, Portillo J, Salinero JJ, Lara B, Abian-Vicen J, Areces F. Caffeinated Energy Drinks Improve High-Speed Running in Elite Field Hockey Players. Int J Sport Nutr Exerc Metab. 2016;26:26–32. doi: 10.1123/ijsnem.2015-0128. [DOI] [PubMed] [Google Scholar]
- 71.Gray B, Das K J, Semsarian C. Consumption of energy drinks: a new provocation test for primary arrhythmogenic diseases? Int J Cardiol. 2012;159:77–78. doi: 10.1016/j.ijcard.2012.05.121. [DOI] [PubMed] [Google Scholar]
- 72.Sanchis-Gomar F, Leischik R, Lippi G. Energy drinks: Increasing evidence of negative cardiovascular effects. Int J Cardiol. 2016;206:153. doi: 10.1016/j.ijcard.2016.01.107. [DOI] [PubMed] [Google Scholar]
- 73.Higgins JP, Tuttle TD, Higgins CL. Energy beverages: content and safety. Mayo Clin Proc. 2010;85:1033–1041. doi: 10.4065/mcp.2010.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]