

Abstract
Introduction
Certain lipids have been shown to be ligands for a subgroup of the nuclear hormone receptor superfamily known as the peroxisome proliferator-activated receptors (PPARs). Ligands for these transcription factors have been used in experimental cancer therapies. PPARs heterodimerize and bind DNA with retinoid X receptors (RXRs), which have homology to other members of the nuclear receptor superfamily. Retinoids have been found to be effective in treating many types of cancer. However, many breast cancers become resistant to the chemotherapeutic effects of these drugs. Recently, RXR-selective ligands were discovered that inhibited proliferation of all-trans retinoic acid resistant breast cancer cells in vitro and caused regression of the disease in animal models. There are few published studies on the efficacy of combined therapy using PPAR and RXR ligands for breast cancer prevention or treatment.
Methods
We determined the effects of selective PPAR and RXR ligands on established human breast cancer cell lines in vitro.
Results
PPAR-α and PPAR-γ ligands induced apoptotic and antiproliferative responses in human breast cancer cell lines, respectively, which were associated with specific changes in gene expression. These responses were potentiated by the RXR-selective ligand AGN194204. Interestingly, RXR-α-overexpressing retinoic acid resistant breast cancer cell lines were more sensitive to the effects of the RXR-selective compound.
Conclusion
RXR-selective retinoids can potentiate the antiproliferative and apoptotic responses of breast cancer cell lines to PPAR ligands.
Keywords: cell cycle, chemotherapy, hormones, nuclear receptors, transcription factors
Introduction
Within the past few years, specific lipids have been shown to be ligands for a subgroup of the nuclear hormone receptor superfamily known as the peroxisome proliferator-activated receptors (PPARs; for review, see Mandrup and Lane [1]). The first of these ligand-dependent transcription factors, namely PPAR-α, was originally cloned as an orphan receptor that was activated by agents that induce peroxisome proliferation in the liver (for review, see Keller and Wahli [2]). Subsequently, PPAR-δ (also known as PPAR-β) and PPAR-γ were cloned by low stringency screening [3]. Like other members of the nuclear receptor superfamily, PPARs have functional domains for DNA and ligand binding [4]. PPARs bind to recognition sequences in the promoter regions of their target genes to regulate transcription. These sequences typically contain a direct repeat of the motif PuGGTCA separated by one nucleotide; this sequence is recognized by other members of the nuclear hormone receptor superfamily, such as retinoid X receptors (RXRs) [5]. These receptors exhibit markedly different tissue distributions, DNA binding affinities, and ligand preferences. PPARs are activated by long-chain fatty acids (for review, see Kliewer and coworkers [6]). PPAR-α can be selectively activated by hydroxyeicosatetraenoic acid (HETE) and fibrates, whereas PPAR-γ preferentially binds γ-linolenic acid (LA), prostaglandin J2 metabolites, and thiazolidinedione drugs (TZDs) [6,7].
Given the ability of PPARs to induce differentiation of a variety of cell types, ligands for these transcription factors have been used in experimental cancer therapies (for review, see Roberts-Thomson [8]). TZD administration to liposarcoma patients induced histologic and biochemical differentiation markers in vivo [9]. TZD treatment of human bladder carcinoma cells in vitro inhibited cellular proliferation via increased cyclin-dependent kinase (Cdk) inhibitor expression and induced cell death [10]. Similarly, the growth of human lung cancer cell lines was inhibited via apoptosis when cultures were treated with TZDs but not the PPAR-γ agonist bezafibrate [11]. TZDs also induced differentiation of human colon cancer cell lines in vitro with concomitant growth arrest [12]. Loss of function mutations in PPARs that impaired ligand binding have also been found in colon cancer [13]. Human breast cancer cell lines express PPARs [14], and fatty acids were able to stimulate activation of a reporter gene via PPAR-γ [15,16]. As seen in other cancers, TZDs induced growth arrest and differentiation in breast carcinoma cells in vitro and in animal models [17,18]. However, a small phase II study of the PPAR-γ ligand troglitazone failed to identify objective responses in patients with refractory breast cancer [19].
PPARs heterodimerize and bind DNA with RXRs, which have homology to other members of the nuclear receptor superfamily (for review, see Mangelsdorf and Evans [20]). The natural ligand for RXRs is 9-cis retinoic acid (RA) [21]. RXR heterodimers typically bind to the 5' half of the DNA direct repeat and may also homodimerize [22]. However, in PPAR/RXR heterodimers, the retinoid receptor occupies the 3' half of the response element [5]. Heterodimerization greatly enhances DNA binding and transcriptional activation [23,24]. RXR overexpression has been reported to enhance the transcriptional response to ligand binding [25].
Retinoids have been effective in vitro against many types of cancer, including that of the breast [26-30]. However, many breast cancers become resistant to the chemotherapeutic effects of these drugs [31-33]. Recently, RXR-selective ligands were discovered that inhibited proliferation of all-trans RA resistant breast cancer cells in vitro and caused regression of the disease in animal models [34]. These RXR-selective ligands did not produce the side effects that are normally associated with retinoid chemotherapy [35]. However, a phase II trial using one of these ligands (bexarotene) showed limited response in patients with metastatic breast cancer [36]. Interestingly, a high-risk lesion for invasive breast cancer, ductal carcinoma in situ, has been shown to overexpress RXR-α [37].
The effects of PPAR and RXR ligands on established breast cancer cell lines require further study. The evaluation of new RXR ligands that cooperate with PPARs may result in important therapeutic breakthroughs. To provide a molecular and cell biologic basis for these studies, we examined the effects of selective PPAR and RXR ligands on established breast cancer cell lines in vitro. We determined that the RXR-selective ligand potentiated the antiproliferative and apoptotic responses to PPAR ligands in human breast cancer cell lines. RA-resistant lines that overexpressed RXR-α were more sensitive to the effects of these ligands, suggesting potential therapeutic uses for these compounds.
Methods
Cell culture and stable transfection
The human breast cancer cell lines used in the present study were purchased from the American Type Culture Collection. Cells were cultured in Dulbecco's modified Eagle's medium, 10% fetal bovine serum, and 40 μg/ml gentamicin at 37°C in a humidified atmosphere of 5% carbon dioxide. T47D cells were transfected with 5 μg expression vector for RXR-α (kindly provided by Dr Ronald Evans) or neomycin resistance plasmid alone using Lipofectamine reagent (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer's recommendations. Cells were selected in 400 μg/ml G418 for 14 days. Resistant clones were picked for expansion and characterization. RXR-α expression was determined by western blot (see below).
Cell death and proliferation assays
The dUTP nick-end labeling (TUNEL) assay for in situ cell death detection was performed using a commercially available kit (Roche Molecular Biochemicals, Indianapolis, IN, USA). Cells were treated with 0.1% dimethyl sulfoxide vehicle, 100 μmol/l HETE, 100 μmol/l LA, or 100 nmol/l of the RXR-selective retinoid AGN194204. Cells were fixed in 4% paraformaldehyde (pH 7.4) and permeabilized with 0.1% Triton X-100, 0.1% sodium citrate for 2 min on ice. A mouse IgM anti-human Fas antibody (Molecular Biology Laboratories, Woburn, MA, USA) that induces apoptosis in sensitive cell lines was used as the positive control. An isotype matched control antibody was used as the negative control. After washing with phosphate-buffered saline (PBS), cells were incubated with fluorescein-dUTP and terminal deoxynucleotidyl transferase for 60 min at 37°C, in accordance with the manufacturer's recommendations (Roche Molecular Biochemicals). After washing three times in PBS, apoptotic cells were visualized by fluorescence microscopy. Proliferation was analyzed by plating 5 × 104 cells in triplicate cultures and treating with vehicle, PPAR, or RXR selective ligands for up to 6 days. Cells were trypsinized and counted with a hemacytometer at 2-day intervals.
To determine G1 to S phase progression, bromodeoxyuridine (BrdU) incorporation analysis was performed. Vehicle and ligand treated cells were incubated with 10 μmol/l BrdU for 1 hour, washed in PBS, and fixed in 70% ethanol containing 50 mmol/l glycine (pH 2) for 20 min at -20°C. After washing in PBS, the cells were incubated with mouse anti-BrdU primary antibody for 30 min at 37°C, in accordance with the manufacturer's recommendations (Roche Molecular Biochemicals). The cells were washed in PBS and incubated with anti-mouse IgG secondary antibody conjugated to fluorescein. Following extensive washing in PBS, the number of BrdU-positive cells were visualized by fluorescence microscopy and expressed as a percentage of the total cells counted in 10 random high-power fields.
Western blot
Total protein (75 μg) from vehicle or ligand treated cells was separated by SDS-PAGE on 10% resolving gels under denaturing and reducing conditions. Separated proteins were electroblotted to polyvinylidene difluoride membranes, in accordance with the manufacturer's recommendations (Bio-Rad, Hercules, CA, USA). Blots were incubated with anti-human primary antibodies to PPARs, retinoic acid receptors (RARs), RXRs, cyclins, Cdks, epidermal growth factor receptor, c-met, extracellular signal-regulated kinases (ERKs), p38, c-myc, AP-1 proteins, caspases, and β-actin (Transduction Laboratories, San Diego, CA, USA and Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 16 hours at 4°C. After washing in Tris-buffered saline containing 0.1% Tween 20 (pH 7.5), blots were incubated for 30 min at room temperature with anti-IgG secondary antibody conjugated to horseradish peroxidase. After extensive washing in Tris-buffered saline containing 0.1% Tween 20 at room temperature, bands were visualized by the enhanced chemiluminescence method (Roche Molecular Biochemicals).
Immunoprecipitation
Cells were lysed in 50 mmol/l HEPES (pH 7.5), 150 mmol/l NaCl, 1 mmol/l EDTA, 2.5 mmol/l EGTA, 1 mmol/l DTT, 1% Nonidet P-40, 10% glycerol, and protease inhibitors for 30 min at 4°C. Lysates were centrifuged at 10,000 g for 10 min and anti-human primary antibody to RXR-α (Santa Cruz Biotechnology) was incubated with the supernatants for 1 hour at 4°C. Antigen–antibody complexes were precipitated by incubation with protein A/G agarose (Santa Cruz Biotechnology) for 1 hour at 4°C. Immunoprecipitated proteins were washed three times with 1 ml lysis buffer. Immunoprecipitated protein complexes were separated by SDS-PAGE as described above. Blots were incubated with anti-PPAR antibodies for 16 hours at 4°C. Blots were stripped and incubated with anti-RXR-α antibody to determine amounts of immunoprecipitated protein in each lane.
Transient transfection and reporter gene assays
Human breast cancer cell lines were plated in triplicate into six-well tissue culture plates. Cells were transiently transfected with 5 μg of a heterologous promoter containing a direct repeat of the PPAR recognition sequence (5'-AGGTCAGAGGTCA-3') cloned in the pGL3 luciferase reporter vector (Promega, Madison, WI, USA) using Lipofectamine (Invitrogen), in accordance with the manufacturer's recommendations. β-Galactosidase expression vector 1 μg was used to normalize for transfection efficiency. Cells were treated with vehicle, 100 μmol/l LA, 100 nmol/l all-trans RA, 100 nmol/l AGN194204, or a combination of LA plus RA or LA plus AGN194204 for 24 hours following transfection. Reporter gene activity was determined using a commercially available kit (Dual-light; Tropix, Foster City, CA, USA). Luciferase activity was normalized to β-galactosidase levels for each sample.
Results
Expression of functional PPAR and RXR proteins
To determine the expression of PPARs, RARs, and RXRs in human breast cancer cells, we analyzed six lines by western blot. As shown in Fig. 1a, all human breast cancer cell lines expressed low levels of PPAR-α. Expression of PPAR-α was sixfold higher in Hs578T cells than in the other five lines. Relative expression of PPAR-γ was considerably higher than that of PPAR-α; however, PPAR-γ expression was threefold lower in MCF7 and Hs578T cells than in other lines. RAR-α protein was detected in all cell lines with the exception of T47D, and expression was generally lower in RA-resistant lines such as MDA-MB-231 and MDA-MB-468. RAR-β expression was below the limit of detection for western blot with the exception of the T47D and SKBR3 lines, in which low levels of the protein were detected. RXR-α expression was low or undetectable in most lines, but it was threefold higher in MDA-MB-468 and SKBR3 cells. We did not detect expression of other PPAR, RAR, or RXR proteins in these cell lines. We concluded that PPARs and RAR-α proteins were expressed in human breast cancer cell lines, with higher RXR-α levels in two out of four estrogen receptor negative lines.
Figure 1.
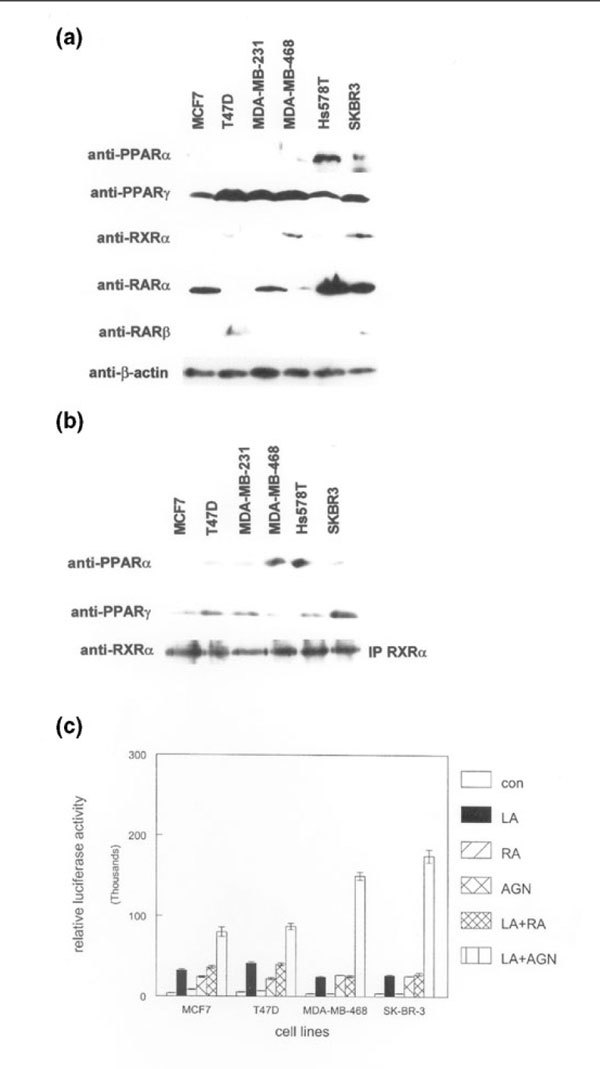
Expression of functionally interacting peroxisome proliferator-activated receptors (PPARs), retinoic acid receptors (RARs), and retinoid X receptors (RXRs) in human breast cancer cell lines. (a) Whole cell lysates from the indicated breast cancer cell lines were subjected to western blot analysis using the anti-PPAR, anti-RAR, anti-RXR, and anti-β-actin antibodies shown on the left. Relative expression of each protein was determined using the same membrane. (b) Human RXR-α protein was immunoprecipitated (IP RXRα) from the indicated breast cancer cell lines. Immunoprecipitated complexes were subjected to western blotting using anti-PPAR-α and anti-PPAR-γ antibodies. Blots were stripped and incubated with anti-RXR-α antibody to determine the relative amounts of immunoprecipitated protein in each lane. These experiments were performed three times, with similar results. Representative blots are shown. (c) An RXR-selective compound potentiates transcriptional activation by a PPAR ligand. The indicated human breast cancer cell lines were transiently transfected with a heterologous PPAR-responsive promoter/reporter construct and treated with 100 μmol/l of the PPAR ligand γ-linolenic acid (LA), 100 nmol/l of the pan RAR agonist all-trans retinoic acid (RA), 100 nmol/l of the RXR-selective ligand AGN194204 (AGN), γ-linolenic acid and retinoic acid (LA+R), or γ-linolenic acid plus AGN194204 (LA+A) for 24 hours before determination of promoter activity. Promoter activity was represented as relative light units from the luciferase reporter. These experiments were performed three times with similar results. Error bars indicate the standard error of the mean.
PPARs have been shown to heterodimerize with RXRs to activate gene transcription [20]. To determine whether PPAR-α and PPAR-γ could dimerize with RXR-α in human breast cancer lines, we immunoprecipitated the retinoid receptor from these cells. As shown in Fig. 1b, RXR-α protein could be detected by the more sensitive immunoprecipitation technique in all cell lines tested. To determine whether PPARs associated with RXR-α in these lines, we incubated these blots with anti-PPAR-α and PPAR-γ antibodies. Both PPARs immunoprecipitated with RXR-α protein in all cell lines. These results indicate that PPARs and RXR-α interact in human breast cancer cell lines.
To determine whether these heterodimers could activate a PPAR-responsive promoter, we transiently transfected four breast cancer cell lines with a heterologous PPAR promoter vector and determined reporter gene activity in response to selective ligands. As shown in Fig. 1c, treatment with 100 μmol/l LA (a selective PPAR-γ ligand) resulted in threefold to fourfold induction of promoter activity in all cell lines. Treatment with 100 nmol/l of the RXR-selective compound AGN194204 produced twofold to threefold increases in promoter activity in these lines. However, treatment with the RAR agonist all-trans RA failed to induce promoter activity, indicating the specificity of this promoter construct. Treatment of transiently transfected breast cancer cell lines with LA and RA simultaneously did not induce activity above that observed with LA alone. However, combining LA with AGN194204 resulted in an eightfold induction of promoter activity in MCF7 and T47D lines, and 15- to 17-fold induction in MDA-MB-468 and SKBR3 cells, which express higher levels of RXR-α. These results indicate that PPAR/RXR heterodimers are functional in human breast cancer lines, and that RXR-selective ligands can potentiate PPAR-induced transcriptional activation in cells with higher levels of RXR-α.
PPAR-γ/RXR synergy results in antiproliferative effects on human breast cancer cell lines
These findings suggest that the RXR-selective ligand AGN194204 may be more biologically active against RXR-α overexpressing breast cancer cell lines. To examine this possibility, we evaluated the proliferative response of this compound as a single agent or in combination with PPAR-selective ligands. As shown in Fig. 2a, treatment with 100 nmol/l AGN194204 slightly inhibited proliferation of T47D cells. The PPAR-γ selective ligand LA was a more effective antiproliferative agent in vitro, producing 30% inhibition of cell growth in these assays. Simultaneous treatment with both ligands resulted in an additive effect, producing 40% growth inhibition of T47D cells. In contrast, AGN194204 was a highly effective antiproliferative compound (70% growth inhibition) when used as a single agent on MDA-MB-468 cells, which express higher levels of RXR-α (Fig. 2b). Similar results were obtained using SKBR3 cells (data not shown). LA treatment of MDA-MB-468 cells resulted in 20% growth inhibition when used as a single agent, but because of the magnitude of the AGN194204 effect we did not observe additional antiproliferative activity when both ligands were used together.
Figure 2.
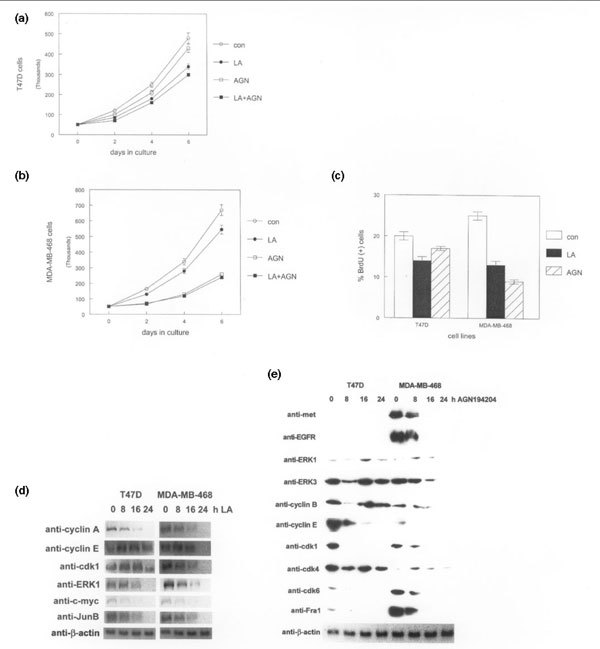
Greater peroxisome proliferator-activated receptor (PPAR)-selective antiproliferative and apoptotic responses potentiated by AGN194204 in retinoid X receptor (RXR)-α-overexpressing human breast cancer cell lines. (a) Triplicate cultures of estrogen receptor positive/retinoic acid (RA)-sensitive T47D cells were treated for 6 days with vehicle (con), 100 μmol/l of the PPAR-γ ligand γ-linolenic acid (LA), 100 nmol/l AGN194204 (AGN), or both compounds simultaneously (LA+AGN). Proliferation was monitored by cell counting at 2-day intervals using a hemacytometer. (b) Triplicate cultures of estrogen receptor negative/RA-resistant MDA-MB-468 cells were treated for 6 days with vehicle (con), 100 μmol/l LA, 100 nmol/l AGN194204, or both compounds simultaneously (LA+AGN). Proliferation was monitored by cell counting at 2-day intervals using a hemacytometer. (c) LA and AGN194204 inhibit S-phase progression in human breast cancer cell lines. The indicated cell lines were treated with 100 μmol/l LA or 100 nmol/l AGN194204 (AGN). The percentage of S-phase cells was determined by bromodeoxyuridine (BrdU) incorporation assay. (d) PPAR- and RXR-selective ligands alter expression of nonoverlapping sets of cell cycle regulatory proteins. Whole cell lysates from the indicated human breast cancer cell lines treated with 100 μmol/l LA for up to 24 hours were subjected to western blot with the antibodies shown on the left. Relative expression of each protein was determined using the same membrane. (e) Whole cell lysates from the indicated human breast cancer cell lines treated with 100 nmol/l AGN194204 for up to 24 hours were subjected to western blot with the antibodies shown on the left. Relative expression of each protein was determined using the same membrane.
To determine whether LA and AGN194204 inhibited S-phase progression in human breast cancer cell lines, we performed BrdU incorporation analysis. As shown in Fig. 2c, LA treatment reduced the number of BrdU positive cells by 30–50%. AGN194204 had little effect on BrdU incorporation in T47D cells, but it inhibited S-phase progression by 70% in the MDA-MB-468 line. These data indicate that the RXR-selective ligand has substantial antiproliferative activity in vitro against breast cancer cell lines expressing higher levels of RXR-α.
To determine changes in expression of genes that regulate cell cycle progression resulting from LA treatment of human breast cancer cell lines, we examined protein levels by western blot. As shown in Fig. 2d, most alterations in the expression of these genes did not occur until 16 hours after LA exposure. Cyclin E levels decreased by twofold at this time point in T47D cells and was undetectable in the MDA-MB-468 line. Cdk1 levels also decreased in both cell lines by the 16-hour and 24-hour timepoints. By 24 hours, markedly decreased protein levels of a number of cell cycle regulatory genes were noted, including ERK1, cyclin A, c-myc, and JunB. The majority of these genes are involved in regulating S-phase progression in many cell types. Regulation of G2 phase proteins such as cyclin B were unaffected by LA treatment (data not shown). These results indicate that LA-induced changes in gene expression correlate with delayed S-phase progression, as shown by BrdU incorporation analysis in human breast cancer cell lines.
Treatment with the RXR-selective ligand AGN194204 altered expression of a larger group of genes, including those encoding growth factor receptors, mitogen-activated protein kinases, cyclins, and Cdks. As shown in Fig. 2e, expression of epidermal growth factor receptor and the scatter factor receptor c-met, which are overexpressed in MDA-MB-468 cells, was markedly inhibited by 16 hours of exposure to AGN194204. Decreased expression of these receptors in MDA-MB-468 cells correlated with reduced levels of downstream signaling proteins in the mitogen-activated protein kinase pathway such as ERK1 and ERK3. In contrast, levels of these proteins were largely unaffected in T47D cells, which are less sensitive to the antiproliferative effects of AGN194204. Cyclin B protein levels were also decreased by 16 hours in the MDA-MB-468 line, but were largely unaffected in T47D cells except for a transient decrease observed at 8 hours. Expression of cyclin E, Cdk1, Cdk6, and Fra-1 was inhibited by twofold to fourfold in both cell lines by 8 hours; these genes represent the earliest observed changes in response to AGN194204 in these cell lines. In contrast, Cdk4 levels were largely unaffected by AGN194204 in either cell line, indicating that the observed changes in gene expression were specific. Therefore, although some of the AGN194204-induced changes in gene expression overlapped with those regulated by LA, others were specific for each selective ligand. These data indicate that increased sensitivity of RXR-α overexpressing human breast cancer cell lines to AGN194204 is reflected in changes in gene expression.
PPAR-α/RXR synergy results in apoptotic effects on human breast cancer cell lines
In contrast to the effects of LA, treatment of breast cancer cell lines with 100 nmol/l of the PPAR-α selective ligand HETE resulted in nuclear condensation, cell rounding, and detachment from the culture dish, which is indicative of apoptosis (data not shown). To confirm this interpretation, we performed TUNEL assays to measure apoptotic cell death in vitro. As shown in Fig. 3a, HETE treatment resulted in 35–50% TUNEL-positive cells after 24 hours. Treatment with 100 nmol/l AGN194204 alone did not produce significant increases in the number of apoptotic cells. However, treatment with a combination of HETE and AGN194204 resulted in 50–75% TUNEL-positive cells after 24 hours. This effect was more pronounced in SKBR3 and MDA-MB-468 cells, which express higher levels of RXR-α protein. These results indicate that selective PPAR activation can produce different cellular responses in human breast cancer cell lines and that the RXR-selective ligand can potentiate both antiproliferative and apoptotic effects.
Figure 3.
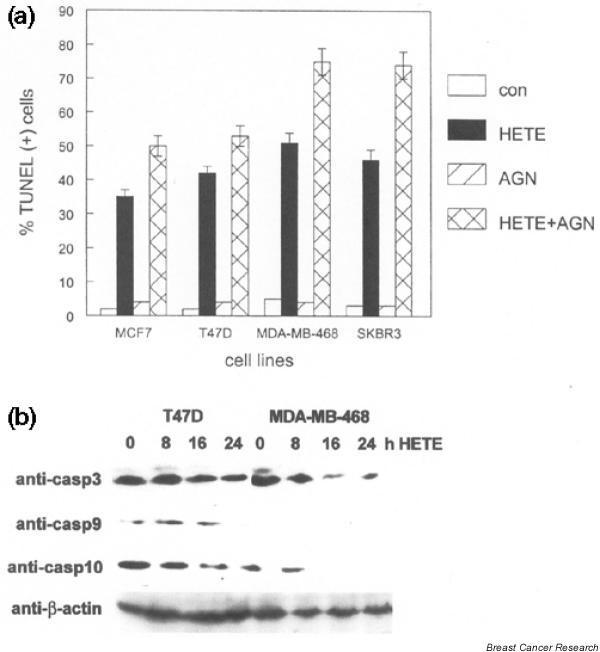
The peroxisome proliferator-activated receptor (PPAR)-α-selective ligand hydroxyeicosatetraenoic acid (HETE) induces apoptosis in human breast cancer cell lines, which is potentiated by AGN194204. (a) The indicated breast cancer cell lines were treated with vehicle (con), 100 μmol/l HETE, 100 nmol/l AGN194204 (AGN), or both compounds simultaneously (HETE+AGN) for 24 hours. The percentage of apoptotic cells was determined by dUTP nick-end labeling (TUNEL) assay and fluorescence microscopy. These experiments were performed three times, with similar results. Error bars indicate the standard error of the mean. (b) The PPAR-α-selective ligand HETE activates the intrinsic apoptotic pathway in human breast cancer cell lines. Whole cell lysates from the indicated breast cancer cell lines treated with 100 μmol/l HETE for up to 24 hours were subjected to western blot, with anti-caspase (casp) and anti-β-actin antibodies shown on the left. Relative expression of each protein was determined using the same membrane. These experiments were performed three times, with similar results. Representative blots are shown.
To determine whether HETE-mediated apoptosis in human breast cancer lines was the result of intrinsic or extrinsic pathways, we examined patterns of caspase activation in these cells. As shown in Fig. 3b, caspase 10 activation in response to HETE (as shown by decreased immunoreactivity to the inactive proenzyme) was observed by 8 hours in both T47D and MDA-MB-468 cells. Activation of the downstream effector caspase 3 was evident by 8–16 hours in MDA-MB-468 cells. Caspase 9 expression was below the detection limit for western blot in this cell line. Activation of caspases 3 and 9 was not evident until 24 hours in T47D cells, which correlated with slightly decreased numbers of apoptotic cells at this time point. Activation of caspases in the extrinsic (death receptor mediated) pathway such as caspase 8 was not observed following HETE treatment (data not shown). These results indicate that selective activation of PPAR-α activates the intrinsic apoptotic pathway in human breast cancer cell lines.
RXR-α overexpression in human breast cancer cell lines results in phenotypic sensitization to the RXR-selective ligand
Our results suggest that RXR-α overexpression may sensitize cells to the antiproliferative effects of AGN194204 and potentiate additional PPAR-regulated responses in human breast cancer cell lines. To test this hypothesis we overexpressed RXR-α by stable transfection in MCF7 and T47D cells, which normally express low levels of the receptor. RXR-α protein levels in these stable clones are shown in Fig. 4a. To determine whether these clones were sensitized to the antiproliferative effects of AGN194204, we performed proliferation assays using ligand or vehicle treated cells. As shown in Fig. 4b, treatment with 100 nmol/l AGN194204 inhibited proliferation of RXR-α overexpressing T47D clones by 50%. Slightly greater inhibition of proliferation (55%) by AGN194204 was observed in RXR-α overexpressing MCF7 clones, reflecting higher receptor expression in these cells (Fig. 4c). In contrast, AGN194204 exposure had less effect on G418 resistant control clones. AGN194204 also increased the percentage of apoptotic cells in cultures of RXR-α overexpressing clones exposed to HETE. As shown in Fig. 4d, AGN194204 increased the percentage of TUNEL-positive cells by more than 50% in HETE-treated RXR-α overexpressing clones. In contrast, treatment with RXR-selective ligand increased the percentage of TUNEL positive cells by only 20% in HETE treated control clones. Treatment with AGN194204 as a single agent had minimal effect on the number of apoptotic cells in cultures of these clones. These results indicate that RXR-α overexpression is sufficient to sensitize breast cancer cell lines to the antiproliferative effects of AGN194204 and potentiates the PPAR-α mediated apoptotic response.
Figure 4.
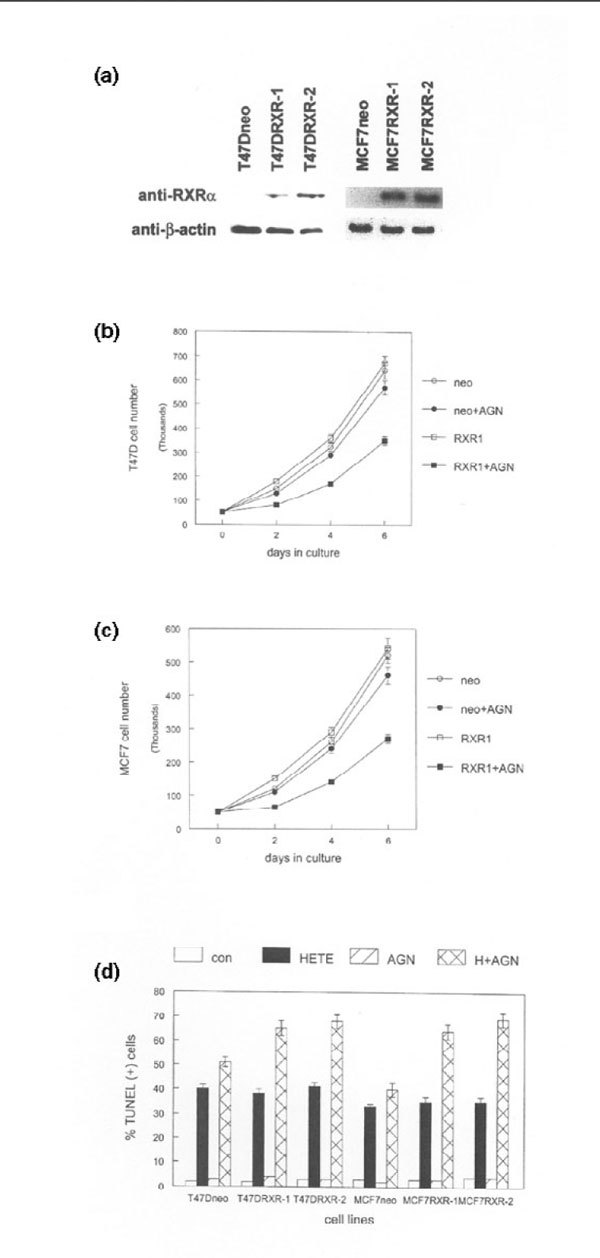
Retinoid X receptor (RXR)-α overexpression induces ligand-selective antiproliferative and apoptotic responses in human breast cancer cell lines. (a) RXR-α expression in T47D and MCF7 G418 resistant control clones (neo) and RXR-α stable clones (RXR-1, RXR-2). Whole cell lysates were subjected to western blot with anti-RXR-α and anti-β-actin antibodies. These experiments were repeated three times with independent clones. Representative blots are shown. (b) RXR-α overexpression induces antiproliferative response to the RXR-selective ligand AGN194204 in T47D stable clones. Triplicate cultures of G418 resistant control cells (neo) and RXR-α overexpressing clones (RXR1) were treated with vehicle or 100 nmol/l AGN194204 (AGN) for 6 days. Cells were counted at 2-day intervals using a hemacytometer. (c) RXR-α overexpression induces antiproliferative response to the RXR-selective ligand AGN194204 in MCF7 stable clones. Triplicate cultures of G418 resistant control cells (neo) and RXR-α overexpressing clones (RXR1) were treated with vehicle or 100 nmol/l AGN194204 (AGN) for 6 days. Cells were counted at 2-day intervals using a hemacytometer. (d) RXR-α overexpression potentiates the hydroxyeicosatetraenoic acid (HETE) apoptotic response to the RXR-selective ligand AGN194204 in T47D and MCF7 clones. G418 resistant control cells (neo) and RXR-α overexpressing clones (RXR-1, RXR-2) were treated with vehicle (con), 100 μmol/l HETE, 100 nmol/l AGN194204, or both compounds simultaneously (H+AGN) for 24 hours. The percentage of apoptotic cells was determined by dUTP nick-end labeling (TUNEL) assay using fluorescence microscopy. These experiments were performed three times, with similar results. Error bars indicate the standard error of the mean.
Discussion
One of the key findings of this study is that PPARs can mediate selective responses in human breast cancer cell lines, namely growth inhibition and apoptosis. Our data indicate that PPAR-α, when activated by the selective ligand HETE, induces apoptosis in breast cancer cell lines. This was characterized by activation of caspases 9 and 10 followed by caspase 3. Activation of caspase 8 was not observed in cells treated with HETE. Future experiments will characterize downstream targets of activated caspases in HETE-treated cells. Conversely, LA-mediated activation of PPAR-γ resulted in decreased growth characterized by S-phase inhibition. PPAR-γ activation resulted in changes in expression of a specific set of cell cycle regulatory genes. Previous studies have indicated that PPAR-γ mediates the action of LA in breast cancer cell lines [15]. These cells have also been shown to contain a functional PPAR response [14]. In addition to HETE and LA, studies have shown that a variety of fatty acids can act as ligands for PPARs, with disparate effects on promoter transactivation [16]. This large group of natural ligands can complicate the interpretation of studies examining the risk for breast cancer development associated with high-fat diets. The results of our experiments suggest that PPAR ligands may affect established breast cancer cell lines differently than normal mammary epithelium in animal or human studies. In the rat mammary carcinogenesis model, a synthetic PPAR-γ ligand inhibited cancer development [18]. Clarification of these effects awaits further experimentation.
Activation of PPAR-γ has been shown to have multiple antitumor effects on breast cancer cells (for review, see Kersten and coworkers [38]). PPAR-γ activation using synthetic ligands such as TZDs was shown to induce lipid accumulation in and differentiation of breast cancer cell lines [17]. In the present study we did not observe these types of changes; rather, PPAR-γ activation resulted in antiproliferative effects. These differences may be due to the fact that different breast cancer cell lines were used in the two studies. The role of PPAR-α ligands as a chemopreventive or chemotherapeutic agent for breast cancer is less clear. However, our studies indicate that PPAR-α activation by HETE is a potent inducer of apoptosis in established human breast cancer cell lines. In vitro studies are inherently limited in determining the effects of PPAR activation on clinical breast cancer, given that a phase II trial of troglitazone showed little effect on refractory tumors [19].
Another important finding of the present study is the antiproliferative effect of the synthetic RXR ligand AGN194204 on human breast cancer cell lines. Of particular interest is the increased sensitivity to this ligand of cell lines expressing higher levels of RXR-α. Some of these estrogen receptor negative cell lines have been shown to be resistant to the antiproliferative effects of the RAR agonist all-trans RA [39]. The RXR ligand mediated effects were associated with inhibition of growth factor signaling and cell cycle regulatory pathways. RXR-α overexpression was sufficient to reproduce the effects of the selective ligand in MCF7 and T47D cells. The effects of AGN194204 on breast cancer cell lines suggest that this ligand may have chemopreventive or chemotherapeutic effects in vivo. In previous studies, synthetic RXR ligands have been effective against mammary carcinogenesis in animal models [33]. However, a phase II clinical trial of oral bexarotene showed limited antitumor effects in patients with metastatic breast cancer [36].
Our findings also demonstrate that AGN194204 potentiated the antitumor effects of PPAR ligands. Combination of PPAR and RXR ligands increased transactivation of a responsive promoter in human breast cancer cell lines, which is in agreement with previous studies [14]. These results suggest that multidrug regimens consisting of PPAR and RXR ligands may provide a therapeutic advantage in breast cancer treatment. The molecular mechanisms regulating these effects are currently unclear. PPAR and RXR ligands have been shown to differentially recruit subsets of transcriptional coactivator proteins to the receptor complex [40]. Combinations of these ligands may result in additional coactivator recruitment, leading to enhanced transcriptional activation and cellular effects. The molecular characterization of these pathways in human breast cancer cells and testing the effects of PPAR and RXR ligands on mammary carcinogenesis in coactivator-null mutant mice will be important in future studies.
Conclusion
PPAR and RXR proteins are expressed by human breast cancer cell lines. These proteins heterodimerize and transactivate a PPAR-responsive promoter. The magnitude of transactivation was greater in breast cancer cell lines expressing higher levels of RXR-α. PPAR-α and PPAR-γ ligands induced apoptotic and antiproliferative responses in these lines, respectively, which were associated with specific changes in gene expression. These responses were potentiated by the RXR-selective ligand AGN194204. RXR-α overexpressing RA-resistant breast cancer cell lines were more sensitive to the effects of the RXR-selective compound. Forced RXR-α overexpression in other breast cancer cell lines resulted in greater sensitivity to the selective ligand.
Competing interests
RASC is a research director at Allergan, which developed the RXR-selective ligand AGN194204. DLC has acted as a consultant for the corporation. Allergan did not contribute funding for this study.
Abbreviations
BrdU = bromodeoxyuridine; Cdk = cyclin-dependent kinase; ERK = extracellular signal-regulated kinase; HETE = hydroxyeicosatetraenoic acid; LA = γ-linolenic acid; PBS = phosphate-buffered saline; PPAR = peroxisome proliferator-activated receptor; RA = retinoic acid; RAR = retinoic acid receptor; RXR = retinoid X receptor; TUNEL = dUTP nick-end labeling; TZD = thiazolidinedione drug.
Acknowledgments
Acknowledgments
We thank Dr Ronald Evans for providing the RXR-α expression vector. This study was supported by American Institute of Cancer Research grant 01B010 to DLC.
Contributor Information
David L Crowe, Email: dcrowe@usc.edu.
Roshantha AS Chandraratna, Email: chandraratna_rosh@allergan.com.
References
- Mandrup S, Lane MD. Regulating adipogenesis. J Biol Chem. 1997;272:5367–5370. doi: 10.1074/jbc.272.9.5367. [DOI] [PubMed] [Google Scholar]
- Keller H, Wahli W. Peroxisome proliferator activated receptors. Trends Endocrinol Metabol. 1993;4:291–296. doi: 10.1016/1043-2760(93)90048-J. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM. PPARγ in monocytes: less pain, any gain? Cell. 1998;93:153–155. doi: 10.1016/S0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. J Biol Chem. 1997;272:20108–20117. doi: 10.1074/jbc.272.32.20108. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Lehmann JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Roberts-Thomson SJ. Peroxisome proliferator activated receptors in tumorigenesis: targets of tumor promotion and treatment. Immunol Cell Biol. 2000;78:436–441. doi: 10.1046/j.1440-1711.2000.00921.x. [DOI] [PubMed] [Google Scholar]
- Demetri GD, Fletcher CDM, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman BM, Singer S. Induction of solid tumor differentiation by the peroxisome proliferator activated receptor γ ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci USA. 1999;96:3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator activated receptor γ (PPARγ) in human transitional bladder cancer and its role in inducing cell death. Neoplasia. 1999;1:330–339. doi: 10.1038/sj.neo.7900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, Inoue K, Hla T, Kondo M. Inhibition of human lung cancer cell growth by the peroxisome proliferator activated receptor γ agonists through induction of apoptosis. Biochem Biophys Res Commun. 2000;270:400–405. doi: 10.1006/bbrc.2000.2436. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator activated receptor γ induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res. 1999;90:75–80. doi: 10.1111/j.1349-7006.1999.tb00668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss of function mutations in PPARγ associated with human cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/S1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- Kilgore MW, Tate PL, Rai S, Sengoku E, Price TM. MCF-7 and T47D human breast cancer cells contain a functional peroxisomal response. Mol Cell Endocrinol. 1997;129:229–235. doi: 10.1016/S0303-7207(97)04057-4. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Redfern A, Bryce RP, Mansel RE. Peroxisome proliferator activated receptor γ (PPARγ) mediates the action of γ-linolenic acid in breast cancer cells. Prostaglandins Leukotrienes Essential Fatty Acids. 2000;62:119–127. doi: 10.1054/plef.1999.0131. [DOI] [PubMed] [Google Scholar]
- Thoennes SR, Tate PL, Price TM, Kilgore MW. Differential transcriptional activation of peroxisome proliferator activated receptor γ by omega-3 and omega-6 fatty acids in MCF-7 cells. Mol Cell. 1998;1:465–470. doi: 10.1016/S1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPARγ. Mol Cell. 1998;1:465–470. doi: 10.1016/S1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, Sporn MB. A new ligand for the peroxisome proliferator activated receptor γ (PPARγ), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–5673. [PubMed] [Google Scholar]
- Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferators activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/A:1024038127156. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Leblanc BP, Stunnenberg HG. 9-cis retinoic acid signaling: changing partners causes some excitement. Genes Dev. 1995;9:1811–1816. doi: 10.1101/gad.9.15.1811. [DOI] [PubMed] [Google Scholar]
- Zhang XK, Lehmann J, Hoffman B, Dawson MI, Cameron J, Graupner G, Hermann T, Tran P, Pfahl M. Homodimer formation of retinoid receptor induced by 9-cis retinoic acid. Nature. 1992;358:587–591. doi: 10.1038/358587a0. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone, and vitamin D3 signaling. Nature. 1992;355:446–449. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XK, Hoffmann B, Tran PBV, Graupner G, Pfahl M. Retinoid X receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature. 1992;355:441–445. doi: 10.1038/355441a0. [DOI] [PubMed] [Google Scholar]
- Wan H, Dawson MI, Hong WK, Lotan R. Overexpressed activated retinoid X receptors can mediate growth inhibitory effects of retinoids in human carcinoma cells. J Biol Chem. 1998;273:26915–26922. doi: 10.1074/jbc.273.41.26915. [DOI] [PubMed] [Google Scholar]
- Crouch GD, Helman LJ. All trans retinoic acid inhibits the growth of human rhabdomyosarcoma cell lines. Cancer Res. 1991;51:4882–4887. [PubMed] [Google Scholar]
- Delia D, Aiello A, Lombardi L, Pelicci PG, Grignani F, Formelli F, Menard S, Costa A, Veronesi U, Pierotti MA. N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993;53:6036–6041. [PubMed] [Google Scholar]
- Rubin M, Fenig E, Rosenauer A, Menendez-Botet C, Achkar C, Bentel JM, Yahalom J, Mendelsohn J, Miller WH. 9-cis retinoic acid inhibits growth of breast cancer cells and downregulates estrogen receptor mRNA and protein. Cancer Res. 1994;54:6549–6556. [PubMed] [Google Scholar]
- Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RAS, Shudo K, et al. Differential effects of synthetic nuclear retinoid receptor selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–4939. [PubMed] [Google Scholar]
- Wu Q, Dawson MI, Zheng Y, Hobbs PD, Agadir A, Jong L, Li Y, Liu R, Lin BZ, Zhang XK. Inhibition of trans retinoic acid resistant human breast cancer cell growth by retinoid X receptor selective retinoids. Mol Cell Biol. 1997;17:6598–6608. doi: 10.1128/mcb.17.11.6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenauer A, Nervi C, Davison K, Lamph WW, Mader S, Miller WH. Estrogen receptor expression activates the transcriptional and growth inhibitory response to retinoids without enhanced retinoic acid receptor α expression. Cancer Res. 1998;58:5110–5116. [PubMed] [Google Scholar]
- Schneider SM, Offterdinger M, Huber H, Grunt TW. Activation of retinoic acid receptor α is sufficient for full induction of retinoid responses in SK-BR-3 and T47D human breast cancer cells. Cancer Res. 2000;60:5479–5487. [PubMed] [Google Scholar]
- Fitzgerald P, Teng M, Chandraratna RAS, Heyman RA, Allegretto EA. Retinoic acid receptor α expression correlates with retinoid induced growth inhibition of human breast cancer cells regardless of estrogen receptor status. Cancer Res. 1997;57:2642–2650. [PubMed] [Google Scholar]
- Bischoff ED, Gottardis MM, Moon TE, Heyman RA, Lamph WW. Beyond tamoxifen: the retinoid X receptor selective ligand LGD1069 (TARGRETIN) causes complete regression of mammary carcinoma. Cancer Res. 1998;58:479–484. [PubMed] [Google Scholar]
- Miller VA, Benedetti FM, Rigas JR, Verret AL, Pfister DG, Straus D, Dris MG, Crisp M, Heyman R, Lowen GR, et al. Initial clinical trial of a selective retinoid X receptor ligand LGD1069. J Clin Oncol. 1997;15:790–795. doi: 10.1200/JCO.1997.15.2.790. [DOI] [PubMed] [Google Scholar]
- Esteva FJ, Glaspy J, Baidas S, Laufman L, Hutchins L, Dickler M, Tripathy D, Cohen R, DeMichele A, Yocum RC, et al. Multicenter phase II study of oral bexarotene for patients with metastatic breast cancer. J Clin Oncol. 2003;21:999–1006. doi: 10.1200/JCO.2003.05.068. [DOI] [PubMed] [Google Scholar]
- Lawrence JA, Merino MJ, Simpson JF, Manrow RE, Page DL, Steeg PS. A high risk lesion for invasive breast cancer, ductal carcinoma in situ, exhibits frequent overexpression of retinoid X receptor. Cancer Epidemiol Biomarkers Prevent. 1998;7:29–35. [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Sheikh MS, Shao ZM, Li XS, Dawson M, Jetten AM, Wu S, Conley BA, Garcia M, Rochefort H, Fontana JA. Retinoid resistant estrogen receptor negative human breast carcinoma cells transfected with retinoic acid receptor α acquire sensitivity to growth inhibition by retinoids. J Biol Chem. 1994;269:21440–21447. [PubMed] [Google Scholar]
- Yang W, Rachez C, Freedman LP. Discrete roles for peroxisome proliferator activated receptor γ and retinoid X receptor in recruiting nuclear receptor coactivators. Mol Cell Biol. 2000;20:8008–8017. doi: 10.1128/MCB.20.21.8008-8017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]