

Abstract
Cytochrome P450 (P450, CYP) 21A2 is the major steroid 21-hydroxylase, converting progesterone to 11-deoxycorticosterone and 17α-hydroxyprogesterone (17α-OH-progesterone) to 11-deoxycortisol. More than 100 CYP21A2 variants give rise to congenital adrenal hyperplasia (CAH). We previously reported a structure of WT human P450 21A2 with bound progesterone and now present a structure bound to the other substrate (17α-OH-progesterone). We found that the 17α-OH-progesterone- and progesterone-bound complex structures are highly similar, with only some minor differences in surface loop regions. Twelve P450 21A2 variants associated with either salt-wasting or nonclassical forms of CAH were expressed, purified, and analyzed. The catalytic activities of these 12 variants ranged from 0.00009% to 30% of WT P450 21A2 and the extent of heme incorporation from 10% to 95% of the WT. Substrate dissociation constants (Ks) for four variants were 37–13,000-fold higher than for WT P450 21A2. Cytochrome b5, which augments several P450 activities, inhibited P450 21A2 activity. Similar to the WT enzyme, high noncompetitive intermolecular kinetic deuterium isotope effects (≥ 5.5) were observed for all six P450 21A2 variants examined for 21-hydroxylation of 21-d3-progesterone, indicating that C–H bond breaking is a rate-limiting step over a 104-fold range of catalytic efficiency. Using UV-visible and CD spectroscopy, we found that P450 21A2 thermal stability assessed in bacterial cells and with purified enzymes differed among salt-wasting- and nonclassical-associated variants, but these differences did not correlate with catalytic activity. Our in-depth investigation of CAH-associated P450 21A2 variants reveals critical insight into the effects of disease-causing mutations on this important enzyme.
Keywords: circular dichroism (CD), cytochrome P450, isotope effect, steroid hormone, X-ray crystallography, congenital adrenal hyperplasia, steroid, steroid biosynthesis
Introduction
Cytochrome P450 (P450 or CYP)5 21A2 (1), an enzyme located in the adrenal cortex, catalyzes the 21-hydroxylation of both progesterone and 17α-hydroxyprogesterone (17α-OH-progesterone), forming 11-deoxycorticosterone and 11-deoxycortisol, respectively (2–4). Deficiencies in the CYP21A2 gene are common and are involved in ∼95% of the cases of congenital adrenal hyperplasia (CAH) (2, 3, 5–7), one of the most common inborn errors of metabolism. The incidence of CAH is ∼1:15,000 worldwide, with well over 100 different genetic variants having been diagnosed. The phenotypes are generally classified as nonclassical (NC), simple virilizing (SV), and salt-wasting (SW), in order of increasing severity (2, 3, 6). The great majority of mutations in CYP21A2 can be traced to the non-functional pseudogene of P450 21A2 (CYP21A1P), which is found in > 70% of all Caucasians. The pseudogene is 98% identical to the functional gene, and crossovers, sequence exchanges, and apparent gene conversion between functional gene and pseudogene contribute to frequent variations in the P450 21A2 protein and the occurrence of CAH (2, 3, 6, 7).
We published a crystal structure of bovine P450 21A2 bound with the substrate 17α-OH-progesterone (8), which provided insight into the roles of some of the residues involved in the enzymatic deficiencies. Using our bovine P450 21A2 structure, New and associates (9) modeled the human P450 21A2 enzyme and discussed how the low activities of some of the variants might be explained. We subsequently crystallized and published a structure of human P450 21A2 bound with its other substrate, progesterone (10). This structure showed some important differences from the bovine enzyme, and we discussed how these could affect the conclusions reached with modeling based on the bovine enzyme (11). However, even with the P450 21A2 structures available, it is not clear what all of the factors causing low catalytic activity are.
We now report a structure of human P450 21A2 bound to its substrate 17α-OH-progesterone. In addition, we expressed and purified 12 human P450 21A2 variants and measured kcat and Km parameters for 21-hydroxylation of both progesterone and 17α-OH-progesterone. Large kinetic deuterium isotope effects were observed with all six of the variants examined, indicating that C–H bond breaking is a rate-limiting step in all cases. The stability of the variants was addressed in several ways. We discuss possible reasons for low activity of the variants, especially those variants not analyzed previously (11).
Results
Structure of wild-type P450 21A2 bound to 17α-OH-progesterone
In the open reading frame of human P450 21A2, the region encoding the N-terminal transmembrane helix (residues 1–29) was replaced by nucleotides coding for the decameric peptide MAKKTSSKGK, and the C terminus was extended by 18 nucleotides encoding six histidines (9).
We determined the crystal structure of the complex between P450 21A2 and 17α-OH-progesterone at 3.3 Å resolution. An illustration of the overall fold of the complex is depicted in Fig. 1A, and selected crystal data, diffraction data collection, and refinement parameters are listed in Table 1. Crystals of the complex with 17α-OH-progesterone were isomorphous to those of the complex with the substrate progesterone and contain three independent copies in the asymmetric unit of space group C2. The conformation of P450 21A2 in complex with 17α-OH-progesterone is very similar to that adopted by the protein with progesterone bound. The root mean square deviation for 439 Cα atom pairs in the overlaid structures is 0.7 Å. The only conformational difference between the two P450 structures concerns residues 411–418 in the extensive random coil region that links helices L and M (Fig. 1C). In the higher-resolution (2.64 Å) complex with progesterone (10), this turn was not defined in the electron density map. In the complex with 17α-OH-progesterone, all residues that are part of the turn in the protein chain could be completely built into the electron density for the three independent molecules. As expected from the lower resolution of the structure of the complex with 17α-OH-progesterone, the side chains of many residues mapping to the surface of the P450 protein were only partially defined in the electron density or were missing completely and were modeled without side chains. Similarly, inspection of the electron density maps for the 17α-OH-progesterone complex did not reveal a second substrate molecule bound at a distal site in the structure of the progesterone complex (10). A closer look at the active sites shows that the orientations and positions of the two substrates are virtually identical (Fig. 1D). The only noteworthy deviation concerns the orientation of the keto group whereby the torsion angles around the C17–C20 bond differ by about 90° in the progesterone and 17α-OH-progesterone substrates. In the case of the latter, the C-20 keto oxygen and 17α-hydroxyl group are pointing in roughly opposite directions. However, in both the progesterone and 17α-OH-progesterone complexes, the C21 methyl group points into the heme system, with distances between C21 and Fe3+ of 4.0 and 4.3 Å, respectively (A molecules).
Figure 1.

Crystal structure of human P450 21A2 in complex with the substrate 17α-OH-progesterone. A, overall view of the complex colored in rainbow style (from N terminus (blue) to C terminus (red)) with individual α-helices and β-strands labeled and every tenth residue numbered. Carbon, nitrogen, and oxygen atoms of the heme moiety are colored in gray, blue, and red, respectively, and Fe3+ is shown as an orange sphere. Carbon and oxygen atoms of 17α-OH-progesterone are colored in salmon and red, respectively. B, quality of the final omit 2Fo − Fc Fourier electron density (1.2σ level) around the substrate. C, overall view of the superimposed progesterone (pink ribbon; molecule A) and 17α-OH-progesterone complexes (green ribbon; molecule A). Conformational differences between the two complexes in a portion (residues 411–418) of the long loop connecting helices L and M are indicated with an arrow. D, close-up view of the active sites of the two superimposed complexes with progesterone and 17α-OH-progesterone bound. Carbon atoms of substrates and heme are colored in gray and light green, respectively.
Table 1.
Selected crystal data, data collection, and refinement parameters for the P450 21A2–17α-OH progesterone complex
| Data collection | |
| Wavelength (Å) | 0.97857 |
| Space group | C2 |
| Resolution (outer shell) (Å) | 43.27 to 3.31 (3.42 to 3.31)a |
| Unit cell constants a, b, c (Å); β (degrees) | 152.43, 88.39, 111.42; 102.1 |
| Unique reflections | 19,819 (1,982) |
| Completeness (%) | 92.3 (93.2) |
| I/σ(I) | 11.8 (1.5) |
| Rpim | 0.060 (0.456) |
| Redundancy | 4.5 (4.4) |
| Refinement | |
| Phasing method | Molecular replacement |
| Rwork | 0.1899 (0.300)b |
| Rfree | 0.2381 (0.296)b |
| No. of amino acids | 1,325c |
| No. of protein atoms | 10,589 |
| No. of heme/ligand atoms | 129/69 |
| Average B-factor, protein atoms (Å2) | 92.5 |
| Average B-factor, ligand/heme atoms (Å2) | 83.8/95.8 |
| Wilson B-factor (Å2) | 91.2 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (degrees) | 2.2 |
| Ramachandran plot: no. of favored/allowed/outlier residues | 1,263/29/15 |
| Protein Data Bank code | 5VBU |
a Numbers in parentheses refer to the outer shell.
b Outer shell for refinement is 3.40 to 3.31 Å.
c For three independent molecules per asymmetric unit. Molecules A and C: residues 29–266/275–325/333–485. Molecule B: residues 29–266/275–325/333–484.
Catalytic efficiencies of variants
Twelve clinically observed variants of P450 21A2 were selected for further study, including three categorized as NC and nine as SW. The UPLC-UV assays for 21-hydroxylation activity are very robust because of the high catalytic efficiency of the wild-type enzyme, and the activity could be measured over a 106-fold range of catalytic efficiency (Fig. 2). Even the NC variants showed at least a 2-order of magnitude decrease in catalytic efficiency, based upon the combined measured P450 plus cytochrome P420 contents of the enzymes. The SW variants showed decreases of > 103-fold, up to 106-fold. It is of interest to note that (i) all of the changes are the result of single-amino acid substitutions, (ii) the amino acid change at a certain site can have a strong effect (e.g. P31L versus P31Q), and (iii) the clinical classification of the severity of the disease (SW versus NC) does not completely match the residual catalytic activity.
Figure 2.

Catalytic efficiencies of wild-type P450 21A2 and some variants. Error bars, S.D.
Heme incorporation into P450 21A2 variants
One issue in the proper folding of P450 21A2 variants is the proper incorporation of the heme prosthetic group. An approximate index of heme incorporation is provided by the A417/A280 ratio of the purified variants, in that only one of the 12 variants examined involved a change in tryptophan (which would lower A280) (see Table 3). All of the mutants showed some decrease in heme content except G292S, with the level decreased to one-tenth in several variants (P31Q, W303R, and R357W).
Table 3.
Heme incorporation into P450 21A2 variants
| Variant | Clinical phenotype | A417/A280 | Fraction relative to wild type |
|---|---|---|---|
| Wild type | 0.63 | 1.0 | |
| P31L | NC | 0.24 | 0.38 |
| P31Q | SW | 0.07 | 0.11 |
| G65E | SW | 0.16 | 0.25 |
| L108R | SW | 0.23 | 0.37 |
| I172N | SV | 0.09 | 0.14 |
| V282L | NC | 0.35 | 0.56 |
| G292S | SW | 0.60 | 0.95 |
| G292C | SW | 0.47 | 0.75 |
| T296N | SV/SW | 0.39 | 0.62 |
| W303R | SW | 0.07 | 0.11 |
| R357W | SW | 0.06 | 0.095 |
| R409C | SW | 0.08 | 0.13 |
The total heme content of the purified P450 21A2 variants includes both P450 and cytochrome P420, a term collectively used for perturbed P450 that is enzymatically inactive (12). The fraction of the heme in the different forms varied considerably, from being almost totally P450 (see Fig. 7A) to almost entirely cytochrome P420 (P31Q, G65E, L108R, W303R, R357W, and R409C; see supplemental Fig. S1).
Figure 7.

Environments of selected variants in the structure of the P450 21A2–17α-OH-progesterone complex. A, I172N (SV); B, V282L (NC); C, G292C (SW); D, G292S (SW); E, R357W (SW); F, R409C (SW). Mutated residues are highlighted with carbon atoms colored in light blue (wild type) and green (mutant), and substrate carbon atoms are colored in cyan. Amino acids in the vicinity of mutated residues are labeled, and contacts with distances of < 3.3 Å are indicated by thin solid lines.
Substrate binding to selected P450 21A2 variants
Four variants were selected for binding analysis, including one NC variant and three SW variants (see Table 4). Wild-type P450 21A2 bound both substrates (progesterone and 17α-OH-progesterone) tightly, with Ks values < 0.1 μm and difficult to estimate accurately using spectral determinations. Although some of the variants still bound the substrates with low-micromolar affinity (see Table 4), the Ks values clearly showed much less affinity. The difference between the NC variant V282L and the three SW variants was not obvious. It should be emphasized that the Ks values do not necessarily reflect productive binding. The extent of the decrease in substrate binding is not enough to explain the attenuated catalytic efficiency (Table 2 and Fig. 2) for the three SW variants (G65E, G292S, and G292C).
Table 4.
Substrate binding by some 21A2 variants
| Variant | Clinical phenotype | Substrate | Ks | Change compared with wild type |
|---|---|---|---|---|
| μm | ||||
| Wild type | Progesterone | 0.010 ± 0.003 | 1 | |
| 17α-OH-progesterone | 0.030 ± 0.003 | 1 | ||
| G65E | SW | Progesterone | 22 ± 3a | 2,200 |
| 17α-OH-progesterone | 2.5 ± 0.7 | 83 | ||
| V282L | NC | Progesterone | 16 ± 2 | 1,600 |
| 17α-OH-progesterone | 6.2 ± 1.7 | 210 | ||
| G292C | SW | Progesterone | 130 ± 40a | 13,000 |
| 17α-OH-progesterone | 1.1 ± 0.4 | 37 | ||
| G292S | SW | Progesterone | 5.4 ± 0.4 | 540 |
| 17α-OH-progesterone | 5.0 ± 0.6 | 170 |
a Subject to more error due to linearity of plots.
Table 2.
Catalytic activities of 21A2 variants
| Variant | Clinical phenotype | Substrate | kcat | Km | kcat/Km | Catalytic efficiency |
|---|---|---|---|---|---|---|
| min−1 | μm | μm−1 min−1 | % wild type | |||
| Wild type | Progesterone | 170 ± 4 | 0.21 ± 0.03 | 800 ± 120 | 100 | |
| 17α-OH-progesterone | 240 ± 5 | 1.5 ± 0.1 | 160 ± 10 | 100 | ||
| P31L | NC | Progesterone | 23 ± 2 | 1.2 ± 3.1 | 19 ± 5 | 2.4 |
| 17α-OH-progesterone | 84 ± 7 | 1.7 ± 0.58 | 49 ± 7 | 30 | ||
| P31Q | SW | Progesterone | 0.0076 ± 0.0005 | 9.6 ± 1.7 | 0.00080 ± 0.0002 | 0.0001 |
| 17α-OH-progesterone | 0.013 ± 0.001 | 7.3 ± 1.1 | 0.0018 ± 0.0003 | 0.0011 | ||
| G65E | SW | Progesterone | 3.6 ± 0.2 | 5.5 ± 0.8 | 0.67 ± 0.10 | 0.082 |
| 17α-OH-progesterone | 1.9 ± 0.2 | 13 ± 2 | 0.14 ± 0.02 | 0.089 | ||
| L108R | SW | Progesterone | 0.0059 ± 0.001 | 2.7 ± 0.2 | 0.0022 ± 0.0002 | 0.00027 |
| 17α-OH-progesterone | 0.002 ± 0.0002 | 1.7 ± 0.5 | 0.0012 ± 0.0007 | 0.00072 | ||
| I172N | SV | Progesterone | 0.087 ± 0.002 | 1.8 ± 1.0 | 0.049 ± 0.027 | 0.0061 |
| 17α-OH-progesterone | 0.18 ± 0.01 | 4.9 ± 0.5 | 0.037 ± 0.004 | 0.023 | ||
| V282L | NC | Progesterone | 62 ± 3 | 3.8 ± 0.6 | 16 ± 4 | 2.0 |
| 17α-OH-progesterone | 2 ± 4 | 14 ± 2 | 3.7 ± 0.6 | 2.3 | ||
| G292C | SW | Progesterone | 0.046 ± 0.004 | 12 ± 3 | 0.0038 ± 0.0009 | 0.00047 |
| 17α-OH-progesterone | 0.0026 ± 0.0002 | 3.6 ± 0.7 | 0.0073 ± 0.0001 | 0.00047 | ||
| G292S | SW | Progesterone | 12 ± 1 | 4.7 ± 0.3 | 2.6 ± 0.2 | 0.32 |
| 17α-OH-progesterone | 3.8 ± 0.3 | 4.6 ± 0.7 | 0.82 ± 0.14 | 0.51 | ||
| T296N | SW | Progesterone | 0.46 ± 0.16 | 1.1 ± 0.2 | 0.42 ± 0.16 | 0.052 |
| 17α-OH-progesterone | 1.7 ± 0.3 | 0.46 ± 0.2 | 6.0 ± 2.7 | 3.7 | ||
| W303R | SW | Progesterone | 0.0079 ± 0.0005 | 11 ± 2 | 0.00072 ± 0.00014 | 0.00009 |
| 17α-OH-progesterone | 0.0023 ± 0.0002 | 2.4 ± 0.5 | 0.00095 ± 0.00021 | 0.0006 | ||
| R357W | SW | Progesterone | 0.016 ± 0.001 | 22 ± 4 | 0.00073 ± 0.00014 | 0.0091 |
| 17α-OH-progesterone | 0.027 ± 0.002 | 22 ± 3 | 0.0012 ± 0.00015 | 0.075 | ||
| R409C | SW | Progesterone | 1.2 ± 0.1 | 0.39 ± 0.08 | 3.0 ± 0.6 | 0.37 |
| 17α-OH-progesterone | 1.8 ± 0.1 | 0.44 ± 0.09 | 4.0 ± 0.9 | 2.5 |
Lack of stimulation by cytochrome b5
Some P450 activities show remarkable stimulatory effects of cytochrome b5 (e.g. P450 17A1 17α,20-lyase activity (13–15)). We examined P450 21A2 for a possible effect, under a variety of conditions of cytochrome b5 concentration. Cytochrome b5 reduced the activity ∼4-fold (Fig. 3). The significance of this attenuation is not clear, but cytochrome b5 was clearly not stimulatory and does not appear to be a factor influencing in vivo activity, in contrast to P450 17A1 (16, 17). In the way of a caveat, we only analyzed the effect of cytochrome b5 under the typical reconstitution conditions used with P450 21A2 and in other systems (13–15) and cannot exclude the possibility that another system might show different effects.
Figure 3.
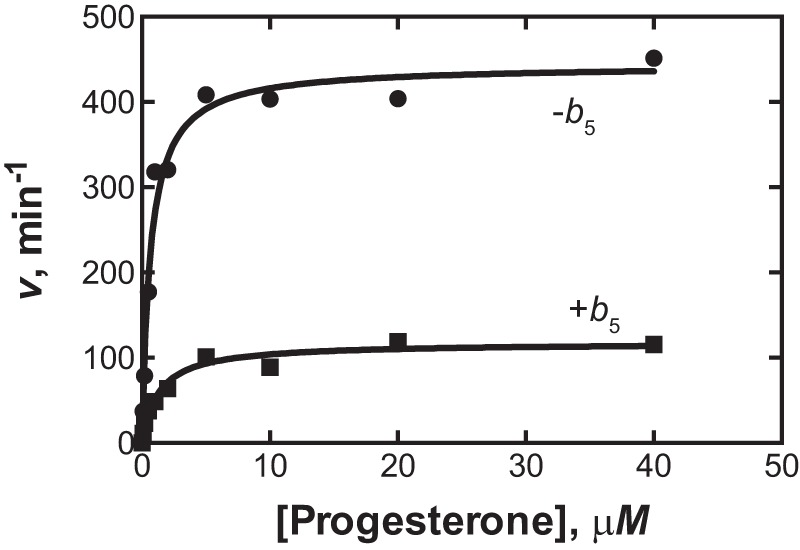
Inhibition of P450 21A2 by cytochrome b5. Progesterone 21-hydroxylation was measured in a reconstituted system containing 2 nm P450 21A2 and 0.60 μm NADPH-P450 reductase in the absence (●) or presence (■) of 0.6 μm cytochrome b5. In the absence of cytochrome b5, kcat was 440 min−1, and the Km was l.7 μm under these conditions. With cytochrome b5 present, the kcat was 120 min−1, and Km was 1.3 μm. Each point on the graph is a mean of duplicate determinations.
Kinetic deuterium isotope effects
Wild-type human P450 21A2 showed high kinetic deuterium isotope effects for the 21-hydroxylation of both progesterone and 17α-OH-progesterone, indicating that C–H bond breaking is one of the rate-limiting steps in the catalytic cycle (10, 18). With all six of the variants we selected for analysis, the 21-hydroxylation of 21-d3-progesterone was considerably slower than that of progesterone (Fig. 4). The results are at least qualitatively similar to those we previously reported with wild-type human P450 21A2 (10) and implicate C–H bond breaking as a rate-limiting step, although the rates vary 104-fold.
Figure 4.

Kinetic isotope effects for several variants of P450 21A2. The results of assays with d0- and 21-d3-progesterone are shown. From the kcat and kcat/Km values (estimated using GraphPad Prism), the following estimates of DV and D(V/K) were calculated: A (G65E), DV = 17, D(V/K) = 13; B (V282L), DV = 31, D(V/K) = 7.8; C (G292C), DV = 8.7, D(V/K) = 5.5; D (G292S), DV = 12, D(V/K) = 7.7; E (T296N), DV = 5.1, D(V/K) = 12; F (R409C), DV = 10, D(V/K) = 5.5 (the conventions DV = Hkcat/Dkcat and D(V/K) = (Hkcat/HKm)/(Dkcat/DKm) of Northrop (1) are used).
Assays of protein stability
We utilized two different assays of temperature-dependent protein stability.
One series of studies was done on temperature dependence of CD spectra of purified P450 21A2 variants. The CD spectra of some of the variants were relatively normal (i.e. similar to the wild-type protein) (Fig. 5) (19, 20). The temperature dependence of protein integrity was also analyzed, and the differences between the wild-type and mutant proteins were found to be rather limited (supplemental Fig. S1). Two of the SW mutants (G292C and L108R) showed very distorted CD spectra, but so did the NC variant V282L.
Figure 5.

Thermal stability of P450 21A2 variants: CD spectra as a function of temperature. A, wild-type P450 21A2; B, G292C; C, G292S; D, V282L; E, R409C; F, P31L; G, W303R; H, L108R. The spectra are labeled with the temperatures. The percentage of α-helicity at 25 °C was calculated as described (19), as modified by Chen and Yang (20): 20% (A), 11% (B), 19% (C), 8% (D), 20% (E), 24% (F), 18% (G), and 14% (H).
We also used an alternative approach that utilized the variant P450s in the Escherichia coli cells, without purification (21) (which might have induced denaturation). Reduced P450–CO complex spectra of P450 variants were acquired in bacterial cells following 10-min exposure to increasing temperatures (Fig. 6). The presence of the 450 nm band is indicative of structurally intact protein, and the band at 420 nm is indicative of cytochrome P420, a denatured form (12). Interestingly, some of the variants with very low catalytic efficiency (Table 2 and Fig. 2) expressed reasonably well, as judged by the spectra. All P450 21A2 variants were denatured to some extent. Although some variants had very low catalytic efficiency (e.g. G292C), they had thermal stabilities similar to wild-type P450 21A2, as judged by the temperatures at which one-half of the 450-nm absorbance spectra were lost. In these assays, the spectra seen with P31Q, G65E, L108R, W303R, R357W, and R409C (supplemental Fig. S2) were not strong enough at 450 nm to apply the assays, reflecting the low intrinsic ability to properly incorporate the heme prosthetic group.
Figure 6.

Thermal stability of P450 21A2 variants: P450 Fe2+-CO spectra in bacterial cells as a function of temperature. A, wild-type P450 21A2; B, P31L; C, G292C; D, G292S; E, V282L; F, T296N. In each case, the spectra are shown, beginning with the red spectrum at 23 °C and with increasing temperature as indicated by the points. The new temperature was held for 10 min after each increase, before scanning. The plots were fit to a sigmoidal model in Prism (GraphPad Software) with the maximum and minimum amount of P450 (normalized to 1.0 and 0). The estimated “T50” values (one-half loss of P450, corresponding to EC50 in the software, Y = bottom + (top − bottom)/(1 + 10∧((log EC50 − X))) were calculated and are shown on the plots.
Discussion
The first structure of P450 21A2 was that of a slightly modified bovine enzyme (T241R/L442A) with the substrate 17α-OH-progesterone (8), and two molecules of this substrate were present, one bound at the active site and the other at a distal site adjacent to the F′ helix. We have not obtained a structure of the bovine enzyme with the substrate progesterone bound. The structures of human P450 21A2 bound with progesterone (10) also revealed a substrate molecule at a distal site that matches the site of the second 17α-OH-progesterone in the structure of the complex with bovine 21A2. Most likely due to the lower resolution of the structure of the human 21A2 enzyme with 17α-OH-progesterone (this work; see Fig. 1), the electron density at this distal site was not defined well enough to identify a second substrate molecule.
The availability of structural, activity, stability, and substrate binding data offers an opportunity to potentially gain a refined understanding of the phenotypes of diverse CAH variants and the different degrees to which SW, SV, and NC mutations affect the P450 21A2 enzyme. Among the variants we investigated in this work are rare mutations as well as common pseudogene-derived ones. The latter include I172N (SV), P31L (NC), and V282L (the most common NC variant).6
Ile-172 (I172N variant, SV) is located in the middle of helix E, which is part of a helical bundle (C, E, I, and M helices; Fig. 1A). Ile-172 is part of a relatively hydrophobic environment including Val-140, Leu-176, and Leu-434 and also a hydrophilic residue, Glu-438 (Fig. 7A). Replacing isoleucine with asparagine affects these hydrophobic interactions to some extent but also allows hydrogen bonds between the amino group of Asn-172 and Glu-438 (Oϵ2; 3.06 Å) and between the side chain carbonyl of Asn-172 and the main-chain N–H of Val-140 (3.60 Å). I172N displayed a considerable loss in catalytic activity (∼2% residual activity with 17α-OH-progesterone; Table 2) and a 7-fold reduction in heme incorporation compared with wild-type P450 21A2 (Table 3).
Val-282 (V282L variant, NC) is located near the N-terminal end of the long I helix and surrounded by hydrophobic residues from the G (Leu-242) and H helices (Met-258, Met-261, and Leu-262) (Fig. 7B). The increased size of the leucine relative to the valine side chain can be accommodated to some degree at this site, consistent with the residual activity and the NC phenotype (Table 2). The activity we measured here is considerably lower than that reported earlier (20% for progesterone and 50% for 17α-OH-progesterone; http://www.cypalleles.ki.se/cyp21.htm)7 (22). Among the variants for which we evaluated the stability change of the Fe2+-CO spectrum based on heating, the V282L mutant showed the greatest reduction in Tm relative to the wild-type protein (Fig. 6E). This variant also displayed a significantly distorted CD spectrum even at 25 °C (Fig. 5D), supporting the notion that even the addition of a single methyl group of the leucine side chain relative to valine is not well-tolerated. This variant also showed very poor affinity for substrates (Table 4).
Gly-292 (G292C variant, SW) maps to the central region of the I helix and lies in close proximity of substrate and heme (Fig. 7C). The cysteine mutation results in steric clashes with ring A of 17α-OH-progesterone and progesterone and potentially heme. The CD spectrum was very distorted even at 25 °C (Fig. 5B). The complete loss of activity in this severe mutant is therefore not surprising (Table 2), despite a heme incorporation that was only marginally affected (75% of wild-type enzyme; Table 3). The effect of temperature was not great, surprisingly (Figs. 5 and 6 and supplemental Fig. S1). Binding of progesterone was very poor (Table 4), but 17α-OH-progesterone bound reasonably well. Interestingly, progesterone binding (relative KS) is dramatically reduced compared with wild-type 21A2 (> 104-fold; Table 4), although the effect is less prominent in the case of 17α-OH-progesterone.
The G292S variant (SW) is less damaging than the cysteine mutant as indicated by activity data that show ∼100-fold reduction compared with wild-type P450 21A2 (Table 2). The serine side chain is slightly smaller, and the γ-hydroxyl may be more easily accommodated, given its vicinity to more polar moieties, such as the 17α-OH group and the main-chain carbonyl groups of Asp-288 and Leu-289 (Fig. 7D). Indeed, this variant binds progesterone about 25-fold more tightly than the P450 G292C variant (Table 4). Neither the G292C nor the G292S variant exhibits a dramatically altered thermal stability. In fact, in the Fe2+-CO spectra, the Tm of the latter was only 2 °C below that of wild-type P450 21A2 (Fig. 6D). However, just like the V282L variant, G292C showed a significantly changed CD spectrum relative to wild-type protein even at 25 °C (Fig. 5B).
The Arg-357 (R357W) variant (SW) is located at the C terminus of the K helix. Methylene groups of the arginine side chain are surrounded by relatively hydrophobic side chains, including Trp-303, Pro-464, and Leu-472, whereas the guanidino moiety faces Asp-44, Gln-390, and Gln-463. The mutation to tryptophan results in a stacking interaction between the side chains of Trp-357 and Trp-303 (∼3.8-Å separation; Fig. 7E). The variant displayed some residual catalytic activity (< 1% with progesterone or 17α-OH-progesterone; Table 2) (but displayed the lowest level of heme incorporation among the investigated CAH variants, < 10% relative to wild-type P450 21A2; Table 3).
Arg-409 (R409C variant, SW) is located in a highly polar environment in the loop region that connects helices L and M and is part of the highly conserved EXXR motif (see Fig. 4D in Ref. 11). The mutation to cysteine introduces a hydrophobic and less sterically demanding entity into this polar environment (Fig. 7F). Thus, the cysteine side chain is unlikely to create any short contacts, and it appears that it can be accommodated between the side chains of Trp-406 and Asn-416. Given the intricate interactions that Arg-409 is engaged in (hydrogen-bonding with four neighboring residues and involving both side and main chain atoms of these; Fig. 7F), none of which can be mimicked by cysteine, it is perhaps surprising that the R409C CAH variant still displays between ∼0.5 and 2.5% of the wild-type activity (Table 2).
We had evaluated potential steric and electrostatic effects of 24 SW, SV, and NC CAH variants based on the previously determined crystal structure of the complex between human P450 21A2 and progesterone (10, 11). Among them were the P31L (NC), P31Q (SW), G65E (SW), L108R (SW), T296N (SW), and W303R (SW) variants, for which we now report catalytic activities with the progesterone and 17α-OH-progesterone substrates (Table 2). Consistent with the severity of the CAH phenotype, the P31L mutant does not hamper catalysis to the same extent as the P31Q mutant (the latter is basically inactive). The deviating magnitude of the activity reductions seen for the two variants are matched by those on heme incorporation (3-fold lower for P31Q; Table 3), and the loss in stability at least for P31L relative to the wild-type protein is quite minor (Fig. 6B). Pro-31, along with an adjacent proline, marks the site of a sharp turn in the direction of the protein chain and helps to anchor the N-terminal hydrophobic tail that serves as the attachment of P450 21A2 to the membrane. The leucine mutation is sterically compromising but less severe, from a structural perspective, than the glutamine mutation that affects both sterics and electrostatics/hydrogen bonding in comparison with proline.
Gly-65 (G65E variant, SW) is close to the surface and at the apex of the short loop that connects the β2 and β3 strands, facing the N-terminal residue of the F′ helix (Pro-214; Fig. 1A). Mutation to glutamate introduces a polar residue into a patch of hydrophobic side chains from residues Leu-38, Leu-40, Leu-64, Leu-66, Val-212, and Ile-213. In addition to disturbing the apolar character of this location, the relatively long glutamate side chain creates steric challenges, and the significant loss of catalytic activity with this variant (∼0.1% of wild-type protein with both substrates; Table 2) is in line with the anticipated structural consequences. The poor incorporation of heme (25% of wild-type 21A2; Table 3) and the significantly reduced binding, particularly of progesterone (some 2,000-fold relative to wild type; Table 4), are perhaps more surprising, given the location of Gly-65 close to the surface and at a considerable distance from active site and heme.
Leu-108 (L108R variant, SW) is located in a loop that precedes helix C (Fig. 1A) and is positioned immediately adjacent to methyl and propionic acid substituents of heme. Introduction of the longer arginine with its positively charged guanidino moiety in place of leucine that sits in a partly hydrophobic region but also has His-120, Lys-121, and Asp-288 among its neighbors was expected to distort the conformational/electronic balance. Consistent with this assessment, the CD spectrum for the L108R variant displays significant changes relative to wild-type protein even at 25 °C (Fig. 5H). Moreover, the complete loss of activity (Table 2) is in line with the structural picture and the severity of the CAH phenotype. The activities based on in vitro kinetic assays and using purified mutant protein reported here are lower than those reported earlier and based on experiments conducted with transiently transfected COS-1 cells (∼0.3% activity with both substrates relative to wild-type protein (23)). Perhaps surprisingly, given the close vicinity of this mutation to heme, the fraction of heme measured relative to wild-type 21A2 still amounts to 40% (Table 3).
In comparison with the above L108R variant, mutation of Thr-296 to asparagine, although classified as an SW CAH phenotype as well, affects the catalytic activity to a smaller degree: 0.05 and 4% of wild-type 21A2 activity with the progesterone and 17α-OH-progesterone substrates, respectively (Table 2). Heme incorporation was also increased relative to the L108R variant (62 and 25%, respectively; Table 3), and the thermal stability was virtually the same as for wild-type protein (Fig. 6F; −1 °C). Thr-296 is located in the long I helix and sits above the side of the heme (5.2-Å distance between Oγ and Fe3+) and in the vicinity of ring A of the substrate molecule (5.5-Å distance between Oγ and O17) (Fig. 1D). In the modeled Asn-296 mutant, the above distances would be reduced to 4.7 and 3.1 Å (Nδ2…Fe3+ and Nδ2…O17, respectively). Apparently, swapping threonine and asparagine does not entirely abolish activity (some 25-fold reduced compared with wild-type protein with 17α-OH-progesterone), an observation that is noteworthy, given how closely it occurs to substrate and heme and given the more drastic consequences of mutations farther away from the active site (e.g. P31Q and L65R).
The consequences of the T296N mutation are also more limited than those seen for another SW CAH variant, W303R, Trp-303 being located farther along the I helix and thus more removed from heme and substrate than Thr-296. Mutation of Trp-303 to arginine destroys the catalytic activity with both substrates (Table 2) and also affects heme incorporation to a very significant extent (11%; Table 3). An inspection of the structures of the human P450 21A2–substrate complexes suggests that an arginine at position 303 is likely to create a repulsion with Arg-357 that sits at the end of the K helix (Fig. 1A). In the model of the mutant, the guanidino moieties of the two arginines are positioned at 3.5 Å from one another. Interestingly, the R357W mutation is also classified as an SW CAH variant, whereby its activity is slightly higher than that of W303R (∼0.01% and 0.1% for progesterone and 17α-OH-progesterone, respectively; Table 2). Moreover, the levels of heme incorporation were quite similar for the two variants, ∼10% (Table 3). The most likely reasons for the detrimental effects on activity and heme incorporation of these two mutations are altered positions and mobility of the I helix that spans the entire core of the protein and runs by the heme/active site. These changes in conformation and dynamic behavior occur either as a consequence of replacement of the Trp-303/Arg-357 pair by Arg/Arg (W303R variant) or Trp/Trp (R357W variant) pairs (in the model of the latter, the two indole moieties are ideally stacked at 3.4 Å, Fig. 7E).
Overall, we find that single amino acid substitutions can have remarkable effects. All of the variants studied here were identified in clinical practice (11). The summaries of catalytic activities of P450 21A2 activities (11) (http://www.cypalleles.ki.se/cyp21.htm)7 are rather crude and contain values measured with many different expression systems and cellular and other assays. We provide here (Table 2 and Fig. 1) and elsewhere (11) a robust set of kcat, Km, and kcat/Km values obtained under precise conditions with UPLC-UV measurements.
Although it is popular to use crystallographic results to consider alterations of substrate binding to explain properties of variants, our results suggest that the losses of catalytic activity (Table 2) are too large to be explained by attenuation of substrate binding (Type I) (24). However, the heme perturbation assay may not necessarily reflect productive substrate binding. Overall, the changes of single amino acids often have dramatic effects on structural properties of P450 21A2. Evidence for this is seen in the incorporation of heme, both total heme (Table 3) and the content of P450 versus inactive cytochrome P420 (Fig. 6 and supplemental Fig. S1). Some of the loss of heme from certain mutants might have occurred during purification, but large differences were also seen in the bacterial cells (Fig. 6 and supplemental Fig. S2).
The CD spectra of some of the purified variants indicated considerable distortion (e.g. G292C, V282L, and L108R) (Fig. 5). However, other variants with catalytic efficiencies just as low did not show the losses of α-helicity (e.g. W303R). The loss of α-helicity with increasing temperature was no worse for the defective mutants than for the wild-type enzyme (supplemental Fig. S1). Overall, the CD results by themselves did not provide explanations for the defective nature of the variants.
The Fe2+-CO results clearly showed less stability of all variants (Fig. 6 and supplemental Fig. S2). Interestingly, the temperature dependence of these was rather invariant (i.e. they started with a certain fraction of cytochrome P420, but the degree to which the residual P450 broke down was similar (V282L was somewhat faster; Fig. 6E)).
Overall, we can conclude that a variety of issues are associated with the different clinical loss-of-function variants. The single amino acid substitutions can produce dramatic losses of functional integrity (Fig. 5), the ability to bind the heme prosthetic group (Table 3, Fig. 6, and supplemental Fig. S2), and the ability to bind substrates (Table 4). Collectively, these problems result in low catalytic efficiency (Fig. 1 and Table 2). Nevertheless, C–H bond breaking appears to be the rate-limiting step across several orders of magnitude of catalytic efficiency (Figs. 1 and 4 and Table 2).
The ability of small changes in structure to influence large changes in catalytic efficiency seems remarkable, but it is of use to consider the Eyring equation,
| (Eq. 1) |
where kB is the Boltzmann constant, h is Planck's constant (25); a mutation yielding a ΔΔG of 1.3 kcal mol−1 leads to a 10-fold change in rate, and a ΔΔG of 6.4 kcal mol−1 leads to a 50,000-fold change in kobs.
In an earlier study of P450 21A2, we had mapped many SW, SV, and NC mutations onto the 3D structure of the enzyme and found patterns of distribution characteristic of the three types of CAH variants (11). Thus, SW-causing mutations that are more common than SV variants were spread throughout the protein, often involving hydrophobic amino acids and dotting the active-site area and heme-binding pocket. By comparison, SV- and NC-causing mutations concern regions farther removed from the center and, particularly in the case of the NC variants, commonly map to areas on or near the enzyme surface, where they could be expected to be less damaging to stability and activity. The comparison of activities of SW, SV, and NC variants, based on various assays, confirmed the greater loss of activity in enzymes exhibiting the SW phenotype relative to those classified as SV and particularly NC (11). The detailed analysis of a dozen variants presented here, using structural, activity, folding stability, and spectroscopic assays to measure heme incorporation, demonstrates in some cases that steady-state kinetic assays using a purified P450 21A2 variant and cell-based measurements of activity can produce considerably different outcomes. Thus, the most common NC variant, V282L, displayed much lower activity in the kinetic assays than in earlier cell-based experiments. Not only did this mutation impact activity to a considerable degree, but it also showed poor substrate binding and the greatest loss in thermodynamic stability among all of the CAH variants tested. Similarly, mutations at the surface can be as damaging as or more damaging than those at the heart of the enzyme, as demonstrated by the P31Q (SW) and the G292S/G292C (SW) variants, respectively. The crystal structure provides some insight into the origins of the relative degree of damage caused by individual mutations. Overall, our in-depth investigation of selected types of CAH variants, relying on an array of experimental approaches, provides new insight into disease-causing mutations in the P450 21A2 enzyme, which is among the metabolic enzymes with the largest number of naturally occurring variations (≫ 100) that alter activity.
Experimental procedures
Chemicals
Progesterone and 17α-OH-progesterone were purchased from Sigma-Aldrich or Steraloids (Newport, RI) and used without further purification. 21-d3-Progesterone was synthesized as described elsewhere (18).
Expression and purification of human P450 21A2
Recombinant P450 21A2 enzyme encompassing amino acids 30–495 was expressed and purified as described previously (10). Briefly, in the CYP21A2 open reading frame, the region coding for the 29-residue-long N-terminal trans-membrane helix (residues 1–29) was replaced by nucleotides encoding the P450 2C3 N-terminal peptide MAKKTSSKGK. At the 3′-end, the construct was extended by 18 nucleotides encoding a His6 tag. The synthesized cDNA was inserted into the pET17b expression vector (EMD Millipore, Billerica, MA), and the plasmid was co-transformed with chaperone pGro12 plasmid into E. coli BL21-Gold DE(3)-competent cells. Following expression, the protein was purified with nickel affinity chromatography and then used for further chromatography in the absence of detergent with an SP Fast-Flow Sepharose column (GE Healthcare). The same basic methods were used for the P450 21A2 variants.
Purification of other enzymes
Rat NADPH-P450 reductase (26) and human cytochrome b5 (27) were expressed in E. coli and purified as described elsewhere.
Assays of catalytic activity
The 21-hydroxylation of progesterone and 17α-OH-progesterone was assayed as described in detail elsewhere (8, 10). P450 concentrations were adjusted accordingly to avoid >20% disappearance of substrate during incubations. The products were separated by UPLC, with detection at 240 nm using a photodiode array system. Rates of product formation were plotted versus substrate concentration to obtain hyperbolic Michaelis–Menten plots, which were subjected to non-linear regression in GraphPad Prism to estimate kcat and Km values.
Spectroscopy
UV-visible spectra were recorded with an Aminco-OLIS DW2a spectrophotometer. Substrate binding assays were done spectrophotometrically as described elsewhere (10, 24).
CD spectroscopy was performed with an Aviv spectrophotometer (model 215, Aviv Biomedical, Lakewood, NJ). The instrument was pre-equilibrated at 25 °C for 10 min before the acquisition of data. The proteins were diluted to a total protein concentration of 2 μm with 50 mm potassium phosphate (pH 7.4) before analysis. Each sample was loaded into a 0.2-cm path length quartz cuvette and allowed to equilibrate inside the cell of a CD spectrophotometer for 5 min at 25 °C. Spectra were recorded from 25 to 85 °C, with increments of 10 °C. Each scan was collected from 185 to 300 nm in the continuous scanning mode at each temperature, with a scan speed of 100 nm min−1 and a 1-nm bandwidth. CD spectral data were smoothed, buffer-subtracted, and corrected to molar ellipticity on the basis of protein concentration and molecular weight. Calculations of α-helix content were made using the formulae of Greenfield and Fasman (19) as modified by Chen and Yang (20).
The experiments in which P450–CO spectra were recorded in bacterial cells were done as described in detail by Johnston and Gillam (21). Bacterial cells were treated with sodium dithionite to establish baseline absorbance, and the CO was then added. The cells were allowed to equilibrate for 10 min at each temperature before measurements. The 450-nm absorbance data were fit to sigmoidal plots in Prism software (GraphPad Software, La Jolla, CA) (Y = bottom + (top − bottom)/(1 + 10∧(log EC50 − X))).
Crystallization of the P450 21A2–substrate complex
The human P450 21A2–17α-OH-progesterone complex was formed by adding substrate (dissolved in ethanol) to P450 21A2 in a 1:5 (enzyme/substrate) ratio, followed by mixing and concentrating to 25 mg of protein/ml in 50 mm potassium phosphate buffer (pH 7.2) containing 5% glycerol (v/v), 0.1 mm DTT, 0.1 mm EDTA, and 100 mm NaCl. Aliquots of the complex solution were frozen in liquid N2 and then stored at −80 °C. Complex crystals were grown using the sitting-drop vapor diffusion technique and mixing equal volumes (200 nl) of complex solution and mother liquor (0.20 m triammonium citrate (pH 7.0) and 20% (w/v) polyethylene glycol 3350) and equilibrating droplets against 60 μl of reservoir solution at 20 °C. Microcrystals were obtained within 2 weeks and, to improve their size, one round of microseeding was performed. Wells containing microcrystals were washed with 10 μl of reservoir buffer and transferred to a microcentrifuge tube with 40 μl of mother liquor. After vortex mixing the seeds for 2 min and diluting 1:1000 with mother liquor, 200 nl of the seed solution was mixed with 200 nl of protein–substrate complex for each drop. Larger crystals of the complex grew from microseeded droplets in about a week. Crystals were mounted in nylon loops, swiped through a droplet of 25% glycerol (v/v) in mother liquor, and flash-frozen in liquid nitrogen.
X-ray data collection, structure determination, and refinement
Diffraction data were collected at 100 K on the insertion device beam line (21-ID-G) of the Life Sciences Collaborative Access Team (LS-CAT), located at Sector 21 of the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), using a wavelength of 0.97857 Å and a Mar300 CCD detector. Data were processed with the program HKL2000 (28), and selected crystal data and refinement statistics are summarized in Table 1. The structure of the P450 21A2–17α-OH-progesterone complex was determined by the molecular replacement technique with the program Phaser (29) using the crystallographic coordinates of the human P450 21A2–progesterone complex as the search model (10) (Protein Data Bank code 4Y8W). Refinement was carried out with the program Refmac5 using the TLS option (29, 30). Manual rebuilding of the model was performed in Coot (31). The root mean square deviation values from the superimposition were calculated with the Superpose routine in CCP4 (29). All structural figures were generated with the program UCSF Chimera (32).
Modeling of selected variants in the structure of the P450 21A2–17α-OH-progesterone complex
For visualization of SW, SV, and NC CAH-causing mutations in the 3D structure of the P450 21A2 complex, we used UCSF Chimera to swap residues and generate illustrations (32). The residue numbers throughout refer to proteins encoded by the human 21-hydroxylase gene with GenBankTM number M26856.1 (WT protein) or the corresponding genes with mutations and are consistent with the numbering used in previous work from our group (10, 11).
Author contributions
C. W. performed the site-directed mutagenesis, purified the variants, ran the catalytic assays, and collected some of the stability data. P. S. P. solved the structure and initiated the CD work. W. Z. acquired the CD spectra. L. L. made the wild-type 21A2 expression vector and purified and crystallized proteins. F. K. Y. synthesized d3-progesterone. M. R. W., F. P. G., and M. E. conceived and directed the studies and analyzed the results. F. P. G. and M. E. wrote the manuscript. All authors agree with the conclusions.
Supplementary Material
Acknowledgments
We thank L. D. Nagy and T. T. N. Phan for purifying NADPH-P450 reductase, E. M. J. Gillam for suggesting the bacterial cell P450–CO binding experiments and technical advice for doing the assays, D. W. Wright for the use of the CD spectrophotometer, A. L. Bitting for technical help with the CD experiments, and K. Trisler for assistance in preparation of the manuscript.
* This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM103937 (to M. E. and F. P. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (code 5VBU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
R. J. Auchus, personal communication.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- P450 (or CYP)
- cytochrome P450
- CAH
- congenital adrenal hyperplasia
- NC
- nonclassical
- OH
- hydroxy
- SV
- simple virilizing
- SW
- salt-wasting
- UPLC
- ultraperformance liquid chromatography.
References
- 1. Northrop D. B. (1982) Deuterium and tritium kinetic isotope effects on initial rates. Methods Enzymol. 87, 607–625 [DOI] [PubMed] [Google Scholar]
- 2. Miller W. L., and Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Auchus R. J., and Miller W. L. (2015) P450 enzymes in steroid processing, in Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 851–879, Springer, New York, [Google Scholar]
- 4. Guengerich F. P. (2015) Human cytochrome P450 enzymes. in Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 523–785, Springer, New York [Google Scholar]
- 5. White P. C., and Speiser P. W. (2000) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 21, 245–291 [DOI] [PubMed] [Google Scholar]
- 6. New M. I. (2003) Inborn errors of adrenal steroidogenesis. Mol. Cell Endocrinol. 211, 75–83 [DOI] [PubMed] [Google Scholar]
- 7. Parsa A. A., and New M. I. (2017) Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J. Steroid Biochem. Mol. Biol. 165, 2–11 [DOI] [PubMed] [Google Scholar]
- 8. Zhao B., Lei L., Kagawa N., Sundaramoorthy M., Banerjee S., Nagy L. D., Guengerich F. P., and Waterman M. R. (2012) Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J. Biol. Chem. 287, 10613–10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haider S., Islam B., D'Atri V., Sgobba M., Poojari C., Sun L., Yuen T., Zaidi M., and New M. I. (2013) Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc. Natl. Acad. Sci. U.S.A. 110, 2605–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pallan P. S., Wang C., Lei L., Yoshimoto F. K., Auchus R. J., Waterman M. R., Guengerich F. P., and Egli M. (2015) Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme-progesterone substrate complex and rate-limiting C–H bond cleavage. J. Biol. Chem. 290, 13128–13143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pallan P. S., Lei L., Wang C., Waterman M. R., Guengerich F. P., and Egli M. (2015) Research resource: correlating human cytochrome P450 21A2 crystal structure and phenotypes of mutations in congenital adrenal hyperplasia. Mol. Endocrinol. 29, 1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 13. Katagiri M., Suhara K., Shiroo M., and Fujimura Y. (1982) Role of cytochrome b5 in the cytochrome P-450-mediated C21-steroid 17,20-lyase reaction. Biochem. Biophys. Res. Commun. 108, 379–384 [DOI] [PubMed] [Google Scholar]
- 14. Katagiri M., Kagawa N., and Waterman M. R. (1995) The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch. Biochem. Biophys. 317, 343–347 [DOI] [PubMed] [Google Scholar]
- 15. Yoshimoto F. K., Gonzalez E., Auchus R. J., and Guengerich F. P. (2016) Mechanism of 17α,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1: 18O labeling and oxygen surrogate evidence for a role of a perferryl oxygen. J. Biol. Chem. 291, 17143–17164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kok R. C., Timmerman M. A., Wolffenbuttel K. P., Drop S. L., and de Jong F. H. (2010) Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J. Clin. Endocrinol. Metab. 95, 994–999 [DOI] [PubMed] [Google Scholar]
- 17. Idkowiak J., Randell T., Dhir V., Patel P., Shackleton C. H., Taylor N. F., Krone N., and Arlt W. (2012) A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J. Clin. Endocrinol. Metab. 97, E465–E475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshimoto F. K., Zhou Y., Peng H. M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., and Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenfield N., and Fasman G. D. (1969) Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y.-H., and Yang J. T. (1971) A new approach to the calculation of secondary structures of globular proteins by optical rotary dispersion and circular dichroism. Biochem. Biophys. Res. Commun. 44, 1285–1291 [DOI] [PubMed] [Google Scholar]
- 21. Johnston W. A., and Gillam E. M. (2013) Measurement of P450 difference spectra using intact cells. Methods Mol. Biol. 987, 189–204 [DOI] [PubMed] [Google Scholar]
- 22. Tusie-Luna M. T., Traktman P., and White P. C. (1990) Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J. Biol. Chem. 265, 20916–20922 [PubMed] [Google Scholar]
- 23. Soardi F. C., Barbaro M., Lau I. F., Lemos-Marini S. H., Baptista M. T., Guerra-Junior G., Wedell A., Lajic S., and de Mello M. P. (2008) Inhibition of CYP21A2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J. Clin. Endocrinol. Metab. 93, 2416–2420 [DOI] [PubMed] [Google Scholar]
- 24. Schenkman J. B., Remmer H., and Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome P-450. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 25. Daniels F., and Alberty R. A. (1966) Physical Chemistry, 3rd Ed., pp. 612–750, John Wiley & Sons, Inc., New York [Google Scholar]
- 26. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 [DOI] [PubMed] [Google Scholar]
- 27. Guengerich F. P. (2005) Reduction of cytochrome b5 by NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 440, 204–211 [DOI] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Borek D., Majewski W., and Minor W. (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 [DOI] [PubMed] [Google Scholar]
- 29. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.