

Abstract
Selenoprotein P (encoded by SELENOP in humans, Selenop in rat), a liver-derived secretory protein, induces resistance to insulin and vascular endothelial growth factor (VEGF) in type 2 diabetes. Suppression of selenoprotein P may provide a novel therapeutic approach to treating type 2 diabetes; however, few drugs inhibiting SELENOP expression in hepatocytes have been identified. The present findings demonstrate that eicosapentaenoic acid (EPA) suppresses SELENOP expression by inactivating sterol regulatory element-binding protein-1c (SREBP-1c, encoded by Srebf1 in rat) in H4IIEC3 hepatocytes. Treatment with EPA caused concentration- and time-dependent reduction in SELENOP promoter activity. EPA activated AMP-activated protein kinase (AMPK); however, the inhibitory effect of EPA on SELENOP promoter activity was not canceled with an AMPK inhibitor compound C and dominant-negative AMPK transfection. Deletion mutant promoter assays and computational analysis of transcription factor-binding sites conserved among the species resulted in identification of a sterol regulatory element (SRE)-like site in the SELENOP promoter. A chromatin immunoprecipitation (ChIP) assay revealed that EPA decreases binding of SREBP-1c to the SELENOP promoter. Knockdown of Srebf1 resulted in a significant down-regulation of Selenop expression. Conversely, SREBP-1c overexpression inhibited the suppressive effect of EPA. These data provide a novel mechanism of action for EPA involving improvement of systemic insulin sensitivity through the regulation of selenoprotein P production independently of the AMPK pathway and suggest an additional approach to developing anti-diabetic drugs.
Keywords: diabetes, hepatocyte, insulin resistance, polyunsaturated fatty acid (PUFA), selenoprotein
Introduction
Selenoprotein P is a secretory protein produced primarily by the liver (1, 2); it functions as a selenium transport protein (3). Selenoprotein P contains 10 selenocysteine residues and is reportedly known to transport selenium, an essential trace element, from the liver to the rest of the body (4, 5). Through comprehensive gene expression analyses in humans, we previously found that hepatic gene expression levels of SELENOP are positively correlated with the severity of insulin resistance and post-glucose-challenge glucose levels in patients with type 2 diabetes (6). According to recent reports, blood levels of selenoprotein P are elevated under conditions of prediabetes (7) and non-alcoholic steatohepatitis (8). Treatment with purified selenoprotein P protein impairs insulin signal transduction in both cell culture and animal models. Conversely, the RNA interference-mediated knockdown of selenoprotein P improves insulin resistance and hyperglycemia in a mouse model of type 2 diabetes, suggesting that the suppression of selenoprotein P production in the liver may be a novel therapeutic target for reducing insulin resistance in type 2 diabetes (6). In addition, physiological concentrations of selenoprotein P inhibits VEGF-stimulated cell proliferation, tubule formation, and migration in human umbilical vein endothelial cells, leading to impaired angiogenesis and delay in wound closure in selenoprotein P-overxpressing mice, suggesting that selenoprotein P causes VEGF resistance (9). More recently, we have demonstrated that selenoprotein P causes exercise resistance by inhibiting reactive species-induced activation of AMPK in the skeletal muscle (10). These findings suggest that selenoprotein P contributes to resistance to insulin, VEGF, and AMP-activated protein kinase (AMPK),2 which are often observed in people with type 2 diabetes, and therefore may be a therapeutic target for type 2 diabetes. However, few drugs have been identified that can inhibit the production of selenoprotein P in hepatocytes. Our previous data showed that the anti-diabetic drug metformin reduces SELENOP gene expression via an AMPK/Forkhead box protein O3a (FoxO3a) pathway in the cultured hepatocytes (11).
Eicosapentaenoic acid (EPA) is a major component of ω-3 polyunsaturated fatty acids (PUFAs) contained in fish oil and is a well known reagent for improving lipid metabolism (12); it is clinically used as a single-agent treatment for hypertriglyceridemia. In addition, ω-3 PUFAs reportedly have beneficial effects in a wide range of health conditions, such as inflammation (13), obesity (14), hepatic steatosis (15), non-alcoholic steatohepatitis (16), insulin resistance, and type 2 diabetes (17–20). ω-3 PUFAs is beneficial against type 2 diabetes as it exerts insulin-sensitizing effects via increased production and secretion of adipocytokines (19, 21) and prevents insulin resistance via anti-inflammatory effects (22). Through modulation of some transcription factors, ω-3 PUFAs can enhance fatty acid oxidation and reduce de novo lipogenesis and consequently reduce hepatic fat accumulation and hepatic insulin resistance (14, 23–25).
Sterol regulatory element-binding proteins (SREBPs) are transcriptional factors of the basic helix-loop-helix leucine zipper family and are considered to be profoundly involved in the transcriptional regulation of cholesterogenic and lipogenic enzymes (26, 27). SREBP-1c, one of three SREBP isoforms and a key transcription factor, is responsible in fatty acid synthesis (28). The PUFAs specific suppression of lipogenic enzymes is reportedly mediated by reduction of nuclear SREBP-1c protein without affecting SREBP-2 in the liver (29–32). In addition, the primary mechanism behind PUFAs suppression of SREBP-1 reportedly functions at the proteolytic processing level and this suppression in turn decreases mRNA transcription through an autoloop regulatory circuit (33).
It has been suggested that AMPK, a member of the serine/threonine kinase family, is a key enzyme in regulating hepatic metabolism of glucose and lipids. Its activation could be involved in EPA-induced improvements in insulin sensitivity (34) by inhibiting SREBP-1 (35, 36). In contrast, another study has suggested that dietary fish oils do not activate AMPK in mouse tissues (37). The role of the AMPK/SREBP-1c pathway in hepatocytes, an important target of EPA, still remains unknown. Herein, we demonstrate that EPA suppresses SELENOP gene expression by inactivating SREBP-1c independently of the AMPK pathway in H4IIEC3 hepatocytes, indicating a possible beneficial effect on the pathology of type 2 diabetes.
Results
EPA suppresses SELENOP expression at the promoter level
EPA caused concentration- and time-dependent suppression in Selenop mRNA expression, similarly to the suppression of Srebf1 and fatty acid synthase (encoded by Fasn in rat), a central enzyme in de novo lipogenesis and an established target of the SREBP-1 pathway, mRNAs (Fig. 1, A and B, respectively). Next, we examined the effects of EPA on SELENOP promoter activity. The human SELENOP promoter region was cloned to a luciferase reporter vector, as previously reported (11). Similar to the results of mRNAs, EPA suppressed SELENOP promoter activity in a concentration- and time-dependent manner (Fig. 1, C and D).
Figure 1.

EPA suppressed Selenop gene expression in H4IIEC3 hepatocytes. A, Selenop, Srebf1, and Fasn gene expression was down-regulated in the low concentration EPA-treated H4IIEC3 hepatocytes. Expression values were normalized to Actb mRNA. Data represent mean ± S.D. (error bars) (n = 4–5). Extreme outliners were excluded. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle-treated cells. B, EPA suppressed Selenop mRNA expression in a time-dependent manner. H4IIEC3 cells were treated with the indicated concentrations of EPA for the indicated times. Expression values were normalized to 18SrRNA mRNA. Data represent mean ± S.D. (error bars) (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle-treated cells. C and D, SELENOP promoter activity was suppressed in a concentration- and time-dependent manner. H4IIEC3 cells were transfected with the SELENOP promoter reporter vector and control reporter vector. Forty-eight hours later, the cells were treated with the indicated concentrations of EPA for the indicated times. Values were normalized to the activity of the control luciferase vector. Data represent mean ± S.D. (error bars) (n = 4). ***, p < 0.001 versus vehicle-treated cells. E, left, intracellular localization of SREBP-1c in H4IIEC3 hepatocytes upon treatment with the indicated concentrations of EPA. Proteins were extracted after the indicated times of EPA treatment. Right, quantitation of precursor SREBP-1 and mature SREBP-1 is normalized to β-actin and Lamin A/C, respectively. Data represent mean ± S.D. (error bars) (n = 3). ***, p < 0.001 versus vehicle-treated cells.
EPA suppresses SELENOP expression via SREBP-1c inactivation
Because previous reports indicated that PUFAs down-regulate SREBP-1 via inhibition of nuclear translocation of SREBP-1 (33, 34), we confirmed the mechanism through which EPA inactivates SREBP-1c by examining the intracellular localization of SREBP-1c in EPA-treated H4IIEC3 hepatocytes. To determine the intracellular localization of SREBP-1c, the cytosolic and nuclear components of the SREBP-1c protein were fractionated (Fig. 1E). EPA treatment reduced SREBP-1c protein levels in the nuclear fraction in a concentration-dependent manner (Fig. 1E). Considering previous reports (31, 34), these results are suggestive of EPA inactivating SREBP-1c by inhibiting its nuclear translocation in H4IIEC3 hepatocytes.
EPA-response element in the SELENOP promoter includes the SREBP-1c-binding site
To determine the nature of the EPA-response element in the SELENOP promoter region, we constructed several deletion mutants of the SELENOP promoter (Fig. 2A). Promoter activity of Mutant (Mut)-A, -B, -C, -D, and Normal, but not Mut-E, was suppressed by EPA treatment (Fig. 2A), indicating the existence of EPA-response element of the SELENOP promoter in Mut-D. Additional deletion mutants of Mut-D were constructed and named Mut-DΔ1 to DΔ3. Promoter activity of Mut-DΔ1 and DΔ2 but not Mut-DΔ3 was suppressed by EPA treatment (Fig. 2B), indicating the existence of EPA-response element of the SELENOP promoter in Mut-DΔ2. Using computational analysis to identify conserved transcription factor-binding sites (TFBSs) among the species as previously reported (11), several putative TFBSs were identified in the Mut-DΔ2 sequence. We focused on the putative SREBP-1c-binding sites (Fig. 2C). Indeed, SREBP-1c is reportedly a key regulator of lipid synthesis and PUFA-specific suppression of nuclear SREBP-1c protein in the liver (29–32).
Figure 2.

SELENOP promoter activity of deletion mutants. A and B, structure and luciferase activity of promoter-deletion mutants. The sequences deleted within the constructs are shown as thin lines. The remaining parts of the SELENOP promoter were fused to a luciferase reporter gene. H4IIEC3 cells were transfected with each reporter vector and control reporter vector at 48 h and then treated with the indicated concentrations of EPA for 24 h. Signals were normalized to the control reporter vector. Data represent mean ± S.D. (error bars) (n = 6). ***, p < 0.001 versus vehicle-treated cells. C, putative SREBP-1c-binding sites of Mut-DΔ2 sequence. Detection of the conserved TFBSs was performed using multiple genome alignments and the highlighted putative transcriptional factor-binding sites.
Next, we examined whether Selenop expression is affected by the specific knockdown of endogenous SREBP-1 in H4IIEC3 hepatocytes. Transfection with Srebf1-specific siRNA reduced Srebf1 mRNA levels by ∼50% (Fig. 3A). This Srebf1 knockdown also significantly down-regulated Selenop expression to a similar extent (Fig. 3A). Conversely, we investigated if SREBP-1c overexpression influences SELENOP promoter activity. The SREBP-1 protein was overexpressed in cells transfected with the precursor SREBP-1a, precursor SREBP-1c, and mature SREBP-1c expression vectors (Fig. 3B). The mature SREBP-1c overexpression especially inhibited the suppressive effect of EPA on SELENOP promoter activity (Fig. 3C); however, SREBP-1a overexpression had no effect (data not shown). These results suggest an association of SREBP-1c activity with the EPA-induced suppression of SELENOP expression.
Figure 3.
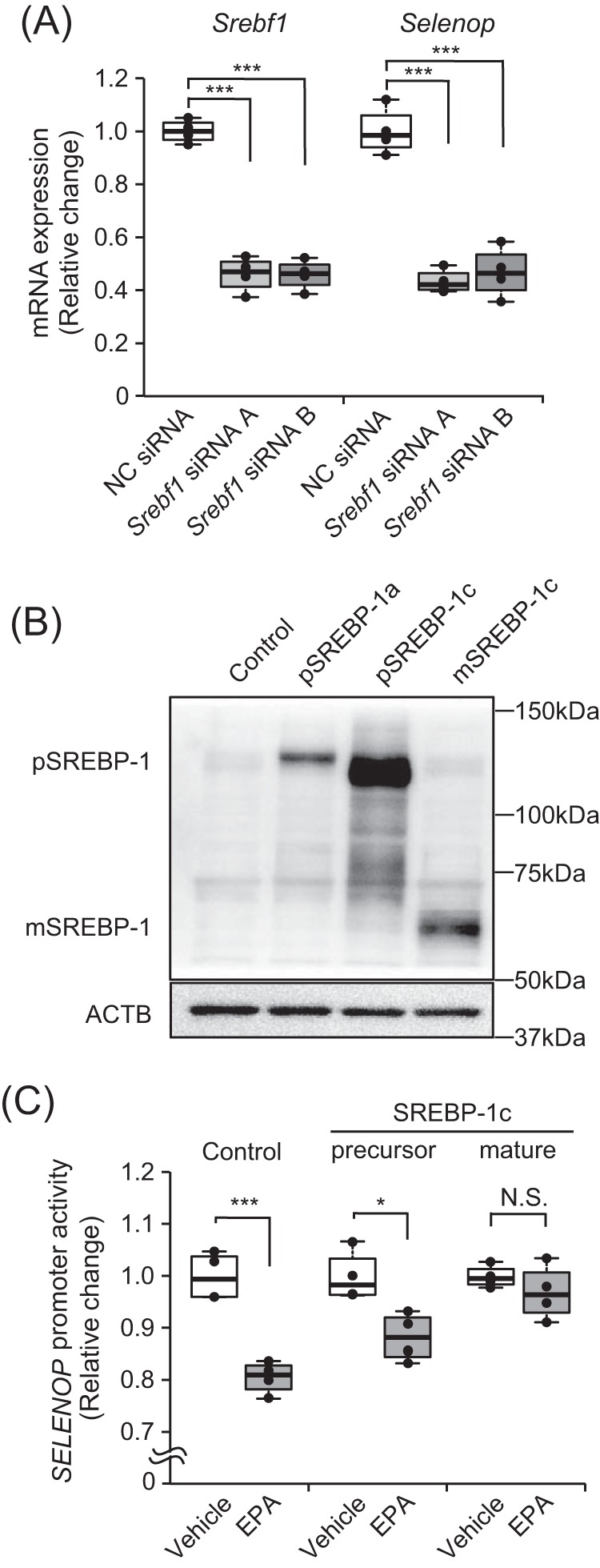
EPA suppressed selenoprotein P expression via SREBP-1c. A, efficiency of Srebf1 siRNA. H4IIEC3 cells were transfected with Srebf1 siRNAs or a negative control (NC) siRNA at 48 h. Knockdown efficiency was assessed by real-time PCR. Expression values were normalized to Actb mRNA. Data represent mean ± S.D. (error bars) (n = 4). ***, p < 0.001 versus negative control siRNA-treated cells. B, protein levels in the presence of the SREBP-1c overexpression vector. H4IIEC3 cells were transfected with the pCMV7-precursor SREBP-1c (pSREBP-1c) or the pCMV7-mature SREBP-1c (mSREBP-1c) vector or the pCMV7 empty vector at 24 h. SREBP-1c protein levels were then assessed by Western blotting. C, SELENOP promoter activity transfected with the precursor SREBP-1c or mature SREBP-1c vector. H4IIEC3 cells were co-transfected with the expression vectors for SREBP-1c, SELENOP promoter reporter, and control reporter at 48 h and then treated with 0.25 mm EPA for 24 h. Data represent mean ± S.D. (error bars) (n = 3–4). *, p < 0.05; ***, p < 0.001; N.S., not significant, versus vehicle-treated cells.
EPA suppressed SELENOP promoter activity through an AMPK-independent pathway
We previously found that metformin suppressed SELENOP expression via the AMPK/FoxO3a pathway (11). Furthermore, previous reports indicated the possible involvement of AMPK activation in EPA-induced improvements in insulin sensitivity (34) and also that AMPK phosphorylation inhibits SREBP-1 (35, 36). Thus, to determine whether AMPK pathways are involved in the EPA-induced suppression of SELENOP promoter activity, we treated H4IIEC3 hepatocytes with compound C, a representative AMPK inhibitor. Consequently, the findings confirmed that the EPA-induced phosphorylation of AMPK and acetyl-CoA carboxylase (ACC) was canceled by the administration of compound C in H4IIEC3 hepatocytes (Fig. 4A); these findings corroborate those of a previous study (34). However, co-administration of compound C and EPA suppressed somewhat the Selenop mRNA expression and SELENOP promoter activity (Fig. 4, B and C). Furthermore, we tested the effects of the dominant-negative form of AMPK on EPA-mediated suppression of Selenop mRNA expression. As shown in Fig. 4D, AMPK dominant-negative had no effect on Selenop mRNA expression. These results suggest that EPA suppressed SELENOP promoter activity independently of the AMPK pathway.
Figure 4.

EPA suppressed SELENOP promoter activity through an AMPK-independent pathway in H4IIEC3 hepatocytes. A, EPA-induced AMPK phosphorylation in the absence or presence of compound C. H4IIEC3 cells were pretreated with 5 μm compound C for 24 h prior to treatment with 0.25 mm EPA and 5 μm compound C for 3 h. AMPK phosphorylation was examined by Western blotting and quantified. B, compound C treatment did not recover EPA-induced suppression of the Selenop mRNA expression. H4IIEC3 cells were treated with 0.25 mm EPA and 5 μm compound C for 24 h. Expression values were normalized to Actb mRNA. Data represent mean ± S.D. (error bars) (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, compound C treatment did not recover EPA-induced suppression of the SELENOP promoter. H4IIEC3 cells were transfected with the SELENOP promoter reporter vector and control reporter vector at 24 h and then pretreated with 5 μm compound C for 1 h before treatment with 0.25 mm EPA and 5 μm compound C for 24 h. Signals were normalized to the control reporter vector. D, influence of adenoviruses carrying dominant-negative (DN) AMPK. H4IIEC3 cells were infected with adenoviruses encoding DN-AMPK or LacZ. Expression values were normalized to Actb mRNA. Data represent mean ± S.D. (error bars) (n = 4). ***, p < 0.001.
EPA suppresses SELENOP gene expression by inhibiting SREBP-1c binding to Selenop promoter DNA
To determine the critical SREBP-1c-binding site for EPA-induced SELENOP suppression, we constructed luciferase vectors that deleted the putative SRE, binding sites of SREBP-1c. It is called Mut-DΔ2-ΔSRE-like (Fig. 5A). Luciferase assay using these vectors revealed that the putative SREBP-1c-binding site was essential for EPA-induced SELENOP suppression (Fig. 5A). Finally, using a ChIP assay, we examined the interaction of SREBP-1c proteins with DNA sequences in the Selenop promoter. Consequently, treatment with EPA was found to decrease the binding of SREBP-1c to the Selenop promoter (Fig. 5B), indicating that EPA decreases SELENOP promoter activity and gene expression via SREBP-1c inactivation in H4IIEC3 hepatocytes (Fig. 5C).
Figure 5.
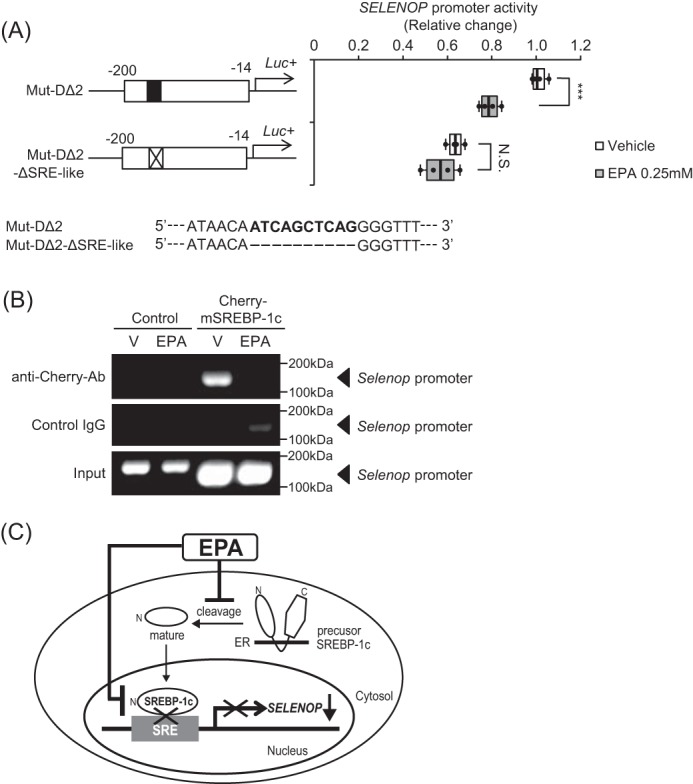
EPA suppresses SELENOP gene expression by inhibiting SREBP-1c binding to Selenop promoter DNA. A, deficiency of the putative SREBP-1-binding site canceled EPA-induced suppression of SELENOP promoter activity. H4IIEC3 cells were co-transfected with each reporter vector and control reporter vector at 48 h and then treated with the indicated concentrations of EPA for 24 h. Signals were normalized to the control reporter vector. Data represent mean ± S.D. (error bars) (n = 4). ***, p < 0.001 versus vehicle-treated cells. B, chromatin immunoprecipitation assay of H4IIEC3 cells treated with EPA. H4IIEC3 hepatocytes were grown in 15-cm dishes and transfected with 30.4 μg of pcDNA3.1(+) mCherry-mature SREBP-1c plasmid vectors per dish together with 91.2 μl of FuGENE6. Forty-eight hours later, cells were treated with 0.25 mm EPA for 24 h. Chromatin samples precipitated with anti-RFP, or anti-normal IgG were amplified using primers for the −200 to −100 bp region of the rat Selenop promoter. C, scheme of selenoprotein P suppression by EPA in the liver. SREBP-1c positively regulates SELENOP promoter activity. EPA suppresses SREBP-1c activity, resulting in suppression of selenoprotein P expression. Thus, the hypoglycemic effects of EPA may be mediated at least in part by selenoprotein P suppression in the liver.
Discussion
To our knowledge, this is the first study to show that a polyunsaturated fatty acid EPA down-regulates diabetes-associated hepatokine SELENOP expression by inactivating SREBP-1c in H4IIEC3 hepatocytes. Several lines of evidence have suggested that ω-3 PUFAs, such as EPA and docosahexaenoic acid (DHA) protect against high-fat diet-induced insulin resistance (38). Neschen et al. (24) demonstrated that EPA protects mice from high-fat diet-induced hepatic insulin resistance by binding directly to peroxisome proliferator-activated receptor α and thus decreasing diacylglycerol levels in the liver of mice. Yahagi et al. (32) found that EPA decreases lipid synthesis by inhibiting nuclear translocation of SREBP-1. Similarly, in this study implementing the use of H4IIEC3 hepatocytes, EPA decreased gene expression of Srebf1 and Fasn (Fig. 1, A and B). In this study we also examined whether some kinds of fatty acids affect gene expression levels of Selenop, Srebf1, and Fasn. Selenop, Srebf1, and Fasn gene expression was down-regulated in the EPA-treated H4IIEC3 hepatocytes at a concentration of 0.01 mm. However, under these conditions, DHA, arachidonic acid, and arachidic acid did not suppress the Selenop expression. DHA and arachidonic acid only slightly suppressed the Selenop expression at a concentration of 0.25 mm, whereas arachidic acid did not. As previously reported (39, 40), EPA, DHA, and arachidonic acid suppressed Srebf1 and Fasn expression. These findings suggest that EPA is a very potent suppressor of selenoprotein P, the hepatokine that causes insulin resistance (Fig. 1A). Recently, Kemuriyama et al. (41) reported that EPA had a greater hepatic triacylglycerol-reducing effect than DHA. This suggests that EPA has a greater suppressive effect on hepatic triacylglycerol accumulation and insulin resistance via inhibition of the Srebp1-Selenop pathway than other fatty acids. This study proposes a novel mechanism underlying EPA-mediated amelioration of systemic insulin resistance.
Then, we examined the mechanisms underlying the inhibitory effect of EPA on SELENOP mRNA expression. The present study has shown that: 1) EPA reduces the gene expression level and the nuclear protein content of SREBP-1c (Fig. 1, A, B, and E), 2) the deletion of the SRE-like element on the promoter cancels the EPA-induced inhibition of SELENOP transcriptional activity (Figs. 3 and 5A) and EPA attenuates the binding of SREBP-1c to the SELENOP promoter (Fig. 5B). Therefore, the inhibitory effect of EPA on SELENOP gene expression is attributed to inactivation of SREBP-1c. SREBP-1c is a membrane protein that binds to the rough endoplasmic reticulum, and the basic helix-loop-helix in the amino-terminal is cut-out and exerts transcriptional activity by migrating into the nucleus. In addition, EPA reportedly exerts inhibitory effects on SREBP-1c due to several mechanisms, such as a suppression of cleavage activity and proteasomal degradation (32, 33). In this study, Western blot analysis and quantitative densitometry analysis clearly showed that EPA reduced SREBP-1c protein levels in the nuclear fraction in a concentration-dependent manner by ∼10–20% (Fig. 1E). We also conducted time course studies (0, 3, 6, 12, 18, and 24 h) to investigate whether EPA inhibited SREBP-1c nuclear translocation in Western blotting. EPA dramatically reduced the nuclear accumulation of mature SREBP-1c within 3 h after EPA treatment. But there was no significant change of precursor SREBP-1c in the cytosolic fraction. Meanwhile, after vehicle treatment, the nuclear accumulation of mature SREBP-1c increased 18 h later, and precusor SREBP-1c was gradually reduced 12 h later (data not shown). These findings indicate that EPA strongly inhibited the nuclear translocation of SREBP-1c and subsequently increased the accumulation of cytosolic SREBP-1c. Therefore, it is estimated that EPA inhibits cleavage and nuclear translocation of SREBP-1c, thus down-regulating SREBP-1c and SELENOP.
Selenop mRNA expression was inhibited by knockdown of Srebf1 mRNA using RNAi (Fig. 3A). Meanwhile, the effect of suppressing the SELENOP promoter activity by EPA was lost because of overexpression of a nuclear active SREBP-1c (Fig. 3C). These results suggest that SREBP-1c positively controls Selenop expression. Hepatic expression of SREBP-1c is associated with insulin resistance and pathology of non-alcoholic fatty liver disease (NAFLD) (42). SREBP-1c may aggravate fatty liver by activating the enzymes involved in fatty acid synthesis and may exacerbate insulin resistance by down-regulating expression of the gene for insulin receptor substrate-2 in the liver (43). Choi et al. (8) reported that serum levels of selenoprotein P are elevated in patients with NAFLD. The present study shows that hepatic expression of SELENOP is positively controlled by SREBP-1c, suggesting that the transcriptional regulation of SELENOP by SREBP-1c exacerbates insulin resistance and pathology of NAFLD.
SELENOP is also known to be up-regulated through the activation of Forkhead box protein O1 (FoxO1) (44) and FoxO3a (11). Insulin down-regulates SELENOP by inactivating FoxO1 (44). Conversely, we previously found that metformin, an anti-diabetic agent, activates AMPK in hepatocytes and thereby down-regulates SELENOP promoter activity and expression (11). Chen et al. (45) showed that inhibition of the AMPK pathway with compound C inhibits the nuclear translocation of FoxO1. AMPK also phosphorylates the Ser372 site of SREBP-1c and inhibits cleavage and nuclear translocation of SREBP-1c (35). As EPA reportedly activates AMPK (46), we initially hypothesized that EPA down-regulates SELENOP expression by activating AMPK. Consistent with a previous report (46), phosphorylation of AMPK and ACC was enhanced by EPA treatment. Moreover, inhibition of the AMPK pathway, using either the AMPK inhibitor compound C or adenovirus-mediated gene transfer of dominant-negative AMPK, did not cancel the suppressive effect of EPA on SELENOP promoter activity and SELENOP mRNA expression (Fig. 4, C and D). These findings clearly indicate that EPA down-regulates SELENOP by inactivating SREBP-1c independently of the AMPK pathway in H4IIEC3 hepatocytes.
Selenoprotein P is a hepatokine that causes hepatic insulin resistance by inactivating AMPK (6). Therefore, EPA may ameliorate hepatic insulin resistance both by inactivating SREBP-1c and by down-regulating selenoprotein P. In addition, selenoprotein P impairs angiogenesis by inducing VEGF resistance (9). Clinically, EPA prevents coronary events in Japanese hypercholesterolaemic patients as shown in the Japan EPA lipid intervention study (JELIS) study (47). Together with the findings of the present study, we can infer that EPA could reduce coronary events, at least partly, by down-regulating the selenoprotein P that causes VEGF resistance.
Experimental procedures
Materials
The antibodies against AMPKα, phospho-AMPKα, ACC, phospho-ACC, fatty acid synthase, β-Actin, and Lamin A/C were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against red fluorescent protein (RFP) were purchased from Medical & Biological Laboratories Co., Ltd. (Nagoya, Japan). Antibodies against SREBP-1, GAPDH, and normal mouse IgG were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Cis-5,8,11,14,17-EPA sodium salt, cis-4,7,10,13,16,19-DHA, arachidonic acid, and arachidic acid, albumin from bovine serum-lyophilized powder (essentially fatty acid free; FFA-free BSA), and 6-[4-(2-piperidin-1-yl-ethoxy)-phenyl)]3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine (compound C) were purchased from Sigma. pCMV7 or pCMV7-precusor or mature SREBP-1c vectors were kindly provided by Dr. Hitoshi Shimano.
Cell culture and fatty acid treatment
Studies were performed using the rat hepatoma cell line H4IIEC3 (American Type Culture Collection, Manassas, VA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies Corp.) and supplemented with 10% fetal bovine serum (Life Technologies), 2 mmol/liter of l-glutamin (Wako Pure Chemical Industries, Ltd., Osaka, Japan), 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin (Wako). The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. All studies were conducted using cells from 80 to 90% confluent cultures; these cells were treated with the indicated concentrations of EPA and DHA, arachidonic acid was dissolved in 99.5% ethanol (final ethanol concentration was 0.25%), arachidic acid was dissolved in 99.0% chloroform (final chloroform concentration was 0.25%) in the presence of 2% FFA-free BSA (Sigma).
Generation of plasmid constructs
The human SELENOP promoter region and several deletion mutants of the SELENOP promoter termed “SELENOP-Promoter-Luc,” “Mut-A,” “Mut-B,” “Mut-C,” “Mut-D,” “Mut-E,” “Mut-DΔ1,” “Mut-DΔ2,” and “Mut-DΔ3” were cloned as previously described (11, 48). Putative SREBP-1c-binding site-deficient vectors were generated using QuikChange Lightning Site-directed Mutagenesis Kits (Agilent Technologies, Santa Clara CA) and the primer pairs as follows: forward, 5′-CCGGGCTCGAGATCTATAACAGGGTTTGCTCT-3′ and reverse 5′-AGAGCAAACCCTGTTATAGATCTCGAGCCCGG-3′, according to the manufacturer's instructions. pcDNA3.1(+) mCherry-mature SREBP-1c plasmid vectors were generated. First, pcDNA3.1(+) mCherry was constructed. Plasmid Cherry was purchased from Addgene ID 39319. pcDNA3.1(+) and pBlueScriptII SK-Cherry were both digested by restrictive endonucleases EcoRI and NotI, respectively; thereafter, Cherry fragments were inserted into the pcDNA3.1(+) vector and cloned in DH5α competent cells. Second, pcDNA3.1(+)-mCherry-mature SREBP-1c was constructed. cDNA fragments encoding mature SREBP-1c were generated by PCR from pcDNA3.1(+)-mature SREBP-1c using the forward primer NotI-SREBP1c (5′-TAGCGGCCGCATATGGATTGCACTTTCG-3′) and the reverse primer SREBP1c-XbaI (5′-CGGCTCTAGACTACTAGTCAGGCTCCGAG-3′). The PCR products and pcDNA3.1(+)-fluorescent protein (mCherry) were both digested by restrictive endonucleases NotI and XbaI, respectively; thereafter, mature SREBP-1c fragments were inserted into the pcDNA3.1(+)-mCherry and cloned in DH5α competent cells. All inserts were confirmed by DNA sequencing.
Adenovirus-mediated gene transfer in H4IIEC3 hepatocytes
Cells were transfected with adenoviruses as described previously (6). Briefly, H4IIEC3 hepatocytes were grown to 90% confluence in 24-well multiplate and transfected with adenoviruses encoding dominant-negative (DN) α1 and α2 AMPK or LacZ for 4 h. The cells were incubated with DMEM for 24 h after removing the adenoviruses; total RNA was then extracted.
Transfection and luciferase reporter gene assay
H4IIEC3 cells were grown in 24-well plates and transfected with 0.4 μg of plasmid DNA per well together with 1.2 μl of FuGENE6 (Promega). For the luciferase reporter gene assays, 0.4 μg of firefly luciferase promoter construct was co-transfected with 0.01 μg of Renilla luciferase control plasmid (pRL-SV40; Promega) and 0.05–0.4 μg of plasmids expressing SREBP-1c or empty control plasmids resulting in a total DNA amount of 0.41–0.81 μg/well. Twenty-four hours later, cells were treated with the indicated reagents, such as EPA, in DMEM, 10% FBS, 2% BSA for the indicated times. After 24 h, luciferase activities were measured using the Dual Luciferase Assay System (Promega), as described previously (49).
siRNA transfection in H4IIEC3 hepatocytes
H4IIEC3 hepatocytes were grown in 24-well plates and transiently transfected with 50 nm small interfering RNA (siRNA) duplex oligonucleotides using 1 μl of LipofectamineTM RNAiMAX (Life Technologies) by the reverse-transfection method according to the manufacturer's instructions. Srebf1-specific siRNAs containing the following sequences were synthesized (Thermo Scientific): Srebf1 A, 5′-CAACAAGCUGACAUGGAU-3′ (sense); Srebf1 B, 5′-GAGCGAGCAUUGAACUGUA-3′ (sense). Negative control siRNA was also utilized (Thermo Scientific). Twenty-four hours after transfection, the cells were followed by the extraction of total RNA.
Quantitative RT-PCR
Total RNA was extracted from cultured H4IIEC3 hepatocytes using a High Pure RNA Isolation Kit (Roche Diagnostics), according to the manufacturer's protocol. The reverse transcription of 100 ng of total RNA was performed using a High-Capacity cDNA Reverse Transcription Kit (Life Technologies), according to the manufacturer's instructions. Quantitative RT-PCR was performed using TaqMan probes (Actb, 4352340E; Selenop, Rn00569905_m1; Srebf1, Rn01495769_m1; Fasn, Rn01463550_m1; phosphoenolpyruvate carboxykinase 1 (encoded by Pck1 in rat), Rn01529014_m1; glucose-6-phosphatase catalytic subunit (encoded by G6pc in rat), Rn00565347_m1) and the 7900HT Fast Real-Time PCR System (Life Technologies), as described previously (50).
Western blotting
Treated cells were collected and lysed as described previously (49). Nuclear extracts were achieved using the NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit according to the manufacturer's protocol (ThermoFisher). Protein samples were subjected to SDS-PAGE and transferred to PVDF membranes using the iBlot Gel Transfer System (Life Technologies). The membranes were blocked in a buffer containing 5% nonfat milk, 50 mm Tris (pH 7.6), 150 mm NaCl, and 0.1% Tween 20 (TBS-T) or PVDF Blocking Reagent for Can Get Signal (Toyobo Co., Ltd., Osaka, Japan) for 1 h at room temperature. Thereafter, the membranes were incubated with specific primary antibodies, washed, and incubated with the secondary, HRP-labeled antibodies. Bands were visualized with the ECL Prime Western Blotting Detection System (GE Healthcare UK Ltd.; Amersham Biosciences Place, Little Chalfont, UK) and LAS-3000 (Fujifilm; Tokyo, Japan) and ChemiDoc Touch Imaging System (Bio-Rad). A densitometric analysis of blotted membranes was performed using ImageJ software.
Detection of the conserved transcription factor-binding sites using multiple genome alignments
The Ensembl 12-way Enredo-Pecan-Ortheus (EPO) eutherian multiple alignments (12-way EPO alignments) (51, 52) were downloaded from ftp.ensembl.org/pub/release-65/emf/ensembl-compara/epo_12_eutherian/.3 The 12-way EPO was excised to obtain the alignment block corresponding to the human genome coordinates from 10 kb upstream of the coding sequence of SELENOP, including the start codon. To predict the conserved TFBSs, the 10-kb upstream genome sequence for each of the 12 species was searched using TRANSFAC (53) MATCHTM program (54) (version 6.1) with varying thresholds. Then, the predicted TFBSs were mapped on the alignments, and the conserved TFBSs for SELENOP were identified.
Chromatin immunoprecipitation assay
A ChIP assay was performed using the ChIP IT Express Enzymatic Kit (Active Motif, Carlsbad, CA), according to the manufacturer's instructions. In brief, H4IIEC3 hepatocytes were grown in 15-cm dishes and transfected with 30.4 μg of pcDNA3.1(+) mCherry-mature SREBP-1c plasmid vectors per dish together with 91.2 μl of FuGENE6 (Promega). Forty-eight hours later, cells were treated with 0.25 mm EPA for 24 h before being fixed and homogenized. Following centrifugation, the supernatant was used for chromatin samples. Chromatin samples were incubated with protein G-coated magnetic beads and RFP antibodies (Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) overnight at 4 °C. Following washing and elution, a reaction solution was used as the template for PCR. PCR primers were set for amplification of the Mut-DΔ2 region of the SELENOP promoter, as follows: forward, 5′-AACATTCTTCTCGTCGCGGCAACCA-3′; and reverse, 5′-AGATCCACAAAGCCACAGGCTGACA-3′.
Author contributions
N. T-S., K-I. I., H. M., and T. T designed the research, analyzed the data, and wrote the manuscript. N. T-S., H. T., T. S., K. C., A. T., F. L., and A. K. performed the experiments. H. I., Y. S., Y. I., Y. T., K. M., S. M., and S. K. interpreted the results of the experiments.
Acknowledgments
We thank Fabienne Foufelle of Université Pierre et Marie Curie for providing the adenovirus vector encoding DN-AMPK and Hitoshi Shimano at the University of Tsukuba for providing the vector encoding SREBP1.
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to H. M., S. K., and T. T.), and Japan Society for the Promotion of Science KAKENHI Grants 16K09740 (to H. M.) and 16K09739 (to Y. T.). The authors declare that they have no conflicts of interest with the contents of this article.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- AMPK
- AMP-activated protein kinase
- EPA
- eicosapentaenoic acid
- SREBP
- sterol regulatory element-binding protein
- TFBS
- transcription factor-binding site
- ACC
- acetyl-CoA carboxylase
- DHA
- docosahexaenoic acid
- NAFLD
- non-alcoholic fatty liver disease
- Mut
- mutant.
References
- 1. Burk R. F., and Hill K. E. (2005) Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 25, 215–235 [DOI] [PubMed] [Google Scholar]
- 2. Carlson B. A., Novoselov S. V., Kumaraswamy E., Lee B. J., Anver M. R., Gladyshev V. N., and Hatfield D. L. (2004) Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 279, 8011–8017 [DOI] [PubMed] [Google Scholar]
- 3. Saito Y., and Takahashi K. (2002) Characterization of selenoprotein P as a selenium supply protein. Eur. J. Biochem. 269, 5746–5751 [DOI] [PubMed] [Google Scholar]
- 4. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., and Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 5. Schomburg L., Schweizer U., Holtmann B., Flohé L., Sendtner M., and Köhrle J. (2003) Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Misu H., Takamura T., Takayama H., Hayashi H., Matsuzawa-Nagata N., Kurita S., Ishikura K., Ando H., Takeshita Y., Ota T., Sakurai M., Yamashita T., Mizukoshi E., Yamashita T., Honda M., et al. (2010) A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 12, 483–495 [DOI] [PubMed] [Google Scholar]
- 7. Yang S. J., Hwang S. Y., Choi H. Y., Yoo H. J., Seo J. A., Kim S. G., Kim N. H., Baik S. H., Choi D. S., and Choi K. M. (2011) Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metab. 96, E1325–E1329 [DOI] [PubMed] [Google Scholar]
- 8. Choi H. Y., Hwang S. Y., Lee C. H., Hong H. C., Yang S. J., Yoo H. J., Seo J. A., Kim S. G., Kim N. H., Baik S. H., Choi D. S., and Choi K. M. (2013) Increased selenoprotein p levels in subjects with visceral obesity and nonalcoholic fatty liver disease. Diabetes Metab. J. 37, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ishikura K., Misu H., Kumazaki M., Takayama H., Matsuzawa-Nagata N., Tajima N., Chikamoto K., Lan F., Ando H., Ota T., Sakurai M., Takeshita Y., Kato K., Fujimura A., Miyamoto K., et al. (2014) Selenoprotein P as a diabetes-associated hepatokine that impairs angiogenesis by inducing VEGF resistance in vascular endothelial cells. Diabetologia 57, 1968–1976 [DOI] [PubMed] [Google Scholar]
- 10. Misu H., Takayama H., Saito Y., Mita Y., Kikuchi A., Ishii K. A., Chikamoto K., Kanamori T., Tajima N., Lan F., Takeshita Y., Honda M., Tanaka M., Kato S., Matsuyama N., et al. (2017) Deficiency of selenoprotein P increases responsiveness to exercise in mice through upregulation of reactive oxygen species and AMP-activated protein kinase in muscle. Nat. Med. 23, 508–516 [DOI] [PubMed] [Google Scholar]
- 11. Takayama H., Misu H., Iwama H., Chikamoto K., Saito Y., Murao K., Teraguchi A., Lan F., Kikuchi A., Saito R., Tajima N., Shirasaki T., Matsugo S., Miyamoto K.-I., Kaneko S., and Takamura T. (2014) Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J. Biol. Chem. 289, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carpentier Y. A., Portois L., and Malaisse W. J. (2006) n-3 Fatty acids and the metabolic syndrome. Am. J. Clin. Nutr. 83, 1499S–1504S [DOI] [PubMed] [Google Scholar]
- 13. Arita M., Yoshida M., Hong S., Tjonahen E., Glickman J. N., Petasis N. A., Blumberg R. S., and Serhan C. N. (2005) Resolvin E1, an endogenous lipid mediator derived from ω-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 7671–7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sato A., Kawano H., Notsu T., Ohta M., Nakakuki M., Mizuguchi K., Itoh M., Suganami T., and Ogawa Y. (2010) Antiobesity effect of eicosapentaenoic acid in high-fat/high-sucrose diet-induced obesity: importance of hepatic lipogenesis. Diabetes 59, 2495–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sekiya M., Yahagi N., Matsuzaka T., Najima Y., Nakakuki M., Nagai R., Ishibashi S., Osuga J., Yamada N., and Shimano H. (2003) Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 38, 1529–1539 [DOI] [PubMed] [Google Scholar]
- 16. Ishii H., Horie Y., Ohshima S., Anezaki Y., Kinoshita N., Dohmen T., Kataoka E., Sato W., Goto T., Sasaki J., Sasaki T., Watanabe S., Suzuki A., and Ohnishi H. (2009) Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J. Hepatol. 50, 562–571 [DOI] [PubMed] [Google Scholar]
- 17. Flachs P., Rossmeisl M., Bryhn M., and Kopecky J. (2009) Cellular and molecular effects of n-3 polyunsaturated fatty acids on adipose tissue biology and metabolism. Clin. Sci. 116, 1–16 [DOI] [PubMed] [Google Scholar]
- 18. Pérez-Echarri N., Pérez-Matute P., Marcos-Gómez B., Martínez J. A., and Moreno-Aliaga M. J. (2009) Effects of eicosapentaenoic acid ethyl ester on visfatin and apelin in lean and overweight (cafeteria diet-fed) rats. Br. J. Nutr. 101, 1059–1067 [DOI] [PubMed] [Google Scholar]
- 19. Pérez-Matute P., Pérez-Echarri N., Martínez J. A., Marti A., and Moreno-Aliaga M. J. (2007) Eicosapentaenoic acid actions on adiposity and insulin resistance in control and high-fat-fed rats: role of apoptosis, adiponectin and tumour necrosis factor-α. Br. J. Nutr. 97, 389–398 [DOI] [PubMed] [Google Scholar]
- 20. Rossi A. S., Lombardo Y. B., Lacorte J. M., Chicco A. G., Rouault C., Slama G., and Rizkalla S. W. (2005) Dietary fish oil positively regulates plasma leptin and adiponectin levels in sucrose-fed, insulin-resistant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, 486–494 [DOI] [PubMed] [Google Scholar]
- 21. Itoh M., Suganami T., Satoh N., Tanimoto-Koyama K., Yuan X., Tanaka M., Kawano H., Yano T., Aoe S., Takeya M., Shimatsu A., Kuzuya H., Kamei Y., and Ogawa Y. (2007) Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arterioscler. Thromb. Vasc. Biol. 27, 1918–1925 [DOI] [PubMed] [Google Scholar]
- 22. Oh D. Y., Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W. J., Watkins S. M., and Olefsky J. M. (2010) GPR120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jump D. B. (2011) Fatty acid regulation of hepatic lipid metabolism. Curr. Opin. Clin. Nutr. Metab. Care. 14, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neschen S., Morino K., Dong J., Wang-Fischer Y., Cline G. W., Romanelli A. J., Rossbacher J. C., Moore I. K., Regittnig W., Munoz D. S., Kim J. H., and Shulman G. I. (2007) n-3 fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-α-dependent manner. Diabetes. 56, 1034–1041 [DOI] [PubMed] [Google Scholar]
- 25. Tanaka N., Zhang X., Sugiyama E., Kono H., Horiuchi A., Nakajima T., Kanbe H., Tanaka E., Gonzalez F. J., and Aoyama T. (2010) Eicosapentaenoic acid improves hepatic steatosis independent of PPARα activation through inhibition of SREBP-1 maturation in mice. Biochem. Pharmacol. 80, 1601–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown M. S., and Goldstein J. L. (1997) The SREBP pathway: regulation review of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 27. Yokoyama C., Wang X., Briggs M. R., Admon A., Wu J., Hua X., Goldstein J. L., and Brown M. S. (1993) SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 [PubMed] [Google Scholar]
- 28. Horton J. D., Shimomura I., Brown M. S., Hammer R. E., Goldstein J. L., and Shimano H. (1998) Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Invest. 101, 2331–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H. J., Takahashi M., and Ezaki O. (1999) Cell biology and metabolism: fish oil feeding decreases mature sterol regulatory element-binding protein 1 SREBP-1c mRNA in mouse liver. J. Biol. Chem. 274, 25892–25898 [DOI] [PubMed] [Google Scholar]
- 30. Mater M. K., Thelen A. P., Pan D. A., and Jump D. B. (1999) Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J. Biol. Chem. 274, 32725–32732 [DOI] [PubMed] [Google Scholar]
- 31. Xu J., Nakamura M. T., Cho H. P., and Clarke S. D. (1999) Regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. J. Biol. Chem. 274, 23577–23583 [DOI] [PubMed] [Google Scholar]
- 32. Yahagi N., Shimano H., Hasty A. H., Amemiya-Kudo M., Okazaki H., Tamura Y., Iizuka Y., Shionoiri F., Ohashi K., Osuga J., Harada K., Gotoda T., Nagai R., Ishibashi S., and Yamada N. (1999) A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem. 274, 35840–35844 [DOI] [PubMed] [Google Scholar]
- 33. Takeuchi Y., Yahagi N., Izumida Y., Nishi M., Kubota M., Teraoka Y., Yamamoto T., Matsuzaka T., Nakagawa Y., Sekiya M., Iizuka Y., Ohashi K., Osuga J., Gotoda T., Ishibashi S., et al. (2010) Polyunsaturated fatty acids selectively suppress sterol regulatory element-binding protein-1 through proteolytic processing and autoloop regulatory circuit. J. Biol. Chem. 285, 11681–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flachs P., Mohamed-Ali V., Horakova O., Rossmeisl M., Hosseinzadeh-Attar M. J., Hensler M., Ruzickova J., and Kopecky J. (2006) Polyunsaturated fatty acids of marine origin induce adiponectin in mice fed a high-fat diet. Diabetologia 49, 394–397 [DOI] [PubMed] [Google Scholar]
- 35. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., and Cohen R. a., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porstmann T., Santos C. R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J. R., Chung Y. L., and Schulze A. (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobrzyn A., Dobrzyn P., Miyazaki M., and Ntambi J. M. (2005) Polyunsaturated fatty acids do not activate AMP-activated protein kinase in mouse tissues. Biochem. Biophys. Res. Commun. 332, 892–896 [DOI] [PubMed] [Google Scholar]
- 38. Ungerfeld R., Dago A. L., Rubianes E., and Forsberg M. (2004) Response of anestrous ewes to the ram effect after follicular wave synchronization with a single dose of estradiol-17β. Reprod. Nutr. Dev. 44, 89–98 [DOI] [PubMed] [Google Scholar]
- 39. Yoshikawa T., Shimano H., Yahagi N., Ide T., Amemiya-Kudo M., Matsuzaka T., Nakakuki M., Tomita S., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., Takahashi A., Sone H., Osuga J. I., et al. (2002) Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem. 277, 1705–1711 [DOI] [PubMed] [Google Scholar]
- 40. Botolin D., Wang Y., Christian B., and Jump D. B. (2006) Docosahexaneoic acid (22:6,n-3) regulates rat hepatocyte SREBP-1 nuclear abundance by Erk- and 26S proteasome-dependent pathways. J. Lipid Res. 47, 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki-Kemuriyama N., Matsuzaka T., Kuba M., Ohno H., Han S. I., Takeuchi Y., Isaka M., Kobayashi K., Iwasaki H., Yatoh S., Suzuki H., Miyajima K., Nakae D., Yahagi N., Nakagawa Y., et al. (2016) Different effects of eicosapentaenoic and docosahexaenoic acids on atherogenic high-fat diet-induced non-alcoholic fatty liver disease in mice. PLoS ONE 11, e0157580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kohjima M., Higuchi N., Kato M., Kotoh K., Yoshimoto T., Fujino T., Yada M., Yada R., Harada N., Enjoji M., Takayanagi R., and Nakamuta M. (2008) SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 21, 507–511 [PubMed] [Google Scholar]
- 43. Ide T., Shimano H., Yahagi N., Matsuzaka T., Nakakuki M., Yamamoto T., Nakagawa Y., Takahashi A., Suzuki H., Sone H., Toyoshima H., Fukamizu A., and Yamada N. (2004) SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 6, 351–357 [DOI] [PubMed] [Google Scholar]
- 44. Walter P. L., Steinbrenner H., Barthel A., and Klotz L. O. (2008) Stimulation of selenoprotein P promoter activity in hepatoma cells by FoxO1a transcription factor. Biochem. Biophys. Res. Commun. 365, 316–321 [DOI] [PubMed] [Google Scholar]
- 45. Chen W. L., Chen Y. L., Chiang Y. M., Wang S. G., and Lee H. M. (2012) Fenofibrate lowers lipid accumulation in myotubes by modulating the PPARα/AMPK/FoxO1/ATGL pathway. Biochem. Pharmacol. 84, 522–531 [DOI] [PubMed] [Google Scholar]
- 46. Suchankova G., Tekle M., Saha A. K., Ruderman N. B., Clarke S. D., and Gettys T. W. (2005) Dietary polyunsaturated fatty acids enhance hepatic AMP-activated protein kinase activity in rats. Biochem. Biophys. Res. Commun. 326, 851–858 [DOI] [PubMed] [Google Scholar]
- 47. Yokoyama M., Origasa H., Matsuzaki M., Matsuzawa Y., Saito Y., Ishikawa Y., Oikawa S., Sasaki J., Hishida H., Itakura H., Kita T., Kitabatake A., Nakaya N., Sakata T., Shimada K., Shirato K., Japan EPA lipid intervention study (JELIS) Investigators (2007) Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 369, 1090–1098 [DOI] [PubMed] [Google Scholar]
- 48. Dreher I., Jakobs T. C., and Köhrle J. (1997) Cloning and characterization of the human selenoprotein P promoter: response of selenoprotein P expression to cytokines in liver cells. J. Biol. Chem. 272, 29364–29371 [DOI] [PubMed] [Google Scholar]
- 49. Honda M., Takehana K., Sakai A., Tagata Y., Shirasaki T., Nishitani S., Muramatsu T., Yamashita T., Nakamoto Y., Mizukoshi E., Sakai Y., Yamashita T., Nakamura M., Shimakami T., Yi M., et al. (2011) Malnutrition impairs interferon signaling through mTOR and FoxO pathways in patients with chronic hepatitis C. Gastroenterology 141, 128–140, 140-e2 [DOI] [PubMed] [Google Scholar]
- 50. Nakamura S., Takamura T., Matsuzawa-Nagata N., Takayama H., Misu H., Noda H., Nabemoto S., Kurita S., Ota T., Ando H., Miyamoto K. I., and Kaneko S. (2009) Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 284, 14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S., Gil L., Gordon L., Hendrix M., Hourlier T., Johnson N., et al. (2012) Ensembl 2012. Nucleic Acids Res. 40, D84–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paten B., Herrero J., Beal K., Fitzgerald S., and Birney E. (2008) Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18, 1814–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wingender E. (2008) The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 9, 326–332 [DOI] [PubMed] [Google Scholar]
- 54. Kel A. E., Gössling E., Reuter I., Cheremushkin E., Kel-Margoulis O. V., and Wingender E. (2003) MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]