

Abstract
Cytochrome c oxidase (CcO) is the last electron acceptor in the respiratory chain. The CcO core is formed by mitochondrial DNA-encoded Cox1, Cox2, and Cox3 subunits. Cox1 synthesis is highly regulated; for example, if CcO assembly is blocked, Cox1 synthesis decreases. Mss51 activates translation of COX1 mRNA and interacts with Cox1 protein in high-molecular-weight complexes (COA complexes) to form the Cox1 intermediary assembly module. Thus, Mss51 coordinates both Cox1 synthesis and assembly. We previously reported that the last 15 residues of the Cox1 C terminus regulate Cox1 synthesis by modulating an interaction of Mss51 with Cox14, another component of the COA complexes. Here, using site-directed mutagenesis of the mitochondrial COX1 gene from Saccharomyces cerevisiae, we demonstrate that mutations P521A/P522A and V524E disrupt the regulatory role of the Cox1 C terminus. These mutations, as well as C terminus deletion (Cox1ΔC15), reduced binding of Mss51 and Cox14 to COA complexes. Mss51 was enriched in a translationally active form that maintains full Cox1 synthesis even if CcO assembly is blocked in these mutants. Moreover, Cox1ΔC15, but not Cox1-P521A/P522A and Cox1-V524E, promoted formation of aberrant supercomplexes in CcO assembly mutants lacking Cox2 or Cox4 subunits. The aberrant supercomplex formation depended on the presence of cytochrome b and Cox3, supporting the idea that supercomplex assembly factors associate with Cox3 and demonstrating that supercomplexes can be formed even if CcO is inactive and not fully assembled. Our results indicate that the Cox1 C-terminal end is a key regulator of CcO biogenesis and that it is important for supercomplex formation/stability.
Keywords: cytochrome c oxidase (complex IV), mitochondria, mitochondrial DNA (mtDNA), translation, yeast, Cox1, Mss51, supercomplex
Introduction
Cytochrome c oxidase (CcO)2 is the last redox multisubunit complex of the mitochondrial respiratory chain. This enzyme couples the transference of electrons from cytochrome c to oxygen with the translocation of protons from the matrix to the intermembrane space. In Saccharomyces cerevisiae, CcO contains at least 12 subunits, where Cox1, Cox2, and Cox3 are encoded by mitochondrial genes and constitute the catalytic core. The corresponding mRNAs are translated by the organelle's ribosomes, and the proteins are subsequently integrated into the CcO. The remaining subunits are encoded by nuclear DNA and imported from the cytosol (1, 2). CcO assembly requires at least 30 factors to correctly coordinate the incorporation of subunits and to add prosthetic groups (1, 2). Yeast CcO is assembled after formation of three subassembly modules, each containing a mtDNA-encoded subunit and a subset of cytosolic subunits (3–6). The Cox1 module stabilizes the Cox3 module and possibly the Cox2 module (6), suggesting that assembly of Cox1 orchestrates CcO assembly. One or two monomers of CcO interact with the dimeric bc1 complex to form III2/IV and III2/IV2 supercomplexes (7, 8). Formation and stabilization of these supercomplexes is dependent on the Rcf1, Rcf2, Aac2, and Cox13 proteins (6, 9–11). Cardiolipin (12), and the proteins Rcf3 and Cox26 are also components of supercomplexes (13–15).
Cox1 is the largest CcO subunit, with 12 transmembrane stretches comprising the bulk of the protein. Its only significant hydrophilic domain is formed by ∼59 residues exposed on the inner side of the inner membrane in the assembled enzyme. Cox1 contains hemes a and a3 plus CuB cofactors for oxygen reduction as well as the channels for proton translocation (1, 2). Partially assembled Cox1 may form pro-oxidant intermediaries containing unassembled heme a3-CuB (16). Indeed, yeast Cox1 synthesis is down-regulated when CcO assembly is defective (for reviews, see Refs. 1 and 17).
Translation of the COX1 mRNA in mitochondria is specifically activated by Pet309 and Mss51 (18–22). According to the current model, newly synthesized Cox1 enters a progressive series of intermediaries named D1–D5 (or COA complexes) in which Mss51 is present together with Cox14, Coa3, and Coa1 and subunits Cox4, Cox5, Cox6, and Cox8 (4, 6). In these subassembly complexes, Shy1, Ssc1, and Cox15 are also present (23–25). Progression of CcO assembly promotes release of Mss51 and Ssc1 from the COA complexes (24, 26), making Mss51 available for further translational activation. At this point, Pet54 is required to render Mss51 competent for efficient translational activation of the COX1 mRNA (27). If CcO assembly is blocked, then Cox1 synthesis decreases because Mss51 is sequestered in the COA complexes and thus is unavailable for translational activation (21, 26).
Interaction of the translational activator/assembly factor Mss51 with COA complexes is stabilized by Cox14 and Coa3. Elimination of these factors disrupts the assembly-mediated down-regulation of Cox1 synthesis (21, 26, 28, 29). We previously demonstrated that deletion of the last 11 or 15 C-terminal residues of Cox1 also disrupted the assembly-mediated regulation of Cox1 synthesis, but not the assembly of active CcO. In the cox1 truncation mutants, the interaction of Mss51 with Cox14 was reduced (30), indicating that, together with Cox14 and Coa3, the Cox1 C-terminal residues are necessary for normal sequestration of Mss51 in COA complexes for assembly-mediated regulation of Cox1 synthesis. In the present study, we examined the effects of site-directed mitochondrial mutations affecting conserved residues near the Cox1 C terminus and identified several that have functional importance. We found that this region of Cox1 was not only essential for assembly-mediated regulation of Cox1 synthesis, but also for supercomplex formation and/or stability. The Cox1 C-terminal end is important to stall supercomplex formation/stability when CcO assembly is blocked. Our results point to important participation of the hydrophilic Cox1 C-terminal domain in regulation of CcO biogenesis.
Results
Mutations P521A/P522A and V524E on the Cox1 C-terminal end disrupt Cox1 synthesis regulation
We previously demonstrated that in site-directed mitochondial mutants lacking the last 15 or 11 residues of Cox1, there was no assembly-mediated regulation of Cox1 synthesis. In contrast, deletion of the last 5 residues did not affect the Cox1 assembly-feedback regulatory loop (30). An alignment of the last 15 residues of S. cerevisiae Cox1 with those of several fungi and mammals revealed that at least Pro-521, Pro-522, His-525, Phe-527, and Pro-530 are conserved in fungi and mammals, despite the fact that their deletion does not prevent respiratory growth of yeast (Fig. 1A). To ask whether these residues have a role in the Cox1 C-terminal regulatory function, we integrated mutated versions of COX1 genes encoding the double mutation P521A/P522A or the single point mutations H525A, F527A, and P530A into mtDNA. In addition, we mutated V524E, which is partially conserved in fungi. None of these mutations affected the steady-state levels of Cox1, Cox2, or Cox3 (Fig. 1B) or growth on non-fermentable medium (Fig. 1C) compared with wild type. Spectrophotometric determination of complex IV activities indicated that all of the Cox1 mutants had activity similar to that of the strain carrying wild-type Cox1, with the exception of the Cox1ΔC15 mutant, which retained 30% CcO activity. Complex III activities were similar in all Cox1 mutants and wild-type Cox1 (Fig. 1D).
Figure 1.

Cox1 C-terminal end mutations do not affect CcO activity. A, alignment of the last 15 residues from S. cerevisiae Cox1 with fungal and mammal Cox1 homologues. The alignment was obtained using the Jalview version 2.8.0b1 software (56). Dark gray boxes, highly conserved residues; light gray boxes, partially conserved residues. Asterisks show the amino acids that were mutated in the present study. Numbers indicate position of the last 15 residues of Cox1 in each species. B, mitochondria from wild type and the Cox1 mutants were separated by SDS-PAGE and analyzed by Western blotting using the indicated antibodies. Citrate synthase (CS) was used as loading control. C, 10-fold serial dilutions from wild type and Cox1 mutants were spotted in complete medium with either glucose or ethanol/glycerol. The plates were incubated for 3 days at 30 °C. D, spectrophotometric measurements of complex IV and complex III activities from mitochondria carrying the indicated Cox1 mutations were normalized to the activity of CS. Statistical significance was determined by one-way analysis of variance and Dunnett's test for multiple comparisons with the WT values (***, p < 0.001; n = 3 independent assays) using Prism version 6 software. Error bars, S.D.
To test whether the Cox1 missense mutations affected the assembly-mediated regulation of Cox1 synthesis, we disrupted CcO assembly in the mutants by deletion of either the Cox4 CcO subunit or Pet111, translational activator of the mitochondrially encoded COX2 mRNA (31). Mitochondrial translation products were pulse-labeled in whole cells with [35S]methionine in the presence of cycloheximide, and the products were detected by SDS-PAGE and autoradiography. As expected, wild-type Cox1 labeling decreased upon deletion of cox4 (Fig. 2A) and pet111 (Fig. 2B). In agreement with previous observations, synthesis of truncated Cox1ΔC15 was not reduced by either the pet111 or cox4 deletions (30). Interestingly, the double COX1 missense mutant P521A/P522A and the single mutant V524E also prevented any reduction in Cox1 synthesis when CcO assembly was disrupted by cox4 or pet111 deletions. In contrast, Cox1 mutants H525A, F527A, and P530A did not affect assembly-mediated regulation of Cox1 synthesis because deletion of cox4 or pet111 decreased labeling of those Cox1 variants. These results suggest that the Cox1 residues Pro-521 and/or Pro-522, as well as Val-524, promote interactions required for sequestration of the translational activator/assembly factor Mss51 in COA complexes.
Figure 2.

Mutations Cox1ΔC15, Cox1-P521A/P522A, and Cox1-V524E are important for assembly feedback regulation of Cox1 synthesis. A and B, cells carrying either wild-type or mutated Cox1 were pulse-labeled with [35S]methionine in the presence of cycloheximide. The Cox1 variants were combined with either cox4Δ (A) or pet111Δ (B) mutations, as indicated. Translation products were separated by SDS-PAGE and analyzed by autoradiography. Cox1 labeling intensity is shown at the right side of each panel. It was quantified using the ImageJ software and normalized to the Cox3/Atp6 (ATPase subunit 6) signal. Labeling intensity was expressed as a percentage of the wild-type Cox1 signal. Error bars, S.D. from three independent experiments. We also compared the signals of Cytb and Var1 (ribosomal protein) to the Cox3/Atp6 signal, showing that the observed pattern in A and B is specific for Cox1 (data not shown). The relevant significant differences between strains (*) were determined by Student's t test. A p value of < 0.01 was considered statistically significant.
The Cox1 P521A/P522A and V524A mutants reduce interaction of Mss51 with the COA complexes
Mss51 sequestration in the COA complexes depends upon interactions with Cox14 that occur when Cox1 is newly synthesized (1, 21, 26). The Cox1ΔC15 mutation compromises the interaction between Mss51 and Cox14, as detected by co-immunoprecipitation (30), making free Mss51 more abundant to promote COX1 mRNA translation (27, 32). We therefore investigated whether binding of Mss51 to Cox14 and other components of the COA complexes was similarly compromised in our Cox1 missense mutants. Mss51 and Cox14 in the mutant strains were tagged with triple hemagglutinin (Mss51-HA) or triple c-MYC (Cox14-Myc) epitopes at their C-terminal ends, respectively. The addition of these epitopes did not affect respiratory growth (data not shown). Purified mitochondria carrying wild-type Cox1, Cox1ΔC15, or the Cox1 missense proteins were solubilized with 0.5% dodecyl maltoside. Mss51-HA was immunoprecipitated from the resulting extracts, and co-precipitated proteins were analyzed by SDS-PAGE and Western blotting. As reported previously (30), the co-precipitation of Mss51-HA and Cox14-Myc decreased in the Cox1ΔC15 mutant as compared with Cox1 wild type (Fig. 3A). In addition, interaction of Mss51-HA with Coa3 was also reduced. The missense mutations P521A/P522A and V524E similarly reduced the interactions of Mss51-HA with Cox14-Myc and Coa3 (Fig. 3A). In contrast, the H525A Cox1 mutation, which does not affect assembly-mediated regulation of Cox1 synthesis (Fig. 2), did not reduce co-precipitation of Mss51 with Cox14-Myc, Cox1, or Coa3 (Fig. 3A). We also immunoprecipitated Cox14-Myc from the mitochondrial extracts and analyzed the presence of co-precipitated Mss51-HA, Cox1, and Coa3. As expected, only Cox1ΔC15, Cox1 P521A/P522A, and Cox1 V524E caused decreased interactions with Mss51-HA, Cox1, or Coa3 (Fig. 3B).
Figure 3.

The interaction between Mss51 and the COA complexes decreased in the Cox1-P521A/P522A, Cox1-V524E, and Cox1ΔC15 mutants. A, mitochondria (500 μg of proteins) from wild type and the Cox1 mutants were solubilized with dodecyl maltoside and incubated with an antibody against HA epitope to immunoprecipitate Mss51-HA. A strain without HA tag in Mss51 was used as a negative control. The total and immunoprecipitated (IP) fractions were separated by SDS-PAGE and transferred to a PVDF membrane for Western blotting analysis with the indicated antibodies. Total fractions represent 3% of the mitochondrial extract, whereas all immunoprecipitated protein was loaded onto the gel. B, mitochondrial proteins from wild type or Cox1 mutants were analyzed as in A, but an antibody against Myc was used to immunoprecipitate Cox14-Myc. C, digitonin-solubilized mitochondria from WT and Cox1 mutants were separated by BN-PAGE (5–13%) and transferred to a PVDF membrane for Western blotting analysis with antibodies against HA epitope (α-HA) or Cox1 (α-Cox1). An antibody against ATP synthase was used as loading control (V and V2). Bands corresponding to supercomplexes (SC), COA complexes containing Cox1, and the translational active form of Mss51 (TA) are indicated. All strains carried the Mss51-HA variant. D, mitochondria from C were separated by a BN-PAGE (4–12%) to resolve supercomplexes III2/IV2 and III2/IV. The separated proteins were analyzed by Western blotting using antibody against Cox1 and by CcO in-gel activity. E, digitonin-solubilized mitochondria from WT and Cox1 mutants bearing wild-type PET111 or a pet111Δ mutation were separated by BN-PAGE and analyzed as in C.
Mss51 molecules are present in distinct complexes within mitochondria that are distinguishable by blue-native gel electrophoresis (BN-PAGE) and sedimentation velocity (24, 27, 29, 33, 34). Mss51 is detected in high-molecular-weight COA complexes that correspond to Cox1 assembly intermediates. Mss51 is also detected in lower-molecular-weight complexes that correspond to the translational activator (TA) form of the protein. If the Cox1 missense mutations P521A/P522A and V524E disrupt assembly-mediated regulation of Cox1 synthesis by reducing sequestration of Mss51 in COA complexes, then mutant mitochondria should contain elevated levels of the lower-molecular-weight TA Mss51 complex relative to wild type. To test this idea, we solubilized mitochondria from the different Cox1 mutants with 1% digitonin and then analyzed the extracts by BN-PAGE and Western blotting. Under these conditions, Mss51 co-migrated with high-molecular-weight COA complexes of ∼250–450 kDa, whereas the translational activator form migrated at ∼120–180 kDa. Mitochondria carrying the Cox1ΔC15, P521A/P522A, and V524E alleles showed elevated accumulation of Mss51 in the translational activator form, although Mss51 in COA complexes was also detected. In contrast, mitochondria carrying H525A, F527A, or P530A Cox1 alleles showed distribution of Mss51 complexes similar to that of mitochondria bearing the wild-type Cox1 protein (Fig. 3C). When the same blot was decorated with antibodies against Cox1, respiratory supercomplexes were evident in strains carrying either wild-type or mutant Cox1 proteins, suggesting that supercomplex formation is not affected by impairing the assembly-mediated regulation. An improved separation of supercomplexes by BN-PAGE indicated that in the Cox1ΔC15 mutant, the supercomplex III2/IV is enriched as compared with mitochondria carrying wild-type Cox1, suggesting that in this mutant supercomplex III2/IV2, formation/stability is compromised. Mitochondria from Cox1-P521A/P522A and Cox1-V524E mutants also showed accumulation of supercomplex III2/IV, but to a lesser extent (Fig. 3D).
CcO assembly mutants overaccumulate Mss51 in the COA complexes and underaccumulate Mss51 in translational activator form, decreasing the rate of Cox1 synthesis (24, 27, 29, 33, 34). We examined the distribution of Mss51 in the Cox1 point mutants when CcO assembly was blocked by deletion of Pet111 (causing the absence of Cox2). As expected, disrupting CcO assembly in mitochondria containing wild-type Cox1 caused Mss51 to overwhelmingly accumulate in COA complexes, whereas the translational activator form was barely detectable (Fig. 3E). In contrast, disrupting CcO assembly in mitochondria containing Cox1ΔC15 did not prevent accumulation of the translational activator form of Mss51. The Mss51 translational activator form was also enriched in the absence of assembly by the Cox1 missense mutations P521A/P522A and V524E, albeit to a lesser extent. Taken together, these data indicate that Cox1ΔC15, Cox1-P521A/P522A, and Cox1-V524E mutations compromise the association of Mss51 with the COA complexes when CcO assembly is blocked. Moreover, because the Cox1 mutants affect Cox1 synthesis similarly to Cox1ΔC15 while accumulating less translational activator Mss51 than Cox1ΔC15, our data suggest that the lower levels of translational activator Mss51 are sufficient to support normal synthesis of Cox1. This conclusion is consistent with the finding that overall levels of Mss51 are not strongly rate-limiting for expression of a reporter gene inserted in the COX1 locus (21).
C-terminal truncation of Cox1 (Cox1ΔC15) induces accumulation of aberrant, nonfunctional supercomplexes
Respiratory supercomplexes comprise one or two CcO monomers associated with one bc1 complex dimer. III2/IV and III2/IV2 supercomplexes can be resolved by BN-PAGE of digitonin-solubilized wild-type yeast mitochondria (7, 8). We investigated supercomplex accumulation in mitochondria from the Cox1 mutants in which assembly-mediated regulation of Cox1 synthesis was impaired. Extracts of mitochondria containing wild-type Cox1, Cox1ΔC15, Cox1-P521A/P522A, or Cox1-V524E were separated by BN-PAGE and analyzed by Western blotting using antibodies against Cox1. Mitochondria from all of the Cox1 variants showed the presence of high-molecular-weight bands associated with III2/IV and III2/IV2 supercomplexes (Fig. 4A). Additional bands corresponding to COA complexes and the assembled CcO enzyme (200–450 kDa) were also detected. Therefore, assembly-mediated regulation of Cox1 synthesis is not necessary for respiratory supercomplex formation.
Figure 4.

In the absence of Pet111, Cox1ΔC15 led to formation of a supercomplex-like band in BN-PAGE. A, digitonin-solubilized mitochondria from wild type and Cox1 mutants carrying either wild-type PET111 (WT) or a pet111Δ mutation (Δ) were separated by BN-PAGE (5–13%) as in Fig. 3. In parallel, a second BN gel was stained for CcO activity using diaminobenzidine and cytochrome c (right). The monomeric form of active CcO is indicated (IV). B, mitochondria from A were separated by SDS-PAGE and analyzed by Western blotting using the indicated antibodies. CS was used as a loading control.
As expected, disruption of CcO assembly by a pet111Δ mutation prevented the accumulation of supercomplexes in otherwise wild-type mitochondria, as well as in those containing missense variants of Cox1 (Fig. 4A). Surprisingly, however, disruption of CcO assembly in mitochondria containing the truncated Cox1ΔC15 protein did not prevent accumulation of a high-molecular-weight species exhibiting similar size as normal supercomplexes. This supercomplex-like band was consistently observed in extracts of Cox1ΔC15 mitochondria, but not in extracts of wild-type Cox1 or Cox1-P521A/P522A mitochondria. In some repeats of this experiment, a faint supercomplex-like band was also visible in Cox1-V524E mitochondria. As expected, CcO assembly-competent (PET111) mitochondria exhibited CcO enzymatic activity, whereas the pet111Δ mutants did not (Fig. 4A, right).
Formation of the observed aberrant high-molecular-weight complex could be promoted if C-terminal truncation of Cox1 makes it unusually stable in the absence of CcO assembly. Steady-state accumulation of the mitochondrially encoded CcO subunits was analyzed by SDS-PAGE and Western blotting. The truncated Cox1ΔC15 protein was indeed present at higher levels than wild-type Cox1, Cox1-P521A/P522A, or Cox1-V524E when assembly was disrupted by the pet111Δ mutation (Fig. 4B). Interestingly, the accumulation of unassembled Cox3 was also increased in mitochondria containing Cox1ΔC15 and, to a lesser extent, Cox1-P521A/P522A or Cox1-V524E, compared with wild-type Cox1.
To test whether the aberrant supercomplex-like species actually contain respiratory complex III (the bc1 complex), we removed cytochrome b by deleting the COB mRNA-specific translational activator Cbs2 (35). Mitochondrial extracts were prepared from pet111Δ and pet111Δ/cbs2Δ double mutants containing either wild-type Cox1 or Cox1ΔC15 and subjected to BN-PAGE. Blots were probed with antibodies against Cox1 or cytochrome b (Fig. 5A). Respiratory supercomplexes III2/IV and III2/IV2 containing either wild-type Cox1 or Cox1ΔC15 were detected using both antibodies when assembly of complexes III and IV was allowed to occur (PET111, CBS2). As expected, the supercomplexes containing wild-type Cox1 disappeared when assembly was disrupted by pet111Δ or pet111Δ/cbs2Δ mutations. The aberrant supercomplex-like species containing Cox1ΔC15 that formed in the absence of CcO assembly (pet111Δ) co-migrated with a species containing cytochrome b and disappeared when cytochrome b synthesis was prevented (cbs2Δ), strongly suggesting that it contains complex III in addition to unassembled subunits of CcO, complex IV. To rule out the possibility that the aberrant supercomplex-like species form as a direct result of the absence of Pet111 rather than the absence of Cox2, we also analyzed a Cox1ΔC15 mutant bearing a mitochondrial cox2 deletion (cox2-62 allele (36)). We obtained similar results with cox2-62 mutants, indicating that the presence of the supercomplex-like band is due to the lack of Cox2 rather than the lack of Pet111 itself (data not shown; Fig. 5C).
Figure 5.

Cox1ΔC15-induced formation of non-functional, bc1 complex-dependent supercomplexes in the absence of Cox2. A, digitonin-solubilized mitochondria of WT, pet111Δ, or pet111Δ/cbs2Δ mutants containing Cox1 or Cox1ΔC15 were separated by BN-PAGE (4–12%) and analyzed by Western blotting using antibodies against Cox1 (α-Cox1), cytochrome b (α-Cytb), or ATP synthase. Supercomplexes III2/IV2 and III2/IV are indicated. B, mitochondria from cells carrying wild-type Cox1, COX2, and a plasmid encoding Rcf1 fused to His6 and HA3 (9) were solubilized with digitonin and separated by BN-PAGE (4–12%). A lane of this gel was further resolved by 2D Tricine-SDS-PAGE (12%) and analyzed by Western blotting with the indicated antibodies. C, mitochondria of cox2-62 cells containing Cox1ΔC15 and Rcf1-His6-HA3 were analyzed as in B.
We further investigated the composition of the aberrant supercomplex-like species using a second Tricine-SDS-PAGE dimension after BN-PAGE. To detect Rcf1, the chaperone involved in supercomplex assembly, we transformed cells with a plasmid carrying Rcf1-His6-HA3 (9). Wild-type mitochondria showed clear co-migration of CcO subunits (Cox1, Cox2, and Cox3), complex III subunits (cytochrome b, cytochrome c1, core 1, and Rip1), and Rcf1 in supercomplexes (Fig. 5B). When CcO assembly of mitochondria carrying Cox1ΔC15 was disrupted by the cox2-62 allele, a small fraction of cytochrome b, cytochrome c1, and core 1 subunits co-migrated with the aberrant supercomplex-like species, together with Cox3, Cox1ΔC15, and Rcf1 (Fig. 5C). These results further support the hypothesis that the high-molecular-weight band containing Cox1ΔC15 but lacking Cox2 is an aberrant supercomplex. Rip1, however, was not observed on aberrant supercomplexes, suggesting that the bc1 complex is not fully assembled on these species. Alternatively, the observed high-molecular-weight complexes could represent genuine intermediates in supercomplex assembly that accumulate as a result of the combined deletion of the Cox1 C-terminal end and Cox2.
Cox3 is required for the formation/stability of the aberrant supercomplexes containing Cox1ΔC15
Each mitochondrially encoded CcO subunit interacts with a subset of cytoplasmic subunits to form intermediate preassembly modules (4, 6). In addition, Cox3 physically interacts with Rcf1, a factor that participates in formation of III2/IV and III2/IV2 supercomplexes (6, 9–11). Indeed, Rcf1 interacts with newly synthesized Cox3, probably before assembly (6, 10). Thus, we investigated whether Cox1ΔC15 could induce formation of aberrant supercomplexes in the absence of Cox3. To remove Cox3, we deleted Pet122, one of the COX3 mRNA-specific translational activators (37, 38). We also deleted the Cox4 subunit, which is proposed to interact with either the Cox1 or Cox3 modules (4, 6). Analysis of mitochondrial extracts by BN-PAGE revealed that the aberrant supercomplex containing Cox1ΔC15 failed to accumulate in the absence of Cox3 but did accumulate in the absence of Cox4 (Fig. 6). These data suggest that Cox3 is essential for the formation of aberrant supercomplexes in the Cox1ΔC15 mutant.
Figure 6.

Supercomplex-like bands induced by Cox1ΔC15 are absent in pet122Δ (and thereof Cox3 deletion) but present in cox4Δ mutants. Digitonin-solubilized mitochondria of pet111Δ, pet122Δ, and cox4Δ cells containing either Cox1 or Cox1ΔC15 were separated by BN-PAGE (4–12%) and analyzed by immunoblotting with a Cox1 antibody. ATPase antibodies were used to detect complex V as loading control.
Cox1 hemylation is necessary for formation of aberrant supercomplexes
We investigated whether the addition of heme a to Cox1 was necessary for formation of aberrant supercomplexes. We eliminated Cox15, a chaperone necessary for heme a synthesis (39, 40), from cells carrying either wild-type Cox1, Cox1-P521A/P522A (both strains are controls of cells unable to form aberrant supercomplexes), or Cox1ΔC15. Digitonin-solubilized mitochondrial extracts were analyzed by BN-PAGE and Western blotting. Only trace amounts of aberrant supercomplexes were detectable in the cox15Δ, Cox1ΔC15 strain (Fig. 7A), indicating that hemylation of Cox1 is a requisite to form/stabilize these complexes. This observation could result from the low levels of Cox1 in the absence of Cox15, making it difficult to detect in supercomplexes (Fig. 7B). Interestingly, Cox1ΔC15, cox15Δ mitochondria accumulated slightly higher levels of Cox3 compared with Cox1, cox15Δ mitochondria (Fig. 7B). This is reminiscent of the observation in Fig. 4B, where Cox3 accumulates in Cox1ΔC15, pet111Δ mitochondria. This result suggests that even when Cox1ΔC15 drives accumulation of Cox3, hemylation is necessary to form/stabilize aberrant supercomplexes.
Figure 7.
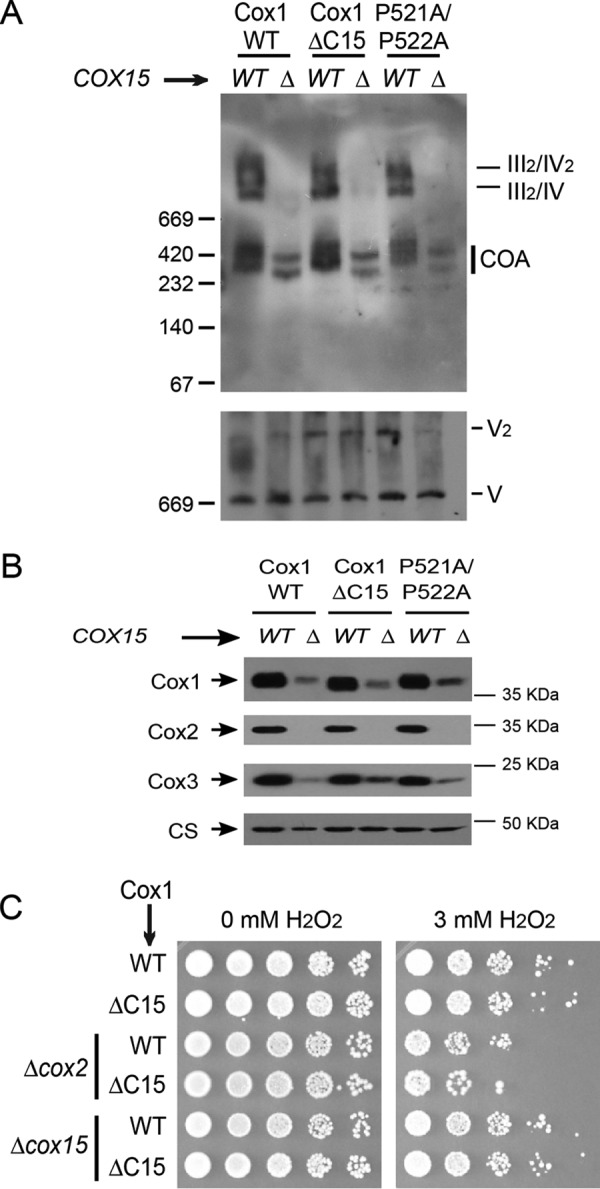
Formation of aberrant supercomplexes requires Cox1 hemylation. A, mitochondria from cells carrying wild-type Cox1, Cox1ΔC15, or Cox1-P521A/P522A and either cox15Δ or wild-type COX15 were separated on BN-PAGE and analyzed as in Fig. 6. B, mitochondria from A were separated by SDS-PAGE and analyzed by Western blotting using the indicated antibodies. CS was used as a loading control. C, cells carrying either the wild-type Cox1 or Cox1ΔC15 and either cox2Δ (allele cox2-62 (36)) or cox15Δ were incubated with 3 mm H2O2 or were mock-treated. 10-fold serial dilutions were grown on YPD plates for 3 days at 30 °C.
Hemylated Cox1 intermediaries produce pro-oxidant species that result in cell sensitivity to H2O2. The peroxide sensitivity is reduced by deletion of Cox1 or inhibition of Cox1 hemylation (16). To test whether Cox1ΔC15 present in aberrant supercomplexes is hemylated, we analyzed the peroxide sensitivity of a strain with the cox2-62 mutation, which drives the formation of aberrant supercomplexes (Cox1ΔC15, cox2-62). This strain was sensitive to 3 mm peroxide, whereas cells carrying Cox1ΔC15 with wild-type COX2 were resistant (Fig. 7C). Moreover, Cox1ΔC15, cox2-62 cells were equally sensitive to H2O2 when compared with a strain carrying wild-type Cox1 and cox2-62. As expected, when Cox15 was deleted from these strains, peroxide resistance increased. Together, these results indicate that Cox1 hemylation is necessary for formation of aberrant supercomplexes and that hemylated Cox1ΔC15 is present in these supercomplexes.
Discussion
Newly synthesized Cox1 plays a central role in the assembly of yeast CcO (3–6), and its synthesis is coupled to assembly at the level of translation (21, 26). CcO assembly and its incorporation into respiratory chain supercomplexes are highly regulated processes that involve a plethora of factors (1, 13–15, 41, 42). In this study, we have examined the effects of altering the sequence of Cox1 C-terminal amino acids on the regulation of its own synthesis, its stability, and the incorporation of CcO into supercomplexes.
We previously demonstrated that deletion of the last 15 residues of the yeast Cox1 C terminus does not prevent assembly of active CcO, as judged by respiratory growth of the mutant, despite the fact that this region includes a number of residues that are highly conserved among fungi and mammals (30). Because these residues do not appear to be critical for function per se, it is likely that these C-terminal Cox1 residues play regulatory roles that confer selective advantage. Indeed, deletion of the last 15 Cox1 residues does impair assembly-mediated regulation of Cox1 synthesis in yeast. Furthermore, this region of the protein is required for a stable association of the translational activator Mss51 with the COA assembly-intermediate complexes during Cox1 biogenesis (30). In the present work, we explored the effects of mutations altering conserved amino acids in this region of Cox1. We identified at least two, and possibly three, missense mutations that similarly impaired regulation of Cox1 synthesis and association of Mss51 with COA complexes: P521A and/or P522A and V524E (Fig. 8 (1)). These mutations also increased the relative amounts of the translational activator form of Mss51, although the effect was less pronounced in the point mutants than in the Cox1ΔC15 truncation. However, mutations altering conserved residues Pro-530, His-525, and Phe-527 to alanine did not detectably alter the regulation of Cox1 synthesis. Thus, because prolines restrict the movement of polypeptide chains (43), it seems likely that Pro-521 and Pro-522 stabilize a conformation of newly synthesized Cox1 that nucleates association of Mss51 and other factors (21) in COA complexes. The nearby V524E mutation could similarly affect a binding structure or simply interfere sterically. In any event, conservation of these residue positions near the Cox1 C terminus in fungi and mammals may be due more to a role for this region in regulation of Cox1 synthesis and assembly than to function of the assembled enzyme.
Figure 8.
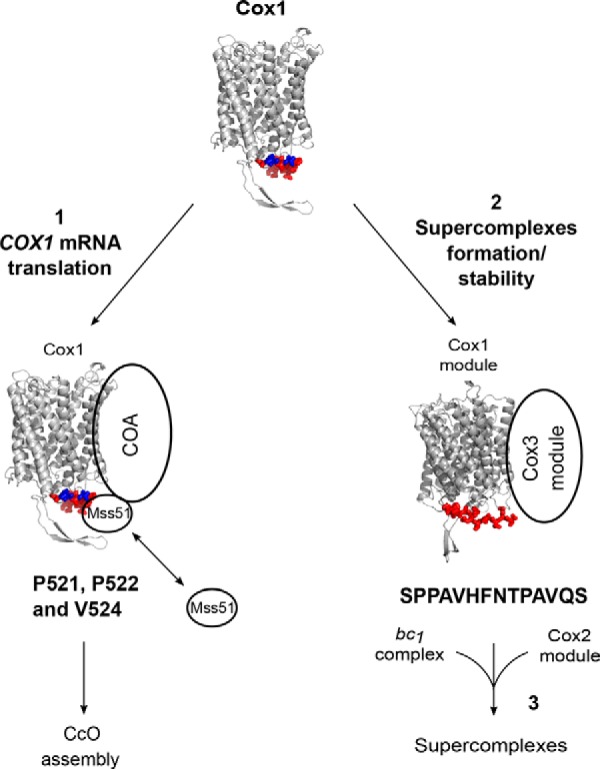
Model depicting the participation of the Cox1 C-terminal end in CcO biogenesis and supercomplex accumulation. A model of S. cerevisiae Cox1 was constructed with SWISS-MODEL (57) based on the crystallographic structure of bovine Cox1 (58). The last 15 residues of Cox1 are indicated in red. Pro-521, Pro-522, and Val-524 are indicated in blue. The last 15 Cox1 residues, and mainly Pro-521/Pro-522 and Val-524, regulate Cox1 synthesis by promoting association of Mss51 with the COA complexes (1). In addition, the Cox1 15-residue C terminus (with sequence SPPAVHFNTPAVQS), rather than Pro-521/Pro-522 and V524E is important to modulate accumulation of respiratory chain supercomplexes (2). According to our results, the Cox1 and Cox3 assembly modules can associate with bc1 complex before the integration of the Cox2 module to form fully functional supercomplexes (3).
Our experiments with the truncated Cox1, lacking the last 15 residues, have revealed new post-translational effects of this mutation, beyond the abrogation of assembly-mediated synthesis control. First, deletion of the C terminus greatly increased the steady-state level of Cox1 under conditions where CcO assembly was blocked by a mutation that prevented Cox2 synthesis. This suggests that degradation of unassembled Cox1 is carried out by proteases that recognize the Cox1 C terminus in defective COA complexes. The increased level of Cox1 under these conditions does not appear to be due to loss of assembly-mediated regulation leading to an increase in Cox1 synthesis because the Cox1-P521A/P522A and Cox1-V524E variants did not have a dramatic effect on the steady-state levels of unassembled Cox1. Interestingly, the Cox1ΔC15 truncation also greatly increased the steady-state level of unassembled Cox3. Assembly of yeast CcO proceeds through parallel formation of three modules, each one containing a mitochondrially encoded subunit and a set of cytoplasmic subunits and chaperones (3–6). Our finding is consistent with the previous observation by others that the Cox1 module stabilizes the Cox3 module (6). The yeast Cox1 C-terminal end also triggers oligomerization of Cox10, the heme o synthase involved in Cox1 hemylation (32).
CcO containing wild-type Cox1 is assembled into respiratory supercomplexes with complex III (bc1) (7, 8). We found that CcO containing truncated Cox1ΔC15 was also assembled into such supercomplexes. Interestingly, we observed that in mitochondria with Cox1ΔC15, the III2/IV supercomplex accumulated, whereas the III2/IV2 supercomplex levels decreased when compared with wild-type mitochondria. This phenotype is similar to that of cells lacking Rcf1, a supercomplex assembly factor (9–11). Pull-down and immunoprecipitation experiments indicate that Cox1, directly or indirectly, interacts with Rcf1 (9–11), suggesting that deletion of the Cox1 C terminus could affect Rcf1 function. In otherwise wild-type cells, disruption of CcO assembly by removal of Cox2 prevented supercomplex formation. However, disruption of CcO assembly in mitochondria synthesizing truncated Cox1ΔC15 led to the accumulation of aberrant supercomplex-like species. Our data demonstrate that these aberrant complexes contain cytochrome b and do not form in its absence, indicating that they contain complex III that associates with preassembly modules of CcO containing Cox1ΔC15 and Cox3. Formation/stability of these aberrant supercomplexes requires hemylation of Cox1. Consistent with this observation, cells that produce the aberrant supercomplexes (Cox1ΔC15, cox2-62) are sensitive to hydrogen peroxide, suggesting that Cox1ΔC15 is hemylated. They also contain Cox3, which is stabilized by the truncated Cox1ΔC15, and do not form in its absence. We propose that the stabilization of unassembled Cox1ΔC15, caused by the C-terminal truncation, leads to aberrant accumulation of CcO preassembly modules in the absence of Cox2, which are then able to associate with complex III to form the aberrant supercomplex-like species we observed (Fig. 8 (2)).
The exact composition of the non-functional, aberrant supercomplexes remains to be determined, whereas it is possible that the bc1 dimer is not fully assembled in them. In our experiments, Rip1 was not detected in these complexes, but core 1 and cytochrome c1 subunits were present, suggesting that bc1 complex is not fully assembled. Indeed, rip1Δ, qcr9Δ, and bcs1Δ mutants accumulate a late-assembly intermediary of bc1 complex, which can interact with assembled CcO to form non-functional supercomplexes (44). CcO assembly chaperones such as Shy1, Pet54, Cox14, Coa3, Cox11, and Cox16 interact with bc1 subunits, probably during assembly of supercomplexes (11, 25, 29). Rcf1, which was present in the aberrant supercomplexes, facilitates formation or stabilization of III2/IV and III2/IV2 supercomplexes and interacts with Cox3 (6, 9–11). This is in agreement with our observation that in the absence of Cox3, the Cox1ΔC15 mutant does not form aberrant supercomplexes, indicating that Cox3 is important for their formation. Thus, the assembly of CcO and bc1 complexes may substantially be interconnected in wild type.
Our data indicate that the Cox1 and Cox3 preassembly modules are able to interact with the bc1 complex in the absence of the Cox2 module (Fig. 8 (3)). Thus, it is possible that in wild type, the Cox2 module could integrate with partially assembled supercomplexes to complete the assembly of CcO. Consistent with this possibility, rapid interactions between newly synthesized pulse-labeled Cox1 and Cox3 are detectable, before interactions of Cox1 with Cox2 and interactions of Cox1 with cytochrome b, which exhibit similar kinetics (4). Interestingly, it was reported that a C-terminal deletion in the human Cox1 was able to assemble in CcO and to form supercomplexes, although these complexes were highly unstable because Cox1 was rapidly degraded (45). It is likely that the Cox1 C terminus is a key regulator of CcO biogenesis in different organisms. As demonstrated in the present work, this region of the protein contains signals that regulate formation/stability of supercomplexes, depending on the assembly state of the CcO.
Experimental procedures
Strains, media, and genetic methods
S. cerevisiae strains used in this study are congenic or isogenic to D273-10B (ATCC24657) and are listed in Table 1. Standard genetic procedures and recipes for media were as described elsewhere (46, 47). Complete fermentable media were YPD or YPGal (1% yeast extract, 2% Bacto-peptone, and 2% glucose or 2% galactose) or synthetic complete media (0.67% yeast nitrogen base, 2% glucose) lacking the indicated amino acids. Non-fermentable medium was YPEG (1% yeast extract, 2% Bacto-peptone, 3% ethanol, and 3% glycerol). The nuclear deletion constructs with URA3, LEU2, or KanMX4 were obtained by PCR. Plasmids containing the cox1 point mutations were transformed by high-velocity microprojectile bombardment into the rho0 strain NAB69 (48). Transformants were selected by their ability to rescue respiratory growth when mated with the strain L74 carrying a G124D mutation in Cox1 (49). Transformants with the cox1 mutant plasmids were mated with XPM10b (containing cox1Δ::ARG8m construct), and cytoductants were selected for their ability to grow in YEPG as haploids. Correct integration of the COX1 constructs into mtDNA was confirmed by PCR and DNA sequencing.
Table 1.
List of strains used in this study
All of these strains are congenic or isogenic to D273-10B. Mitochondrial genotypes are shown in parenthesis. ΔΣai refers to an intronless COX1 gene.
| Strain | Genotype nuclear (mitochondrial) | Reference/source |
|---|---|---|
| LSR2 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2 (ρ+, ΔΣai, COX1ΔC15) | Ref. 30 |
| TF258 | Matα, lys2, ura3-52 or Δ, his4-519, leu2-3, 112, MSS51::3xHA, COX14::3xMYC, (ρ+, ΔΣai) | Ref. 21 |
| TF272 | Matα, lys2, ura3-52 or Δ, his4-519, leu2-3, 112, MSS51::3xHA, COX14::3xMYC, (ρ+, ΔΣai, COX1ΔC15) | Ref. 21 |
| JPM49 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet122Δ::KANMX (ρ+, ΔΣai) | Ref. 27 |
| JPM57 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet122Δ::KANMX (ρ+, ΔΣai, COX1ΔC15) | Ref. 27 |
| YC146 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, cox4Δ::LEU2, (ρ+, ΔΣai) | This work |
| YC151 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA,cox4Δ::LEU2, (ρ+, ΔΣai, COX1ΔC15) | This work |
| XPM51 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2 (ρ+, ΔΣai) | Ref. 30 |
| XPM201 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai) | Ref. 30 |
| XPM202 | Matα, lys2, arg8::hisG, ura3-52, his3::HindIII, (ρ+, ΔΣai, cox2-62) | Ref. 30 |
| XPM209 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1ΔC15) | Ref. 30 |
| XPM210 | Matα, lys2, arg8::hisG, ura3-52, his3::HindIII, (ρ+, ΔΣai, COX1ΔC15, cox2-62) | Ref. 30 |
| XPM295 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai) | Ref. 30 |
| XPM298 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1ΔC15) | Ref. 30 |
| MS62 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| MS63 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1-V524E) | This work |
| MS64 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1-H525A) | This work |
| MS65 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1-F527A) | This work |
| MS66 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, (ρ+, ΔΣai, COX1-P530A) | This work |
| RGV22 | Matα, lys2, ura3-52 or Δ, his4-519, leu2-3, 112, MSS51::3xHA, COX14::3xMYC, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| RGV23 | Matα, lys2, ura3-52 or Δ, his4-519, leu2-3, 112, MSS51::3xHA, COX14::3xMYC, (ρ+, ΔΣai, COX1-V524E) | This work |
| RGV24 | Matα, lys2, ura3-52 or Δ, his4-519, leu2-3, 112, MSS51::3xHA, COX14::3xMYC, (ρ+, ΔΣai, COX1-H525A) | This work |
| RGV73 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, (ρ+, ΔΣai) | This work |
| RGV74 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, (ρ+, ΔΣai, COX1ΔC15) | This work |
| RGV75 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1-H525A) | This work |
| RGV77 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| RGV78 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1-V524E) | This work |
| RGV79 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1-F527A) | This work |
| RGV80 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, (ρ+, ΔΣai, COX1-P530A) | This work |
| RGV95 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| RGV96 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2, (ρ+, ΔΣai, COX1-H525A) | This work |
| RGV97 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2, (ρ+, ΔΣai, COX1-F527A) | This work |
| RGV98 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2, (ρ+, ΔΣai, COX1-P530A) | This work |
| RGV99 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, cox4Δ::LEU2, (ρ+, ΔΣai, COX1-V524E) | This work |
| RGV101 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai) | This work |
| RGV102 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1ΔC15) | This work |
| RGV103 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| RGV104 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1-V524E) | This work |
| RGV105 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1-H525A) | This work |
| RGV106 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1-F527A) | This work |
| RGV107 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, pet111Δ::URA3, (ρ+, ΔΣai, COX1-P530A) | This work |
| RGV108 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, (ρ+, ΔΣai, COX1-V524E) | This work |
| RGV109 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, (ρ+, ΔΣai, COX1-P521A/P522A) | This work |
| RGV112 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, cbs2Δ::KanMX4, (ρ+, ΔΣai) | This work |
| RGV113 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, pet111Δ::URA3, cbs2Δ::KanMX4, (ρ+, ΔΣai, COX1ΔC15) | This work |
| YCV163 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, cox15Δ::LEU2, (ρ+, ΔΣai, COX1-PP521AA) | This work |
| YCV164 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, cox15Δ::LEU2, (ρ+, ΔΣai) | This work |
| YCV165 | Matα, lys2, arg8::hisG, ura3-52, leu2-3, 112, MSS51-3xHA, cox15Δ::LEU2, (ρ+, ΔΣai, COX1ΔC15, cox2-62) | This work |
Construction of cox1 mutant genes
The 3′ half of the COX1 coding region was amplified with primers that incorporated the mutations encoding P521A/P522A, V524E, H525A, F527A, or P530A. As template, we used plasmid pXPM57 containing the full-length, intronless COX1 gene (30). These products were digested with NdeI and AflII and ligated into pXPM57 equally digested. Mutations were confirmed by PCR and DNA sequencing.
Analysis of mitochondrial proteins
Yeast cells were grown in YPGal to late exponential phase. Crude mitochondria were obtained by treating the cells with Zymolyase 20T or with glass bead disruption, as described elsewhere (50). Immunoprecipitation was performed as described previously (30). Briefly, mitochondria (500 μg) were solubilized with 0.5% (w/v) dodecyl maltoside and centrifuged, and the cleared supernatant was incubated with anti-HA or anti-Myc antibodies coupled to protein A-Sepharose beads. Total (3% of sample) and immunoprecipitated fractions were separated by SDS-PAGE and transferred to PVDF membrane. Western blots were probed with antibodies against HA (Roche Applied Science), Myc (Roche Applied Science), Cox1, Coa3 (A. Barrientos), Cox2 (Mitosciences), Cox3 (Mitosciences), and citrate synthase. In vivo pulse labeling of cells or in organello labeling of crude mitochondria with [35S]Methionine was performed as described previously (51). The radiolabeled proteins were separated by SDS-PAGE, and the gel was dried before analysis with a Typhoon 8600 PhosphorImager (GE Healthcare).
BN-PAGE and 2D Tricine-SDS-PAGE
BN-PAGE and 2D Tricine-SDS-PAGE were performed as described previously (52). The mitochondrial pellet (100 μg of proteins) was resuspended in 50 μl of sample buffer (750 mm 6-aminocaproic acid, 50 mm BisTris, pH 7, 1% digitonin) and centrifuged. The supernatant was loaded in either 5–13% or 4–12% polyacrylamide gradient gels. When indicated, BN-PAGE lanes were excised and separated by 2D Tricine-SDS-PAGE using 12% polyacrylamide gels. Gels were transferred to PVDF membranes and probed with antibodies against HA (Roche Applied Science), Cox1, Cox2 (Mitosciences), Cox3 (Mitosciences), Cytb, and ATPase (Diego González-Halphen). The high-mass protein markers were obtained from GE Healthcare. In-gel CcO activity was performed after BN-PAGE using 0.04% diaminobenzidine (Sigma-Aldrich) and 0.02% horse heart cytochrome c (Sigma-Aldrich) in phosphate buffer, pH 7.4 (53).
Spectrophotometric enzyme assays
Complex III and complex IV activities were determined by following the absorbance changes resulting from the reduction and oxidation of cytochrome c, respectively, at 550 nm (ϵ550 nm =18.5 mm−1·cm−1) in a SPECTRAmax PLUS384 microplate reader (Molecular Devices) according to Ref. 54. Briefly, complex III activities were measured in a buffer containing 25 mm potassium phosphate (pH 7.5), 75 μm oxidized cytochrome c from horse heart, 0.1 mm EDTA, 1 mm sodium cyanide, and 10 μg of protein/ml of mitochondrial membranes. The reaction was started with 100 μm decylubiquinol. Complex IV activities were measured in a buffer containing 25 mm potassium phosphate (pH 7) and 50 μm reduced cytochrome c from horse heart. The reaction was started by adding mitochondrial membranes to a final concentration of 10 μg of protein/ml. Citrate synthase (CS) activity was measured at 412 nm (ϵ = 13.6 mm−1 cm−1) in a buffer containing 10 mm Tris-HCl (pH 8), 0.3 mm acetyl-CoA, 0.1 mm 5,5′-dithio-bis-2-nitrobenzoic acid, 0.1% Triton X-100, and 10 μg of protein/ml of mitochondrial membranes. The reaction was started with 0.5 mm oxaloacetate (55).
Peroxide sensitivity assays
Cells were grown overnight on YPD until exponential phase, at an A600 of 0.6. H2O2 was added to a 3 mm final concentration, and cultures (10 mg/ml of wet weight cell pellet) were incubated for 2 h at 30 °C with gentle agitation. Alternatively, mock-treated cells were incubated with H2O and treated equally. Cells were washed with sterile water and plated on YPD in 10-fold serial dilutions for 2 days at 30 °C.
Author contributions
X. P.-M. conceived and coordinated the study, analyzed the results, and wrote the manuscript. R. G.-V. performed and analyzed the experiments from Figs. 1–7 and contributed to the writing of the manuscript. M. A. S.-V. and T. D. F. created the mitochondrial Cox1 point mutants. T. D. F. also contributed to the writing of the manuscript. Y. C.-V. constructed yeast strains, performed the experiment in Fig. 7C, and provided technical assistance for all of the experiments. A. C.-O. and S. U.-C. bioenergetically characterized the Cox1 mutants. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Luisa Sandoval-Romero, Jared Rutter, Gabriel del Río-Guerra, and Teresa Lara-Ortíz for the gift of yeast strains and plasmids; Miriam Vázquez-Acevedo, Liesbeth Wintjes Guadalupe Códiz-Huerta, Minerva Mora-Cabrera, and Laura Ongay-Larios for technical assistance; Antoni Barrientos, Rosemary Stuart, and Diego González-Halphen for the gift of antisera; Prof. Dr. Ulrich Brandt for kindly sharing equipment; and Claudia Rivera-Cerecedo and Héctor Malagón-Rivero for technical assistance obtaining antisera.
This work was supported by Consejo Nacional de Ciencia y Tecnología Research Grant 47514 (to X. P.-M.) and Fellowships 250726 (to R. G.-V.) and 289024 (to A. C.-O.) and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT) UNAM Grants IN204414 and IN209217 (to X. P.-M.). This work is part of the Ph.D. thesis of R. G.-V. from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México. The authors declare that they have no conflicts of interest with the contents of this article.
- CcO
- cytochrome c oxidase
- BN-PAGE
- blue-native gel electrophoresis
- TA
- translational activator
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- CS
- citrate synthase.
References
- 1. Mick D. U., Fox T. D., and Rehling P. (2011) Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 12, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soto I. C., Fontanesi F., Liu J., and Barrientos A. (2012) Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 1817, 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McStay G. P., Su C. H., Thomas S. M., Xu J. T., and Tzagoloff A. (2013) Characterization of Cox1p assembly intermediates in Saccharomyces cerevisiae. J. Biol. Chem. 288, 26546–26556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McStay G. P., Su C. H., and Tzagoloff A. (2013) Modular assembly of yeast cytochrome oxidase. Mol. Biol. Cell 24, 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McStay G. P., Su C. H., and Tzagoloff A. (2013) Stabilization of Cox1p intermediates by the Cox14p-Coa3p complex. FEBS Lett. 587, 943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Su C. H., McStay G. P., and Tzagoloff A. (2014) The Cox3p assembly module of yeast cytochrome oxidase. Mol. Biol. Cell 25, 965–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cruciat C. M., Brunner S., Baumann F., Neupert W., and Stuart R. A. (2000) The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275, 18093–18098 [DOI] [PubMed] [Google Scholar]
- 8. Schägger H., and Pfeiffer K. (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y. C., Taylor E. B., Dephoure N., Heo J. M., Tonhato A., Papandreou I., Nath N., Denko N. C., Gygi S. P., and Rutter J. (2012) Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 15, 348–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strogolova V., Furness A., Robb-McGrath M., Garlich J., and Stuart R. A. (2012) Rcf1 and Rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol. Cell. Biol. 32, 1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vukotic M., Oeljeklaus S., Wiese S., Vögtle F. N., Meisinger C., Meyer H. E., Zieseniss A., Katschinski D. M., Jans D. C., Jakobs S., Warscheid B., Rehling P., and Deckers M. (2012) Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 15, 336–347 [DOI] [PubMed] [Google Scholar]
- 12. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., and Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 13. Levchenko M., Wuttke J. M., Römpler K., Schmidt B., Neifer K., Juris L., Wissel M., Rehling P., and Deckers M. (2016) Cox26 is a novel stoichiometric subunit of the yeast cytochrome c oxidase. Biochim. Biophys. Acta 1863, 1624–1632 [DOI] [PubMed] [Google Scholar]
- 14. Römpler K., Müller T., Juris L., Wissel M., Vukotic M., Hofmann K., and Deckers M. (2016) Overlapping role of respiratory supercomplex factor Rcf2 and its N-terminal homolog Rcf3 in Saccharomyces cerevisiae. J. Biol. Chem. 291, 23769–23778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strecker V., Kadeer Z., Heidler J., Cruciat C. M., Angerer H., Giese H., Pfeiffer K., Stuart R. A., and Wittig I. (2016) Supercomplex-associated Cox26 protein binds to cytochrome c oxidase. Biochim. Biophys. Acta 1863, 1643–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khalimonchuk O., Bird A., and Winge D. R. (2007) Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J. Biol. Chem. 282, 17442–17449 [DOI] [PubMed] [Google Scholar]
- 17. Ott M., Amunts A., and Brown A. (2016) Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 85, 77–101 [DOI] [PubMed] [Google Scholar]
- 18. Decoster E., Simon M., Hatat D., and Faye G. (1990) The MSS51 gene product is required for the translation of the COX1 mRNA in yeast mitochondria. Mol. Gen. Genet. 224, 111–118 [DOI] [PubMed] [Google Scholar]
- 19. Manthey G. M., and McEwen J. E. (1995) The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J. 14, 4031–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez-Martinez X., Broadley S. A., and Fox T. D. (2003) Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 22, 5951–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perez-Martinez X., Butler C. A., Shingu-Vazquez M., and Fox T. D. (2009) Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol. Biol. Cell 20, 4371–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zamudio-Ochoa A., Camacho-Villasana Y., García-Guerrero A. E., and Pérez-Martínez X. (2014) The Pet309 pentatricopeptide repeat motifs mediate efficient binding to the mitochondrial COX1 transcript in yeast. RNA Biol. 11, 953–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bareth B., Dennerlein S., Mick D. U., Nikolov M., Urlaub H., and Rehling P. (2013) The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell. Biol. 33, 4128–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fontanesi F., Soto I. C., Horn D., and Barrientos A. (2010) Mss51 and Ssc1 facilitate translational regulation of cytochrome c oxidase biogenesis. Mol. Cell. Biol. 30, 245–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mick D. U., Wagner K., van der Laan M., Frazier A. E., Perschil I., Pawlas M., Meyer H. E., Warscheid B., and Rehling P. (2007) Shy1 couples Cox1 translational regulation to cytochrome c oxidase assembly. EMBO J. 26, 4347–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barrientos A., Zambrano A., and Tzagoloff A. (2004) Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 23, 3472–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mayorga J. P., Camacho-Villasana Y., Shingú-Vázquez M., García-Villegas R., Zamudio-Ochoa A., García-Guerrero A. E., Hernández G., and Pérez-Martínez X. (2016) A novel function of Pet54 in regulation of Cox1 synthesis in Saccharomyces cerevisiae mitochondria. J. Biol. Chem. 291, 9343–9355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fontanesi F., Clemente P., and Barrientos A. (2011) Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 286, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mick D. U., Vukotic M., Piechura H., Meyer H. E., Warscheid B., Deckers M., and Rehling P. (2010) Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 191, 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shingú-Vázquez M., Camacho-Villasana Y., Sandoval-Romero L., Butler C. A., Fox T. D., and Pérez-Martínez X. (2010) The carboxyl-terminal end of Cox1 is required for feedback assembly regulation of Cox1 synthesis in Saccharomyces cerevisiae mitochondria. J. Biol. Chem. 285, 34382–34389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poutre C. G., and Fox T. D. (1987) PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics 115, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khalimonchuk O., Kim H., Watts T., Perez-Martinez X., and Winge D. R. (2012) Oligomerization of heme o synthase in cytochrome oxidase biogenesis is mediated by cytochrome oxidase assembly factor Coa2. J. Biol. Chem. 287, 26715–26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khalimonchuk O., Bestwick M., Meunier B., Watts T. C., and Winge D. R. (2010) Formation of the redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase. Mol. Cell. Biol. 30, 1004–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pierrel F., Bestwick M. L., Cobine P. A., Khalimonchuk O., Cricco J. A., and Winge D. R. (2007) Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 26, 4335–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rödel G. (1986) Two yeast nuclear genes, CBS1 and CBS2, are required for translation of mitochondrial transcripts bearing the 5′-untranslated COB leader. Curr. Genet. 11, 41–45 [DOI] [PubMed] [Google Scholar]
- 36. Dunstan H. M., Green-Willms N. S., and Fox T. D. (1997) In vivo analysis of Saccharomyces cerevisiae COX2 mRNA 5′-untranslated leader functions in mitochondrial translation initiation and translational activation. Genetics 147, 87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kloeckener-Gruissem B., McEwen J. E., and Poyton R. O. (1988) Identification of a third nuclear protein-coding gene required specifically for posttranscriptional expression of the mitochondrial COX3 gene is Saccharomyces cerevisiae. J. Bacteriol. 170, 1399–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown N. G., Costanzo M. C., and Fox T. D. (1994) Interactions among three proteins that specifically activate translation of the mitochondrial COX3 mRNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 14, 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barros M. H., Carlson C. G., Glerum D. M., and Tzagoloff A. (2001) Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 492, 133–138 [DOI] [PubMed] [Google Scholar]
- 40. Glerum D. M., Muroff I., Jin C., and Tzagoloff A. (1997) COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J. Biol. Chem. 272, 19088–19094 [DOI] [PubMed] [Google Scholar]
- 41. Fontanesi F., Soto I. C., and Barrientos A. (2008) Cytochrome c oxidase biogenesis: new levels of regulation. IUBMB Life 60, 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chaban Y., Boekema E. J., and Dudkina N. V. (2014) Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 1837, 418–426 [DOI] [PubMed] [Google Scholar]
- 43. Kay B. K., Williamson M. P., and Sudol M. (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14, 231–241 [PubMed] [Google Scholar]
- 44. Cui T. Z., Conte A., Fox J. L., Zara V., and Winge D. R. (2014) Modulation of the respiratory supercomplexes in yeast: enhanced formation of cytochrome oxidase increases the stability and abundance of respiratory supercomplexes. J. Biol. Chem. 289, 6133–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hornig-Do H. T., Tatsuta T., Buckermann A., Bust M., Kollberg G., Rötig A., Hellmich M., Nijtmans L., and Wiesner R. J. (2012) Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 31, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burke D., Dawson D., and Stearns T. (2000) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 47. Guthrie C., and Fink G. R. (eds) (2002) Guide to Yeast Genetics and Molecular and Cell Biology, Vol. 350, pp. 3–42, Academic Press, Inc., San Diego [Google Scholar]
- 48. Bonnefoy N., Remacle C., and Fox T. D. (2007) Genetic transformation of Saccharomyces cerevisiae and Chlamydomonas reinhardtii mitochondria. Methods Cell Biol. 80, 525–548 [DOI] [PubMed] [Google Scholar]
- 49. Meunier B., Lemarre P., and Colson A. M. (1993) Genetic screening in Saccharomyces cerevisiae for large numbers of mitochondrial point mutations which affect structure and function of catalytic subunits of cytochrome-c oxidase. Eur. J. Biochem. 213, 129–135 [DOI] [PubMed] [Google Scholar]
- 50. Diekert K., de Kroon A. I., Kispal G., and Lill R. (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65, 37–51 [DOI] [PubMed] [Google Scholar]
- 51. Westermann B., Herrmann J. M., and Neupert W. (2001) Analysis of mitochondrial translation products in vivo and in organello in yeast. Methods Cell Biol. 65, 429–438 [DOI] [PubMed] [Google Scholar]
- 52. Wittig I., Braun H. P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 53. Wittig I., Karas M., and Schägger H. (2007) High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteomics 6, 1215–1225 [DOI] [PubMed] [Google Scholar]
- 54. Spinazzi M., Casarin A., Pertegato V., Salviati L., and Angelini C. (2012) Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7, 1235–1246 [DOI] [PubMed] [Google Scholar]
- 55. Barrientos A., Fontanesi F., and Diaz F. (2009) Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. Genet. 10.1002/0471142905.hg1903s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., and Yoshikawa S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272, 1136–1144 [DOI] [PubMed] [Google Scholar]